
Abstract
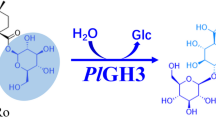
An eco-friendly and convenient preparation method for notoginsenoside ST-4 has been established by completely transforming vina-ginsenoside R7 using a recombinant glycosidase hydrolyzing enzyme (HaGH03) from Herpetosiphon aurantiacus. This enzyme specifically hydrolyzed the glucose at the C-20 position but not the external xylose or two inner glucoses at position C-3. Protein sequence BLAST revealed that HaGH03, composed of 749 amino acids and presumptively listed as a member of the family 3 glycoside hydrolases, has highest identity (48 %) identity with a thermostable β-glucosidase B, which was not known of any functions for ginsenoside transformation. The steady state kinetic parameters for purified HaGH03 measured against p-nitrophenyl β-D-glucopyranoside and vina-ginsenoside R7 were K M = 5.67 ± 0.24 μM and 0.59 ± 0.23 mM, and k cat = 69.2 ± 0.31/s and 2.15 ± 0.46/min, respectively. HaGH03 converted 2.5 mg/mL of vina-ginsenoside R7 to ST-4 with a molar yield of 100 % and a space-time yield of 104 mg/L/h in optimized conditions. These results underscore that HaGH03 has much potential for the effective preparation of target ginsenosides possessing valuable pharmacological activities. This is the first report identifying an enzyme that has the ability to transform vina-ginsenoside R7 and provides an approach to preparing rare notoginsenoside ST-4.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
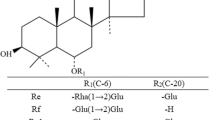
Ginsenosides are a group of dammarane-type triterpenoid saponins mainly found in plants of the genus Panax (Araliaceae family), such as Panax ginseng, Panax quinquefolium, Panax notoginseng, and Panax vietnamensis. Ginsenosides show a wide spectrum of pharmacological activities, including in antihypertension, cardioprotection, immunomodulation, and as an antiatherosclerotic (Attele et al. 1999; Ng 2006; Lee and Kim 2014). Ginsenosides are categorized into two major classes according to the aglycone position, shown in Fig. 1, namely protopanaxadiols (PPDs) with sugar moieties attached to positions C-3 and/or C-20, and protopanaxatriols (PPTs) with attachment at C-3 and/or C-6, and/or at C-20 (Wang et al. 2006; Dan et al. 2008; Liu 2012).

Chemical structures of ginsenosdies with PPD (Nos. 1-11) and PPT types (Nos. 12-19). Glc, β-D-glucopyranosyl; Xyl, β-D-xylopyranosyl; Rha, α-L-rhamnopyranosyl; Ara(p), α-L-arabinopyranosyl; and Ara(f), α-L-arabinofuranosyl. Values represent the mean of three replicates ± standard deviation
Recently, rare deglycosylated ginsenosides have attracted much attention owing to more potent physiological activities in vivo and favorable physical and/or chemical properties in terms of crossing cell membranes (Tawab et al. 2003). However, the rare ginsenoside content is extremely low or undetectable in raw plant materials, which imposes restrictions on their availability and exploitation of their novelty and pharmacological activities. During the past few decades, great efforts have been made in accomplishing large-scale preparation of rare ginsenosides by means of chemical hydrolysis, heating, and microbial/enzymatic conversion, in attempts to discover new compounds and potential drug candidates. In terms of specificity and high productivity, microbial or enzymatic methods have proved most effective and provided insight into obtaining desirable ginsenosides and other valuable chemicals of natural origin by selective cleavage of specific sugar moieties (Su et al. 2006, 2009; Chang et al. 2009; Ye et al. 2010; Park et al. 2010; Groussin and Antoniotti 2012; Jin et al. 2013; Liu et al. 2014). Many ginsenoside-hydrolyzing enzymes, mainly belonging to the glycoside hydrolase family (EC 3.2.1.-), have been identified, expressed, and applied to the transformation of predominant ginsenosides into rare ones. Recombinant ginsenoside-hydrolyzing enzymes are briefly summarized in Table 1 and are most frequently used to transform Rg1, Rg2, Rg3, Re, Rc, Rb1, Rb2, Rd, Rf, and R1.
Notoginsenoside ST-4, a promising agent against herpes simplex viral infection, was isolated from steam-treated notoginseng with a yield of just 0.00066 % (Pei et al. 2011). No other pharmacological activities of this rare notoginsenoside have been investigated yet because of the extremely cumbersome preparation process and the trace yield. In our previous work, notoginsenoside Ft1, the C-20 isomer of ST-4, was shown to have activities in enhancing platelet aggregation (Gao et al. 2014a), promoting angiogenesis (Shen et al. 2012), activating both glucocorticoid and estrogen receptors (Shen et al. 2014), and having a pro-apoptotic effect on human neuroblastoma SH-SY5Y cells (Gao et al. 2014b). It should therefore be of interest to determine the biological effects of ST-4 and elucidate the structure-activity relationships of both ST-4 and Ft1. However, there is no convenient approach to prepare notoginsenoside ST-4 on a batch scale. Of the reported ginsenosides, vina-ginsenoside R7 is undoubtedly the best choice as a substrate for biotransformation into the target compound ST-4. First, vina-ginsenoside R7 was identified as being of relatively high content in Vietnam ginseng and notoginseng (Minh et al. 1994; Wang et al. 2008). Second, and more importantly, it has a similar structure to notoginsenoside ST-4, except for a glucose substituted at the C-20 position. To the best of our knowledge, neither enzymes nor microorganisms have been investigated for the biotransformation of vina-ginsenoside R7 into notoginsenoside ST-4. Consequently, in this work, an eco-friendly and convenient preparation method for this conversion has been established using a recombinant, newly identified ginsenosidase from Herpetosiphon aurantiacus. Detailed enzymatic properties of this ginsenosidase were characterized to establish its substrate specificity and the substrate spectrum.
Materials and methods
Chemicals and materials
Vina-ginsenoside R7, notoginsenoside ST-4, and other ginsenoside standards were supplied by the Shanghai R&D Centre for Standardization of Traditional Chinese Medicine (Shanghai, China). Methanol and acetonitrile of HPLC grade were obtained from Fisher Scientific Co. (Santa Clara, CA, USA). p-Nitrophenyl (pNP), β- D -glucopyranoside, and oNP-β-D-galactopyranoside were purchased from Sigma (St. Louis, MO, USA). Deionized water was prepared by the Milli-Q system (Millipore, Bedford, MA, USA). All other chemicals and reagents were of analytical grade or better.
Molecular cloning, expression, and purification
H. aurantiacus DSM 785 (DSMZ, Brauschweig, Germany), Escherichia coli DH5α, and E. coli BL21 (DE3) were used as the source of the β-glycosidase gene, the host for gene cloning, and the host for recombinant protein expression, respectively. The gene encoding the ginsenoside hydrolase (2250 bp) was amplified by polymerase chain reaction (PCR) using the following primers with BamHI and HindIII restriction sites (in italics): forward 5′-CGCGGATCCATGACCGCGAGCGATCAAC-3′ and reverse 5′-CCCAAGCTTCTAGCCCTGATTGACCTTGGC-3′. The amplified DNA fragment was purified, digested with appropriate restriction enzymes, and ligated into the expression vector pET-28a (Novagen).
E. coli BL21 (DE3) strains harboring the expression plasmid were grown in LB medium containing kanamycin (50 μg/mL) at 37 °C to an absorbance at 600 nm (OD600) of 0.4. Expression was induced with isopropyl-β-D-thiogalactopyranoside (IPTG) at a final concentration of 0.2 mM for 20 h at 16 °C, before centrifugation (12,000×g at 4 °C for 10 min). Harvested cells were washed two times using 0.9 % (w/v) sodium chloride, suspended in 20 mM sodium phosphate buffer (pH 7.4, 500 mM NaCl, 20 mM imidazole), and disrupted by ultrasonication. Cell debris was removed two times by centrifugation (15,000×g at 4 °C for 25 min). Purification of the N-terminal His-tagged fusion protein was executed using a His trap Ni–NTA FF column (GE Healthcare), which was eluted with a gradient of imidazole from 20 to 500 mM. The protein homogeneity was confirmed by 12 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE).
Enzymatic activities and properties of H. aurantiacus family 3 ginsenoside-hydrolyzing enzyme (HaGH03)
Specific activity of the purified HaGH03 was measured at 37 °C in a 100-μL reaction mixture containing 1.5 mg/mL of ginsenoside, or in a 500-μL mixture with 2 mM aryl-glycoside, as well as a certain amount of HaGH03 and 50 mM sodium phosphate buffer (pH 6.5). The hydrolysis of aryl-glycoside was terminated after 5 min by the addition of 500 μL 1 mM Na2CO3, and the amount of liberated chromogenic p-nitrophenol (pNP) or o-nitrophenol (oNP) was immediately determined by the optical absorbance at 405 nm (Larsbrink et al. 2014). One unit (U) of β-glycosidase activity was defined as the amount of protein required to produce 1 μmol of pNP (oNP), produce 1 μmol notoginsenoside ST-4 from vina-ginsenoside R7, or decrease 1 μmol of other ginsenosides per minute, in the given conditions. The effect of pH on the activity of HaGH03 was investigated at 37 °C using 1.35 mM of vina-ginsenoside R7 in the following buffers: sodium acetate (pH 5.0), sodium phosphate (pH 6.0–8.0), and glycine-sodium hydroxide (pH 9.0). The pH stability of HaGH03 was assayed by analyzing its residual activity after incubation in each buffer for 12 h at 4 °C. The thermostability of HaGH03 was investigated by determining the residual activity after incubation of the enzyme (5 mg/mL) in 50 mM sodium phosphate buffer for 1 h at various temperatures. Purified HaGH03 was incubated with metal ions and chemical reagents at a final concentration of 1 or 10 mM for 1 h at 30 °C. The activity without reagents or metal ions was used as the control with relative activity defined as 100 %. Kinetic parameters were determined with vina-ginsenoside R7 and pNPG in different concentrations at pH 6.5 and 40 °C.
Biotransformation of vina-ginsenoside R7 and ginsenosides by HaGH03
Briefly, catalyzed reactions were initiated by mixing different concentrations (2.5–5.5 mg/mL) of vina-ginsenoside R7 and 0.6 mg/mL HaGH03 in 200 μL of 50 mM sodium phosphate buffer (pH 6.5). Samples were withdrawn periodically to determine the amount of conversion and were analyzed quantitatively by ultra-high performance liquid chromatography–mass spectrometry (UHPLC-MS). The reactions were quenched with methanol to precipitate the protein. After vortexing for 30 s, the resulting mixture was centrifuged at 20,000×g for 20 min. The supernatant was diluted by methanol with digoxin as an inner standard (IS).
To prepare notoginsenoside ST4 produced by HaGH03, biocatalytic resolution of 0.7 mg/mL vina-ginsenoside R7 was conducted with a catalyst loading of 1.2 g of lyophilized cell-free extract in 300 mL of pH 6.5 sodium phosphate buffer for 24 h at 37 °C. For preparation of ST-4, the reaction solution was extracted two times with 100 mL of n-butanol. The resultant n-butanol layer was then combined and concentrated in a rotary evaporator at 55 °C.
Analysis method
Quantitative analysis was performed using an Agilent 1290 series UHPLC (Agilent Technologies, Waldbronn, Germany) and an Agilent 6410 Triple Quadrupole mass spectrometer equipped with an electrospray ionization source (ESI). Chromatographic separation was achieved on an ACQUITY UPLC HSS T3 column (100 mm × 2.1 mm i.d., 1.8 μm; Waters Co., Milford, MA, USA) at 45 °C using a mobile phase of 0.1 % formic acid with 5 mM ammonium acetate (A) and acetonitrile (B) at a flow rate of 0.4 mL/min. The gradient elution (B) steps applied to determine R7, Fe, Rg3, Fd, Rb1, Rb2, Rb3, Rc, and XVII were set as follows: 0–1 min (15–37 %), 1–2 min (37 %), 2–2.5 min (37–40 %), 2.5–3 min (40–45 %), 3–5 min (45–80 %), and 5–6 min (90 %). The gradient elution (B) steps applied to measure R1, Rg1, Rg2, F1, Rh1, and Re were set as follows: 0–2 min (15–29 %), 2–2.5 min (29–32 %), 2.5–4 min (32–34 %), 4–6 min (34–70 %), 6–6.5 min (70–90 %), 6.5–7.5 min (90–95 %), and 7.5–8.5 min (15 %). Mass spectrometric analysis was performed in the negative ion multiple reaction monitoring (MRM) mode with 3.4-kV capillary voltage for all experiments. The m/z of precursor/product ions, fragment electric (FE) voltages, and collision energies (CEs) are summarized in Table 2. NMR spectra were obtained on a Bruker AV 400 NMR spectrometer (Faellanden, Switzerland) in C5D5N at 25 °C.
Sequence analysis of HaGH03
Database searching used BLAST of Uniprot and published papers on ginsenoside-hydrolyzing enzymes belonging to the GH3 family. Detailed sequence alignment analyses were conducted using ENDscript 3 (Robert and Gouet 2014) and a multiple protein sequence alignment website (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Results
Cloning, expression, and purification of recombinant glycosidase HaGH03
The gene from H. aurantiacus consisting of a 2250-bp fragment encoding a presumptive family 3 glycoside hydrolase was ligated into the pET28a vector and expressed solubly in E. coli BL21 (DE3) under IPTG induction. The fusion protein HaGH03 with a His-tagged N-terminus was purified by Ni–NTA affinity chromatography, resulting in a single band of about 81 kDa on SDS-PAGE (Fig. 2).

SDS-PAGE analysis of recombinant HaGH03. Lane M, molecular weight standard; lane 1, soluble fraction of crude extract of induced recombinant BL21 (DE3); lane 2, purified recombinant HaGH03 after His trap Ni–NTA FF column
Biochemical properties of the newly identified ginsenosidase HaGH03
The newly identified ginsenosidase HaGH03 was subjected to detailed characterization of its biochemical properties to establish its substrate specificities, estimated by catalysis of aryl-glycosides (pNP β Glc, pNP β Gal, pNP β Xyl, pNP α Glc, oNP β Glc, and oNP β Gal) and ginsenosides (Re, Rg1, Rg2, Rf, Rh1, F1, Rb1, Rb2, Rc, Rd, Rg3, vina-ginsenoside R7, notoginsenoside Fd, Fe, R1, and gypenoside XVII). Table 3 shows that the specificity of HaGH03 for aryl-glycosides was dependent on whether they were β-(1→2) or β-(1→4) linked glucopyranosides or galactopyranosides. No activity was observed toward the α-(1→4) linked glucopyranoside. The order of specific activity toward PPD-type ginsenosides listed in Table 3 was as follows: ginsenoside Rb1 > gypenoside XVII > notoginsenoside Fd > ginsenoside Rd > notoginsenoside Fe > ginsenoside Rg3 > ginsenoside Rc > ginsenoside Rb3 > vina-ginsenoside R7 > ginsenoside Rb2. No hydrolysis activity was determined for ginsenoside Rg2, and the order toward PPT-type ginsenosides was ginsenoside Rh1 > ginsenoside Re > ginsenoside R1 > ginsenoside F1 > ginsenoside Rg1 > ginsenoside Rf. The catalytic properties of HaGH03 were assessed to examine its potential as a candidate biocatalyst for the transformation of glycosides. The effect of pH on the hydrolytic activity of HaGH03 was investigated using vina-ginsenoside R7 and pNPG as substrates. The maximum activity was observed around pH 6.5 (in 50 mM sodium phosphate buffer at 37 °C) (Fig. 3a and Supplementary Fig. S1a). After incubation in different buffers for 12 h at 4 °C, the enzyme retained more than 70 % of the initial activity (pH 6.5, 37 °C) from pH 5.0 to 9.0. Maximum hydrolytic activity was observed at 50 °C using vina-ginsenoside R7 and pNPG as substrates, while the enzyme lost about 35 % of the initial activity (pH 6.5, 37 °C ) at 55 °C (Fig. 3b and Supplementary Fig. S1b). The thermal inactivation of HaGH03 followed first-order kinetics with half-lives of 47.7, 35.1, and 1.1 h at 30, 40, and 50 °C, respectively (Supplementary Fig. S1c). Irreversible alterations of proteins may take place in the presence of metal ions or chemical reagents. Supplementary Table S1 shows that the recombinant HaGH03 activities were inhibited slightly on the addition of Pb2+, Cu2+, or Fe2+ at a concentration of 10 mM, as well as by EDTA. Sodium dodecyl sulfate (SDS) had the strongest inhibition effect on HaGH03 activity even at a relatively low concentration (1 mM). No significant activation was observed for either the metal ions or the chemical reagents tested. In terms of steady state kinetic parameters, K M values for the purified HaGH03 measured against pNPG and vina-ginsenoside R7 were 5.67 ± 0.24 μM and 0.59 ± 0.23 mM, and k cat values were 69.2 ± 0.31/s and 2.15 ± 0.46/min, respectively.

a Effect of pH on the activity and stability of HaGH03 at 37 °C using vina-ginsenoside R7 as a substrate. b Effect of temperature on the activity and stability of recombinant HaGH03 at pH 6.5 using vina-ginsenoside R7 as a substrate. Data represent the mean of three replicates; error bar represents the standard deviation
Biotransformation of vina-ginsenoside R7 and other ginsenosides by HaGH03
To detect the effect of substrate concentration on the product formation, the bioconversion of vina-ginsenoside R7 by HaGH03 was performed with varied concentrations of R7 at pH 6.5 and 37 °C. In addition, the time courses of notoginsenoside ST-4 production were monitored quantitatively by MRM mode UHPLC-ESI-MS to optimize the transformation conditions. Figure 4 shows that HaGH03 transformed vina-ginsenoside R7 at 2.5 mg/mL to notoginsenoside ST-4 with a molar conversion of 100 % and a productivity of 104 mg/L/h in the optimum conditions. However, only 80 % was converted at the maximum substrate concentration tested (5.5 mg/mL) within 36 h. For a complete transformation, it is an objective demand to lengthen the reaction time with the increase of the substrate loading, which suggested us to use a moderate substrate concentration with a reasonable dose of enzyme. Finally, approximately 0.15 g notoginsenoside ST-4 with a purity of up to 99 % was produced through biocatalysis by HaGH03, as monitored by UPLC-MS (Fig. 5b).

The time course for notoginsenoside ST-4 production catalyzed by recombinant HaGH03

a Mass spectrum of notoginsenoside ST-4. b The UPLC-MS chromatogram of prepared notoginsenoside ST-4 by HaGH03. c Major fragmentation of sugar side chain in notoginsenoside ST-4 by Q-TOF-MS
In the negative ion mode, the characteristic ion of protopanaxadiol at m/z 459.3858 [aglycone-H]− was observed in the mass spectra of ST-4. Figure 5a, c shows that the fragment m/z 915.5322 [M-H]−, corresponding to the elemental formula C47H80O17, was consistent with the loss of one glucose residue from the substrate (m/z 1077.5830, [M-H]−) at position C-20. The product was tentatively identified using mass spectrometry, and the absolute configuration was verified by 1H NMR and 13C NMR spectroscopy (Supplementary Fig. S2); NMR data were assigned by comparison with notoginsenoside ST-4.
It was of interest to preliminarily investigate whether HaGH03 could specifically transform other notoginsenosides and ginsenosides. Here, we sought to investigate a set of substrates including two types of ginsenosides and identify the products using UPLC-Q/TOF-MS (Supplementary Fig. S3). In our experimental conditions, HaGH03 could hydrolyze the glucose residues of F1 and Re at the C-20 position to PPT and Rg2 as similar as the cleaving regulation on R7. In addition, HaGH03 could convert Rb2 and Rc to ginsenoside C-Y and Mc by further cleaving the glucoses at position C-3. The enzyme also showed 100 % conversion of notoginsenosides Fe and Fd into Mc and Mx by hydrolyzing the only glucose at C-3, and it could partially hydrolyze Rf and Rg1 to PPT via Rh1.
Sequence analysis of HaGH03
The enzymatic activity of HaGH03 (UniprotKB accession number A9B3B2) from H. aurantiacus DSM 785 has not been characterized before. A protein BLAST search against the Uniprot Knowledgebase revealed that HaGH03 has 48 % identity to a thermostable β-glucosidase B (P14002) from Clostridium thermocellum, which belongs to the GH3 family and does not have a known function in ginsenoside transformation. A comprehensive comparison of primary to quaternary structure prediction was conducted using multiple sequence alignment and ENDscript 3, with a GH3 from Thermotoga neapolitana (Q0GC07) as the template. Figure 6 shows multiple sequence alignment of HaGH03 with enzymes of the GH3 family, including four ginsenosidases, two enzymes of known structure (Q0GC07 and D1GCC6), and two characterized enzymes (P27034 and O30713), processed by ClustalW2. It has been determined through structural and functional analyses that in β-glucosidase 3B (Q0GC07) from Thermotoga neapolitana, domain 1 contains the nucleophile residue D242 and domain 2 has a α/β sandwich fold and contains the acid/base amino acid residue E458 (Pozzo et al. 2010). β-Glucosidases (D1GCC6 and O30713) from Kluyveromyces marxianus and Agrobacterium tumefaciens have also been analyzed and contain these conserved catalytic residues (Castle et al. 1992; Yoshida et al. 2010). HaGH03 shares the putative active site nucleophile and the acid/base, Asp233 and Glu423, respectively, marked with red triangles in Fig. 6.

Multiple detail sequence alignment of HaGH03 (A9B3B2) with selected GH3 enzymes by ClustalW2 and ENDscript 3, including four ginsenosidases (UniprotKB accession number: H2DGJ4, J9Y082, E7CY69, and F6C6C1), two known structures enzymes (Q0GC07 and D1GCC6), and two characterized enzymes (P27034 and O30713). Putative catalysis active sites for nucleophiles and the acid/base (Asp233 and Glu423) were marked with red triangles (Color figure online)
Discussion
The K M value of HaGH03 against pNPG is about 600 times lower than that of other ginsenoside-transforming enzymes belonging to glycoside hydrolase family 3, such as bglSk from Sanguibacter keddieii and bglAm from Actinosynnema mirum KACC 2008T, indicating that HaGH03 had a better affinity to the usual substrate (Kim et al. 2012; Cui et al. 2013a). From the ratio of kinetic parameters k cat/K M (the specificity constant), HaGH03 showed much higher activity toward pNPG than vina-ginsenoside R7.
Theoretically, there are a lot of pathways for vina-ginsenoside R7 hydrolysis by HaGH03, including hydrolysis of the outer or inner sugar moiety at C-3, and the glucose at C-20. This uncertainty in cleavage positions has increased the difficulty in finding appropriate enzymes with specific transformation activity to produce notoginsenoside ST-4. However, the HaGH03-mediated hydrolysis of vina-ginsenoside R7 only generated notoginsenoside ST-4 by cleaving the outer glucose at C-20 without affecting the outer xylose or the two inner glucoses at position C-3. In terms of specific activities toward ginsenosides (Table 3), HaGH03 has its strongest hydrolyzing ability for ginsenoside Rb1 among the substrates tested in our research. HaGH03 showed similar hydrolytic activities in the transformation of Rb1, Rb2, and Rc as the β-glucosidase from Dictyoglomus turgidum (Lee et al. 2012); however, HaGH03 exhibited a different order of hydrolytic activity toward ginsenosides, Rb1 > Rd > Rc > Rb2. Besides, the hydrolytic pathways of Rf1→Rh1→PPT, Rb2→C-Y and Rc→C-Mc of HaGH03 were the same as β-glucosidase from Penicillium aculeatum, which hydrolyzed exo-, 3-O-, and 6-O-β-glucosides but not 20-O-β-glucoside and other glycosides of ginsenosides (Lee et al. 2013). The pathway of Re→Rg2 is identical to that of bglAm from Actinosynnema mirum KACC 20028T, which has only 13.82 % amino acid sequence identity, through the cleavage of glucose at position C-20 (Cui et al. 2013a). In the light of our experimental data, the most parsimonious conclusion is that HaGH03 shows a higher preference for the glucose residue at position C-20 than at position C-3. As shown in Supplementary Fig. S3, HaGH03 does not hydrolyze other outer glycosides such as xylose or rhamnose linked to positions C-3 and C-6, or arabinopyranose or arabinofuranose linked to C-20, which is similar to the β-glucosidase from Thermus thermophilus. Because of the ability to cleave the glucose attached at positions C-3 and C-6, H. aurantiacus β-glucosidase has a wider substrate spectrum than the latter which applied to produce ginsenoside F2 from gypenoside XVII due to the highly selective hydrolysis of the outer glucose at the C-20 position (Shin et al. 2014).
As listed in Table 1, the characterized ginsenoside-hydrolyzing enzymes exhibit promiscuous activities toward diverse substrates, although they all belong to the glycoside hydrolase family 3. The extensive applications of less polar ginsenosides in pharmacotherapy are prompting researchers to identify enzymes that can produce minor ones by hydrolyzing major ginsenosides. With the development of biotechnology, more and more enzymes other than glucosidases, such as α-l-arabinofuranosidases and β-galactosidases cloned from microorganisms, have been used for complete conversion of major ginsenosides to rare ones (Shin et al. 2013). However, no one has investigated the transformation of vina-ginsenoside R7 to notoginsenoside ST-4 before this study. The present research provides useful guidance for the promising application of HaGH03 in the enzymatic production of rare ginsenosides.
In summary, we have successfully established an eco-friendly and convenient method to obtain notoginsenoside ST-4 by completely converting vina-ginsenoside R7 using a recombinant glycosidase, HaGH03, from H. aurantiacus. HaGH03 was overexpressed in E. coli BL21 (DE3) in a completely soluble form, and its detailed enzymatic properties were characterized to establish substrate specificities. The results underscore that HaGH03 has much potential for the effective preparation of various types of rare ginsenosides possessing valuable pharmacological activities, especially notoginsenoside ST-4.
References
An DS, Cui CH, Lee HG, Wang L, Kim SC, Lee ST, Jin FX, Yu HS, Chin YW, Lee HK, Im WT, Kim SG (2010) Identification and characterization of a novel Terrabacter ginsenosidimutans sp nov β-Glucosidase that transforms Ginsenoside Rb1 into the rare Gypenosides XVII and LXXV. Appl Environ Microbiol 76:5827–5836
Attele AS, Wu JA, Yuan CS (1999) Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol 58:1685–1693
Castle LA, Smith KD, Morris RO (1992) Cloning and sequencing of an Agrobacterium tumefaciens beta-glucosidase gene involved in modifying a vir-inducing plant signal molecule. J Bacteriol 174:1478–1486
Chang KH, Jee HS, Lee NK, Park SH, Lee NW, Paik HD (2009) Optimization of the enzymatic production of 20(S)-ginsenoside Rg3 from white ginseng extract using response surface methodology. New Biotechnol 26:181–186
Cui CH, Kim SC, Im WT (2013a) Characterization of the ginsenoside-transforming recombinant β-glucosidase from Actinosynnema mirum and bioconversion of major ginsenosides into minor ginsenosides. Appl Microbiol Biotechnol 97:649–659
Cui CH, Liu QM, Kim JK, Sung BH, Kim SG, Kim SC, Im WT (2013b) Identification and characterization of a Mucilaginibacter sp Strain QM49 β-glucosidase and its use in the production of the pharmaceutically active minor Ginsenosides (S)-Rh1 and (S)-Rg2. Appl Environ Microbiol 79:5788–5798
Dan M, Su M, Gao X, Zhao T, Zhao A, Xie G, Qiu Y, Zhou M, Liu Z, Jia W (2008) Metabolite profiling of Panax notoginseng using UPLC–ESI-MS. Phytochemistry 69:2237–2244
Gao B, Huang L, Liu H, Wu H, Zhang E, Yang L, Wu X, Wang Z (2014a) Platelet P2Y12 receptors are involved in the haemostatic effect of notoginsenoside Ft1, a saponin isolated from Panax notoginseng. Br J Pharmacol 171:214–223
Gao B, Shi HL, Li X, Qiu SP, Wu H, Zhang BB, Wu XJ, Wang ZT (2014b) p38 MAPK and ERK1/2 pathways are involved in the pro-apoptotic effect of notoginsenoside Ft1 on human neuroblastoma SH-SY5Y cells. Life Sci 108:63–70
Groussin AL, Antoniotti S (2012) Valuable chemicals by the enzymatic modification of molecules of natural origin: terpenoids, steroids, phenolics and related compounds. Bioresour Technol 115:237–243
Jin S, Luo M, Wang W, Zhao CJ, Gu CB, Li CY, Zu YG, Fu YJ, Guan Y (2013) Biotransformation of polydatin to resveratrol in Polygonum cuspidatum roots by highly immobilized edible Aspergillus niger and Yeast. Bioresour Technol 136:766–770
Kim JK, Cui CH, Yoon MH, Kim SC, Im WT (2012) Bioconversion of major ginsenosides Rg1 to minor ginsenoside F1 using novel recombinant ginsenoside hydrolyzing glycosidase cloned from Sanguibacter keddieii and enzyme characterization. J Biotechnol 161:294–301
Kim JK, Cui CH, Liu Q, Yoon MH, Kim SC, Im WT (2013) Mass production of the ginsenoside Rg3(S) through the combinative use of two glycoside hydrolases. Food Chem 141:1369–1377
Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, Creagh AL, Haynes CA, Kelly AG, Cederholm SN, Davies GJ, Martens EC, Brumer H (2014) A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506:498–502
Lee CH, Kim JH (2014) A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. J Ginseng Res 38:161–166
Lee GW, Kim KR, Oh DK (2012) Production of rare ginsenosides (compound Mc, compound Y and aglycon protopanaxadiol) by β-glucosidase from Dictyoglomus turgidum that hydrolyzes β-linked, but not α-linked, sugars in ginsenosides. Biotechnol Lett 34:1679–1686
Lee GW, Yoo MH, Shin KC, Kim KR, Kim YS, Lee KW, Oh DK (2013) β-Glucosidase from Penicillium aculeatum hydrolyzes exo-, 3-O-, and 6-O-β-glucosides but not 20-O-β-glucoside and other glycosides of ginsenosides. Appl Microbiol Biotechnol 97:6601
Lee HJ, Shin KC, Lee GW, Oh DK (2014) Production of aglycone protopanaxatriol from ginseng root extract using Dictyoglomus turgidum β-glycosidase that specifically hydrolyzes the xylose at the C-6 position and the glucose in protopanaxatriol-type ginsenosides. Appl Microbiol Biotechnol 98:3659–3667
Liu ZQ (2012) Chemical insights into ginseng as a resource for natural antioxidants. Chem Rev 112:3329–3355
Liu C, Jin Y, Yu H, Sun C, Gao P, Xiao Y, Zhang T, Xu L, Im WT, Jin F (2014) Biotransformation pathway and kinetics of the hydrolysis of the 3-O- and 20-O-multi-glucosides of PPD-type ginsenosides by ginsenosidase type I. Process Biochem 49:813–820
Minh DN, Kasai R, Ohtani K, Ito A, Thoi Nham N, Yamasaki K, Tanaka O (1994) Saponins from Vietnamese ginseng, Panax vietnamensis Ha et Grushv. Collected in central Vietnam. II. Chem Pharm Bull 42:115–122
Ng TB (2006) Pharmacological activity of sanchi ginseng (Panax notoginseng). J Pharm Pharmacol 58:1007–1019
Park CS, Yoo MH, Noh KH, Oh DK (2010) Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl Microbiol Biotechnol 87:9–19
Pei Y, Du Q, Liao PY, Chen ZP, Wang D, Yang CR, Kitazato K, Wang YF, Zhang YJ (2011) Notoginsenoside ST-4 inhibits virus penetration of herpes simplex virus in vitro. J Asian Nat Prod Res 13:498–504
Pozzo T, Pasten JL, Karlsson EN, Logan DT (2010) Structural and functional analyses of β-glucosidase 3B from Thermotoga neapolitana: a thermostable three-domain representative of glycoside hydrolase 3. J Mol Biol 397:724–739
Quan LH, Min JW, Yang DU, Kim YJ, Yang DC (2012) Enzymatic biotransformation of ginsenoside Rb1 to 20(S)-Rg3 by recombinant β-glucosidase from Microbacterium esteraromaticum. Appl Microbiol Biotechnol 94:377–384
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. doi:10.1093/nar/gku316
Shen KK, Ji LL, Gong CY, Ma YB, Yang L, Fan Y, Hou MQ, Wang ZT (2012) Notoginsenoside Ft1 promotes angiogenesis via HIF-1α mediated VEGF secretion and the regulation of PI3K/AKT and Raf/MEK/ERK signaling pathways. Biochem Pharmacol 84:784–792
Shen KK, Leung SWS, Ji LL, Huang Y, Hou MQ, Xu AM, Wang ZT, Vanhoutte PM (2014) Notoginsenoside Ft1 activates both glucocorticoid and estrogen receptors to induce endothelium-dependent, nitric oxide-mediated relaxations in rat mesenteric arteries. Biochem Pharmacol 88:66–74
Shin KC, Oh HJ, Kim BJ, Oh DK (2013) Complete conversion of major protopanaxadiol ginsenosides to compound K by the combined use of α-l-arabinofuranosidase and β-galactosidase from Caldicellulosiruptor saccharolyticus and β-glucosidase from Sulfolobus acidocaldarius. J Biotechnol 167:33–40
Shin KC, Seo MJ, Oh HJ, Oh DK (2014) Highly selective hydrolysis for the outer glucose at the C-20 position in ginsenosides by β-glucosidase from Thermus thermophilus and its application to the production of ginsenoside F2 from gypenoside XVII. Biotechnol Lett 36:1287–1293
Su JH, Xu JH, Lu WY, Lin GQ (2006) Enzymatic transformation of ginsenoside Rg3 to Rh2 using newly isolated Fusarium proliferatum ECU2042. J Mol Catal B Enzym 38:113–118
Su JH, Xu JH, Yu HL, He YC, Lu WY, Lin GQ (2009) Properties of a novel glucosidase from Fusarium proliferatum ECU2042 that converts ginsenoside Rg3 into Rh2. J Mol Catal B Enzym 57:278–283
Tawab MA, Bahr U, Karas M, Wurglics M, Schubert ZM (2003) Degradation of ginsenosides in humans after oral administration. Drug Metab Dispos 31:1065–1071
Wang CZ, McEntee E, Wicks S, Wu JA, Yuan CS (2006) Phytochemical and analytical studies of Panax notoginseng (Burk.) F.H. Chen. J Nat Med 60:97–106
Wang XY, Wang D, Ma XX, Zhang YJ, Yang CR (2008) Two new dammarane-type bisdesmosides from the fruit pedicels of Panax notoginseng. Helv Chim Acta 91:60–66
Ye L, Zhou CQ, Zhou W, Zhou P, Chen DF, Liu XH, Shi XL, Feng MQ (2010) Biotransformation of ginsenoside Rb1 to ginsenoside Rd by highly substrate-tolerant Paecilomyces bainier 229-7. Bioresour Technol 101:7872–7876
Yoshida E, Hidaka M, Fushinobu S, Koyanagi T, Minami H, Tamaki H, Kitaoka M, Katayama T, Kumagai H (2010) Role of a PA14 domain in determining substrate specificity of a glycoside hydrolase family 3 β-glucosidase from Kluyveromyces marxianus. Biochem J 431:39–49
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (U1032604) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1071).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 409 kb)
Rights and permissions
About this article

Cite this article
Wang, RF., Zheng, MM., Cao, YD. et al. Enzymatic transformation of vina-ginsenoside R7 to rare notoginsenoside ST-4 using a new recombinant glycoside hydrolase from Herpetosiphon aurantiacus . Appl Microbiol Biotechnol 99, 3433–3442 (2015). https://doi.org/10.1007/s00253-015-6446-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6446-z