
Abstract
A wide range of promoters with different strengths and regulatory mechanisms are valuable tools in metabolic engineering and synthetic biology. While there are many constitutive promoters available, the number of inducible promoters is still limited for pathway engineering in Saccharomyces cerevisiae. Here, we constructed aromatic amino-acid-inducible promoters based on the binding sites of Aro80 transcription factor, which is involved in the catabolism of aromatic amino acids through transcriptional activation of ARO9 and ARO10 genes in response to aromatic amino acids. A dynamic range of tryptophan-inducible promoter strengths can be obtained by modulating the number of Aro80 binding sites, plasmid copy numbers, and tryptophan concentrations. Using low and high copy number plasmid vectors and different tryptophan concentrations, a 29-fold range of fluorescence intensities of enhanced green fluorescent protein (EGFP) reporter could be achieved from a synthetic U4C ARO9 promoter, which is composed of four repeats of Aro80 binding half site (CCG) and ARO9 core promoter element. The U4C ARO9 promoter was applied to express alsS and alsD genes from Bacillus subtilis for acetoin production in S. cerevisiae, resulting in a gradual increase in acetoin titers depending on tryptophan concentrations. Furthermore, we demonstrated that γ-aminobutyrate (GABA)-inducible UGA4 promoter, regulated by Uga3, can also be used in metabolic engineering as a dose-dependent inducible promoter. The wide range of controllable expression levels provided by these tryptophan- and GABA-inducible promoters might contribute to fine-tuning gene expression levels and timing for the optimization of pathways in metabolic engineering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Controlling the expression of genes in metabolic pathways or in regulatory networks is an essential component in metabolic engineering and synthetic biology (Andrianantoandro et al. 2006; Keasling 2010). Although gene expression can be regulated at multiple points, promoter-driven transcriptional initiation is a key regulatory step in determining gene expression levels and timing (Blazeck and Alper 2013). Successful pathway engineering requires diverse range of constitutive and inducible promoters, which allow sophisticated transcriptional regulation of each gene participating in the pathway (Blazeck and Alper 2013; Da Silva and Srikrishnan 2012). Therefore, numerous efforts have been made to isolate native promoters (Mumberg et al. 1995; Sun et al. 2012) or to develop synthetic promoters suitable for genetic engineering (Alper et al. 2005; Blazeck et al. 2013; Blount et al. 2012; Hartner et al. 2008).
Saccharomyces cerevisiae is a well-studied eukaryotic model system with great potential as microbial cell factories for the production of fuels and chemicals (Nevoigt 2008; Nielsen et al. 2013). Strong constitutive promoters in the glycolytic pathway, P TDH3 (also known as P GPD ), P PGK1 , P TPI1 , and P PDC1 , and the promoter of translation elongation factor (P TEF1 ) have been widely used for gene expression in S. cerevisiae, along with other weaker constitutive promoters such as P CYC1 and P ADH1 (Cartwright et al. 1994; Da Silva and Srikrishnan 2012; Mumberg et al. 1995; Sun et al. 2012). Although constitutive promoters are convenient to maintain gene expression without additional manipulation, they are not suitable for the metabolic pathway containing toxic intermediates or for the expression of target genes at a specific time point (Da Silva and Srikrishnan 2012). Inducible or regulated promoters can complement these problems. The galactose-inducible P GAL1 and P GAL10 promoters have been mostly used in metabolic engineering applications, although other inducible promoters such as P CUP1 , P PHO5 , and P MET25 are also available in S. cerevisiae (Hottiger et al. 1995; Macreadie et al. 1991; Mumberg et al. 1994; Rudolph and Hinnen 1987). The GAL promoters are tightly repressed in the presence of glucose, resulting in about 1000-fold induction by galactose (Adams 1972; Blazeck et al. 2012). Recently, a series of synthetic galactose-inducible promoters with higher basal activity and dynamic range of galactose-induced expression levels have been generated by combining various upstream activation sequences (UASs) and core promoter elements (Blazeck et al. 2012). However, the GAL promoters have several disadvantages. Because of the glucose repression effect, GAL promoters cannot be induced by direct addition of galactose if the culture medium contains glucose (Johnston 1987; Lohr et al. 1995). Therefore, complete medium exchange is necessary for cells grown in glucose. In addition, galactose is used not only as an inducer but also as a carbon source. Since galactose is a less preferred carbon source than glucose, shifting the glucose-grown cells into galactose medium reduces cell growth rate, and the galactose-induced expression levels decrease as galactose is consumed during growth (Lee and DaSilva 2005).
The GAL promoters are activated by Gal4 transcription factor, which belongs to the Zn2Cys6 family of fungal-specific transcription factors (MacPherson et al. 2006; Todd and Andrianopoulos 1997). These transcription factors form homodimers and each Zn2Cys6 domain binds to CGG half sites aligned in various orientations (inverted repeat, CGGNxCCG; everted repeat, CCGNxCGG; direct repeat, CGGNxCGG; and reverse direct repeat, CCGNxCCG) and spacing (MacPherson et al. 2006; Todd and Andrianopoulos 1997). Although galactose-dependent activation of Gal4 is mediated by relieving the Gal80-dependent repression of Gal4, other Zn2Cys6 transcription factors are activated by direct binding of specific inducers (MacPherson et al. 2006). For example, proline directly binds and activates Put3 transcription factor, involved in proline utilization (Des Etages et al. 1996), and Leu3, involved in branched chain amino acids biosynthesis, is activated by binding of α-isopropylmalate, a pathway-specific intermediate (Hahn and Young 2011; Sze et al. 1992). Therefore, the promoters regulated by other Zn2Cys6 family proteins might have the potential to be developed as inducible promoters for genetic engineering applications. When selecting inducible promoters for biotechnological purposes, several factors have to be considered, which include the basal activity and induction fold of the promoter, and the cost and side effects of the inducer (Nevoigt et al. 2007).
Aro80, a member of Zn2Cys6 family, is involved in the utilization of aromatic amino acids as nitrogen sources through transcriptional activation of ARO9 and ARO10 in the presence of aromatic amino acids (Iraqui et al. 1999; Lee and Hahn 2013). Aro9 and Aro10 act as transaminase and decarboxylase, respectively, in the degradation of aromatic amino acids through Ehrlich pathway (Hazelwood et al. 2008). ARO9 and ARO10 genes are also regulated by nitrogen catabolite repression (NCR), thus activated by GATA transcription activators (Gat1 and Gln3) in the absence of good nitrogen sources (Lee and Hahn 2013). However, the availability of aromatic amino acids is the major determinant for the transcriptional activation of these genes (Lee and Hahn 2013). Aro80 is believed to act as a dimer like other Zn2Cyc6 family of transcription factors (MacPherson et al. 2006). Aro80 binding site consists of two CCG direct repeats separated by 7 base pairs (CCGN7CCG), and the binding sites are repeated twice in the promoters of ARO9 and ARO10, allowing binding of up to two Aro80 dimers. S. cerevisiae genome contains two additional genes containing Aro80 binding sites; ARO80 itself and ESBP6 encoding a protein homologous to a monocarboxylate permease (Eden et al. 2007). However, these genes, containing only one copy of the CCGN7CCG element in their promoters, are largely insensitive to the induction by aromatic amino acids (Lee and Hahn 2013).
In this study, we used Aro80 binding site to design promoters inducible by aromatic amino acids. A wide range of tryptophan-induced expression levels could be achieved by modulating the number of Aro80 binding sites, plasmid copy numbers, and the concentrations of inducer. The effectiveness of the tryptophan-inducible promoters was demonstrated by applying this promoter system to express heterologous genes for the production of acetoin in S. cerevisiae. In addition, we demonstrated the possibility of using γ-aminobutyrate (GABA)-inducible UGA4 promoter for metabolic engineering. The UGA4 promoter is regulated by Uga3, another member of the Zn2Cyc6 family of transcription factors.
Materials and methods
Strains and media
Escherichia coli strain DH5α [F− Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (r K −, m K +) phoA supE44 λ − thi-1 gyrA96 relA] was used for genetic manipulations. S. cerevisiae BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and bdh1Δ (BY4741, bdh1Δ::KanMX6) were obtained from EUROSCARF. Yeast cells were cultured in YPD medium (20 g/L glucose, 10 g/L yeast extract, and 20 g/L bacto-peptone) or synthetic defined (SD) medium (20 or 50 g/L glucose, 6.7 g/L yeast nitrogen base without amino acids) supplemented with auxotrophic requirements (120 μg/mL Leu and 50 μg/mL each of His, Met, and Ura).
Plasmid construction
The DNA fragment encoding EGFP was prepared by PCR amplification and cloned into p416GPD and p416ADH, resulting p416GPD-EGFP and p416ADH-EGFP, respectively. To construct the synthetic promoters, PCR amplifications were performed using the primers containing additional Aro80 binding sites, F_UnC ARO80 /R_C ARO80 for ARO80 core element (−129 to −1) and F_UnC ARO9 /R_C ARO9 for ARO9 core element (−132 to −1), generating [UnC ARO80 ] and [UnC ARO9 ] (n = 2, 3, 4). The ARO9 promoter (P ARO9 ) was amplified by PCR using the primers, F_P ARO9 /R_C ARO9 . The TDH3 promoter of p416GPD-EGFP was removed by cutting the plasmid with SacI and BamHI, and replaced with the DNA fragments [UnC ARO80 ], [UnC ARO9 ], or P ARO9 resulting in p416[UnC ARO80 ]-EGFP, p416[UnC ARO9 ]-EGFP, and p416[P ARO9 ]-EGFP, respectively. The selected tryptophan-inducible promoter, [U4C ARO9 ], was replaced the TDH3 promoter of p413GPD, p423GPD, p416GPD, and p426GPD, generating p413[U4C ARO9 ], p423[U4C ARO9 ], p416[U4C ARO9 ], and p426[U4C ARO9 ], respectively. The alsS gene from Bacillus subtilis was amplified by PCR using genomic DNA and cloned into p413[U4C ARO9 ] and p423[U4C ARO9 ], generating p413[U4C ARO9 ]-alsS and p423[U4C ARO9 ]-alsS. The alsD gene from B. subtilis was amplified by PCR using genomic DNA and cloned into p416[U4C ARO9 ] and p426[U4C ARO9 ], generating p416[U4C ARO9 ]-alsD and p426[U4C ARO9 ]-alsD. To construct GABA-inducible acetoin producing pathway, UGA4 promoter sequence (−460 to −1) was prepared by PCR amplification and replaced the [U4C ARO9 ] promoter of p413[U4C ARO9 ]-alsS and p426[U4C ARO9 ]-alsD, creating p413[P UGA4 ]-alsS and p426[P UGA4 ]-alsD. Plasmids and primers used in this study are listed in Tables 1 and 2.
Culture conditions
For the experiments investigating the promoter inducibility by aromatic amino acids, overnight culture cells were diluted to OD600 of 0.45, incubated for 6 h in SD-Ura containing 20 g/L glucose, and then induced with 200 μg/mL tryptophan, phenylalanine, or tyrosine. Tryptophan and phenylalanine were dissolved in distilled water at a concentration of 10 mg/mL and used as stock solution. For tyrosine induction, instead of treatment of tyrosine using stock solution, the culture medium was exchanged with SD-Ura containing 200 μg/mL tyrosine because of the low solubility of tyrosine in water.
Yeast cells harboring alsS and alsD genes in various plasmids were precultured in SD-His-Ura containing 20 g/L glucose and then inoculated to OD600 of 0.2 in SD-His-Ura containing 50 g/L glucose with indicated concentrations of tryptophan or GABA for acetoin production.
RNA preparation and quantitative reverse transcription PCR
One milliliter of cells was harvested and frozen at −80 °C in 300 μL of lysis buffer [10 mM Tris–HCl (pH 7.4), 10 mM EDTA, 0.5 % SDS]. Acidic phenol (300 μL) was added to each sample and incubated at 65 °C for 20 min with occasional vortexing. Prior to chloroform extraction, the solution was chilled on ice for 10 min. After centrifugation and ethanol precipitation, the resulting RNA pellets were dissolved in RNase-free water. The relative amount of messenger RNA (mRNA) was determined by quantitative reverse transcription PCR (qRT-PCR) as previously described (Lee and Hahn 2013). Reverse transcription (RT) of 2 μg of total RNA was carried out with 0.1 μg of oligo-(dT) for 1 h at 42 °C using M-MLV reverse transcriptase (M-biotech, Inc., Korea), followed by heat inactivation for 10 min at 75 °C. PCR mixture containing 1 of 20 μL RT reaction solution, 1× SYBR master mix (Roche Diagnostics), and gene-specific primers was subjected to qPCR reaction with 45 cycles of 95 °C for 10 s, 55 °C for 20 s, and 72 °C for 20 s using Roche LightCycler 480 real-time PCR system (Roche Diagnostics). Primer sequences used for qRT-PCR are shown in Table 2.
Measurement of EGFP fluorescence intensity
To measure the EGFP fluorescence intensity, cells were harvested at appropriate point and resuspended with phosphate-buffered saline (PBS). The EGFP fluorescence intensity was measured by a TECAN GeNios Pro microplate reader (TECAN, USA) at excitation wavelength of 485 nm and emission wavelength of 535 nm. Cell density was measured using the microplate reader Multiskan GO (Thermo Scientific, USA) at 600 nm.
High-performance liquid chromatography
To quantify the concentration of acetoin, 1 mL of culture supernatants was collected and filtered through a 0.22-μm syringe filter. High-performance liquid chromatography (HPLC) analysis was performed in UltiMate 3000 HPLC system (Thermo fishers scientific) equipped with a BioRad Aminex HPX-87H column (300× 7.8 mm, 5 μm) at 60 °C with 5 mM H2SO4 as a flow rate of 0.6 mL/min and refractive index (RI) detector.
Results
Construction of aromatic amino-acid-inducible synthetic promoters
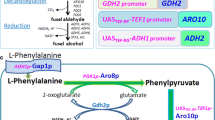
We investigated the possibility of using Aro80 binding site to design synthetic promoters inducible by aromatic amino acids. ARO80 and ARO9 promoters contain two and four CCG repeats, respectively, and the binding of Aro80 to these promoters has been confirmed by chromatin immunoprecipitation assays (Lee and Hahn 2013). However, aromatic amino acids induce transcription of only ARO9, but not ARO80 (Lee and Hahn 2013). Therefore, we hypothesized that the number of Aro80 binding site in the promoter might affect the inducibility by aromatic amino acids. To prove this, we fused one or two CCG half sites to the native ARO80 promoter consisting of UAS ARO80 (U2) and the core promoter (C ARO80 ), generating U3C ARO80 and U4C ARO80 (Fig. 1a). The synthetic promoters were fused to an EGFP reporter gene, and then cloned into p416, a CEN/ARS-based low copy number plasmid vector containing CYC1 terminator, to test tryptophan-dependent transcriptional induction. However, the addition of more Aro80 binding sites to the ARO80 promoter failed to induce transcriptional activation in the presence of tryptophan (Fig. 1b). On the contrary, when the same Aro80 binding sites were fused to the ARO9 core promoter (C ARO9 ), the transcription of EGFP was induced by tryptophan depending on the increasing number of Aro80 binding sites, without affecting the basal expression levels (Fig. 1b). The U4C ARO9 promoter showed about 20-fold induction after the treatment of 200 μg/mL tryptophan for 1 h. These results suggest that tryptophan-inducible synthetic promoters with different induction folds can be constructed by controlling the number of CCG repeating units, but the core promoter region also plays an important role for the transcriptional activation by Aro80. It needs further studies to elucidate which features of the ARO80 core promoter prevent the Aro80-dependent activation.

Construction of promoters inducible by aromatic amino acids. a Schematic representation of the promoter constructs containing core promoter region and additional upstream activating sequences (UAS). The EGFP gene was used as a reporter. The sequence details of UAS elements are shown below. CCG triplets, the binding sites of Aro80, are underlined. UAS ARO9 is upstream activating sequence of native ARO9 promoter and the sequences different from the synthetic UAS are represented in lowercase letters. b Tryptophan-dependent induction of synthetic promoters. Cells harboring p416-based plasmid expressing EGFP from the indicated promoter elements were treated with 200 μg/mL tryptophan for 1 h, and the EGFP mRNA levels were determined by qRT-PCR normalized to ENO1. Error bars represent standard deviation from triplicates
Next, we compared the effects of different aromatic amino acids on the induction of the U4C ARO9 promoter by measuring EGFP fluorescence intensities. Although all three aromatic amino acids induced EGFP expression, tyrosine was less effective than tryptophan and phenylalanine (Fig. 2a). Moreover, because of its low solubility (<0.5 g/L), tyrosine might not be suitable as a practical inducer of the U4C ARO9 promoter. The EGFP expression levels decreased after 13 h of induction, which might reflect the degradation or utilization of the inducers. Although tryptophan exerted a little lower induction fold (8.1-fold) than did phenylalanine (9.1-fold) up to 4 h, tryptophan served as a better inducer than phenylalanine after 13 h. Note that since we used 200 μg/mL amino acids, the molar concentration of phenylalanine (1.21 mM) is slightly higher than that of tryptophan (0.98 mM). In the case of tryptophan, its degradation product tryptophol is also known as an activator of Aro80, which might be in part responsible for the longer induction period in the presence of tryptophan. Because the U4C ARO9 promoter sequence is very similar to that of the native ARO9 promoter (P ARO9 ) (Fig. 1a), we also investigated the inducibility of P ARO9 by aromatic amino acids. As expected, the aromatic amino-acid-dependent induction pattern of the P ARO9 promoter was comparable to that of the U4C ARO9 promoter (Fig. 2b).

Effect of different aromatic amino acids on EGFP expression levels from the U4C ARO9 promoter (a) and the native ARO9 promoter (P ARO9 ) (b). Cells harboring p416[U4C ARO9 ]-EGFP or p416[P ARO9 ]-EGFP were treated with 200 μg/mL tryptophan (Trp), phenylalanine (Phe), or tyrosine (Tyr) for the indicated time period, and the fluorescence intensities (RFU) were detected and normalized to the cell densities (OD600)
We also tested whether further addition of Aro80 binding sites to the native ARO9 promoter can enhance the tryptophan-dependent induction levels. However, fusion of additional two or four half sites to the upstream of USA ARO9 rather reduced transcription levels in the presence of tryptophan (data not shown). Therefore, for Aro80-dependent activation, most efficient activation seems to be achieved by binding of two Aro80 dimers.
Regulation of tryptophan-induced expression levels by plasmid copy numbers and tryptophan concentrations
Next, we examined tryptophan-induced expression levels from the U4C ARO9 promoter depending on various tryptophan concentrations and plasmid copy numbers. Cells harboring low copy number plasmid, p416[U4C ARO9 ]-EGFP, or high copy number plasmid, p426[U4C ARO9 ]-EGFP, were treated with 50–800 μg/mL tryptophan, and transcription and protein expression levels were determined by qRT-PCR and fluorescence detection, respectively. The EGFP mRNA levels increased gradually depending on tryptophan concentrations, resulting in up to 20- and 15-fold induction levels in low and high copy number plasmids, respectively, compared with each uninduced control (Fig. 3a). Because of the leaky basal expression from the U4C ARO9 promoter, both uninduced and induced EGFP mRNA levels expressed from high copy number plasmid were about 10- to 15-fold higher than those expressed from low copy number plasmid. The activities of commonly used constitutive promoters, P ADH1 (in p416ADH-EGFP) and P TDH3 (in p416GPD-EGFP), were not affected by tryptophan. The U4C ARO9 -controlled tryptophan-induced expression levels were comparable to the P ADH1 -driven expression levels when expressed from low copy number plasmid.

Effect of plasmid copy numbers and tryptophan concentrations on EGFP expression levels from the U4C ARO9 promoter. Cells harboring the indicated plasmids were treated with the indicated concentrations of tryptophan for 1 h, and transcription and protein levels of EGFP were investigated. The constitutive promoters, P ADH1 and P TDH3 , were used as controls. a EGFP mRNA levels were determined by qRT-PCR normalized to ENO1. Error bars represent standard deviation from triplicates. b The RFU/OD600 value was normalized to that of untreated cells harboring p416[U4C ARO9 ]-EGFP, and represented as relative fluorescence intensity
The EGFP protein expression levels detected by fluorescence intensities reflected the transcription induction patterns (Fig. 3b). Cells harboring p416[U4C ARO9 ]-EGFP plasmid showed a gradual increase in fluorescence intensities as increasing tryptophan concentrations from 50 to 800 μg/mL with a maximum induction fold of 6 (Fig. 3b). Expression of [U4C ARO9 ]-EGFP from high copy number plasmid resulted in about 5-fold higher uninduced and induced expression levels, while keeping the tryptophan concentration-dependent induction profile (Fig. 3b). Using these two vectors and different tryptophan concentrations, a 29-fold range of expression levels could be achieved from the U4C ARO9 promoter. Taken together, a wide range of tryptophan-inducible promoter strengths can be obtained by modulating the number of Aro80 binding sites, plasmid copy numbers, and tryptophan concentrations, thereby enabling the fine tuning of transcription levels for metabolic engineering applications.
Acetoin production using the U4C ARO9 promoter
To verify the effectiveness of the tryptophan-inducible promoters in metabolic engineering, we applied this system to the biosynthetic pathway of acetoin, a potential high-value platform chemical for a broad range of applications such as food, flavor, and pharmaceutical industries (Xiao and Lu 2014). For effective production of acetoin from pyruvate, a heterologous pathway consisting of acetolactate synthase (AlsS) and acetolactate decarboxylase (AlsD) from B. subtilis were introduced into S. cerevisiae. In addition, endogenous BDH1 gene, encoding 2,3-butanediol dehydrogenase, was deleted to prevent the formation of 2,3-butanediol from acetoin (Fig. 4a).

Application of the U4C ARO9 promoter to metabolic engineering for acetoin production. a Metabolic pathway for acetoin production. Two molecules of pyruvate are converted α-acetolactate by acetolactate synthase (AlsS), and then acetolactate decarboxylase (AlsD) converts α-acetolactate to acetoin. To block the 2,3-butanediol production from acetoin, BDH1 gene encoding 2,3-butanediol dehydrogenase was deleted. b Cells carrying alsS expression vector, p413[U4C ARO9 ]-alsS (Low) or p423[U4C ARO9 ]-alsS (high), and alsD expression vector, p416[U4C ARO9 ]-alsD (low) or p426[U4C ARO9 ]-alsD (high), in four different combinations were grown for 48 h in the absence or presence of 200 μg/mL tryptophan, and acetoin production levels were monitored. c Acetoin production depending on tryptophan concentrations in cells harboring p413[U4C ARO9 ]-alsS and p426[U4C ARO9 ]-alsD
The alsS and alsD genes were expressed under the control of U4C ARO9 , and the required balance between AlsS and AlsD was simply tested by cloning the genes into both low copy number (p413 or p416) and high copy number (p423 or p426) plasmids, and examining acetoin production levels in cells harboring four different combinations of plasmid types (Fig. 4b). In the presence of 200 μg/mL tryptophan, cells expressing alsS from low copy number plasmid (p413) and alsD from high copy number plasmid (p426) produced 2.5 g/L acetoin after 48 h, the highest concentration among the four combinations. Expressing both alsS and alsD from high copy number plasmids did not give the best result, exemplifying the importance of regulating and balancing gene expression levels in pathway engineering. Because of the basal activity of the U4C ARO9 promoter, lower levels of acetoin production were also observed even in the absence of tryptophan (Fig. 4b). The acetoin titers increased gradually as increasing tryptophan concentrations, faithfully reflecting the tryptophan concentration-dependent increase in U4C ARO9 promoter activity (Fig. 4c). As a result, up to 3.4 g/L acetoin was produced in the presence of 800 μg/mL tryptophan. Although promoters stronger than the U4C ARO9 promoter could be more effective in maximizing acetoin production levels, these results demonstrate the usefulness of the tryptophan-inducible promoter in modulating metabolic flux simply by changing the concentrations of tryptophan.
Application of the GABA-inducible UGA4 promoter to metabolic engineering
Since we demonstrated that Aro80-dependent transcriptional regulation can be successfully used to design novel inducible promoters, we searched for other Zn2Cys6 family member of transcription factors that are regulated by inducers suitable for genetic engineering. Uga3 transcription factor is involved in the utilization of GABA as a nitrogen source by activating transcription of UGA1, UGA2, and UGA4 genes in response to GABA (Andre 1990; Cardillo et al. 2011; Idicula et al. 2002; Talibi et al. 1995). In the UGA4 promoter, the region from −404 to −386 was identified as UASGABA (Idicula et al. 2002; Talibi et al. 1995), where CGG half sites are aligned in everted orientation (CCGN4CGG) (Fig. 5a). It has been known that Uga3-dependent activation of UASGABA requires Uga35, another Zn2Cys6 protein with a pleiotropic function (Garcia et al. 2000).

Application of GABA-inducible UGA4 promoter for acetoin production. a Construction of GABA-inducible system for acetoin production. b Cells harboring p413[P UGA4 ]-alsS and p426[P UGA4 ]-alsD were tested for acetoin production depending on GABA concentrations
We investigated whether the GABA-inducible UGA4 promoter can be applied to metabolic engineering for acetoin production. The alsS and alsD genes were expressed under the control of UGA4 promoter (−460 to −1) from low and high copy number plasmids, respectively. Cells harboring the two plasmids produced 0.1 g/L acetoin in the absence of GABA (Fig. 5b), which is about 10-fold lower than that produced in cells expressing alsS and alsD from the U4C ARO9 promoter (Fig. 4c). Therefore, the UGA4 promoter might have a lower basal activity than that of U4C ARO9 under our experimental conditions. However, acetoin production increased in correlation to GABA concentrations, resulting up to 5-fold increase in acetoin titer in the presence of 800 μg/mL GABA. These results demonstrate that GABA can be used as a dose-dependent modulator of the UGA4 promoter activity in metabolic engineering.
Discussion
Promoters, the key determinants of transcriptional initiation, are essential components for controlling gene expression in metabolic engineering and synthetic biology (Blazeck and Alper 2013; Nevoigt et al. 2007). In this study, we demonstrated that promoters regulated by Aro80 transcription factor can be used as tryptophan-inducible promoters for pathway engineering in S. cerevisiae. The tryptophan-induced expression levels can be modulated by changing the number of Aro80 binding sites, plasmid copy numbers, and the concentrations of inducer, providing a dynamic range of promoter strengths available for fine-tuning gene expression levels for pathway optimization. Furthermore, we showed that GABA-inducible UGA4 promoter, regulated by Uga3, can also be used in metabolic engineering.
The tryptophan- or GABA-inducible promoters have advantages in that their promoter strengths can be easily modulated by adding different concentrations of inducers directly into the culture medium. Therefore, these promoters might be useful in regulating gene expression levels at specific time points during the growth. Among the inducible promoters available in S. cerevisiae, the CUP1 promoter can also be regulated by Cu2+ concentration-dependent manner, but the toxicity of Cu2+ can be a problem when using high concentrations of Cu2+ (Hottiger et al. 1995). In the case of GAL promoters, complete medium exchange is necessary to prevent glucose repression effect (Johnston 1987; Lohr et al. 1995). In addition, because galactose is used as a carbon source, it is not convenient to modulate galactose concentrations as a way to regulate expression levels. Other regulated promoters such as P ADH2 , P PHO5 , and P MET25 are repressed in the presence of glucose (Price et al. 1990), inorganic phosphate (Rudolph and Hinnen 1987), and methionine (Mumberg et al. 1994), respectively. Therefore, these promoters are useful for inducing gene expression when such nutrients or metabolite are depleted during the cultivation, but not appropriate for dose-dependent regulation by the regulating chemicals. As dose-dependent inducible systems, synthetic transcription factors, constructed by fusing DNA binding domains with transcription activating domains, have been developed, which are regulated by tetracycline analog doxycycline or hormones such as β-estradiol (Belli et al. 1998; Liang et al. 2013; McIsaac et al. 2013). The tryptophan-induced expression levels form the U4C ARO9 promoter was within a similar range of the P ADH1 -deriven expression levels. Since P ADH1 promoter is weaker than other widely used strong promoters such as P TDH3 and P TEF1 (Mumberg et al. 1995; Sun et al. 2012), the tryptophan-inducible promoters might be suitable for genes requiring low- to intermediate-level expression in the metabolic pathway. The activity of tryptophan- or GABA-inducible promoters could be further enhanced by various promoter engineering strategies, which include combining the UAS with different core promoters and terminators (Blazeck et al. 2012; Curran et al. 2013), or introducing poly(dA:dT) tracks that disfavor nucleosome assembly (Raveh-Sadka et al. 2012).
Taken together, we newly introduced tryptophan- and GABA-inducible promoters as useful tools for metabolic engineering in S. cerevisiae. The wide range of controllable expression levels of these promoter systems might contribute to fine-tuning gene expression levels and timing for pathway optimization.
References
Adams BG (1972) Induction of galactokinase in Saccharomyces cerevisiae: kinetics of induction and glucose effects. J Bacteriol 111(2):308–315
Alper H, Fischer C, Nevoigt E, Stephanopoulos G (2005) Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A 102(36):12678–12683
Andre B (1990) The UGA3 gene regulating the GABA catabolic pathway in Saccharomyces cerevisiae codes for a putative zinc-finger protein acting on RNA amount. Mol Gen Genet 220(2):269–276
Andrianantoandro E, Basu S, Karig DK, Weiss R (2006) Synthetic biology: new engineering rules for an emerging discipline. Mol Syst Biol 2:0028
Belli G, Gari E, Piedrafita L, Aldea M, Herrero E (1998) An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res 26(4):942–947
Blazeck J, Alper HS (2013) Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol J 8(1):46–58
Blazeck J, Garg R, Reed B, Alper HS (2012) Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol Bioeng 109(11):2884–2895
Blazeck J, Reed B, Garg R, Gerstner R, Pan A, Agarwala V, Alper HS (2013) Generalizing a hybrid synthetic promoter approach in Yarrowia lipolytica. Appl Microbiol Biotechnol 97(7):3037–3052
Blount BA, Weenink T, Vasylechko S, Ellis T (2012) Rational diversification of a promoter providing fine-tuned expression and orthogonal regulation for synthetic biology. PLoS One 7(3):e33279
Cardillo SB, Correa Garcia S, Bermudez Moretti M (2011) Common features and differences in the expression of the three genes forming the UGA regulon in Saccharomyces cerevisiae. Biochem Biophys Res Commun 410(4):885–889
Cartwright CP, Li Y, Zhu YS, Kang YS, Tipper DJ (1994) Use of beta-lactamase as a secreted reporter of promoter function in yeast. Yeast 10(4):497–508
Curran KA, Karim AS, Gupta A, Alper HS (2013) Use of expression-enhancing terminators in Saccharomyces cerevisiae to increase mRNA half-life and improve gene expression control for metabolic engineering applications. Metab Eng 19:88–97
Da Silva NA, Srikrishnan S (2012) Introduction and expression of genes for metabolic engineering applications in Saccharomyces cerevisiae. FEMS Yeast Res 12(2):197–214
Des Etages SA, Falvey DA, Reece RJ, Brandriss MC (1996) Functional analysis of the PUT3 transcriptional activator of the proline utilization pathway in Saccharomyces cerevisiae. Genetics 142(4):1069–1082
Eden E, Lipson D, Yogev S, Yakhini Z (2007) Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol 3(3):e39
Garcia SC, Moretti MB, Batlle A (2000) Constitutive expression of the UGA4 gene in Saccharomyces cerevisiae depends on two positive-acting proteins, Uga3p and Uga35p. FEMS Microbiol Lett 184(2):219–224
Hahn S, Young ET (2011) Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189(3):705–736
Hartner FS, Ruth C, Langenegger D, Johnson SN, Hyka P, Lin-Cereghino GP, Lin-Cereghino J, Kovar K, Cregg JM, Glieder A (2008) Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res 36(12):e76
Hazelwood LA, Daran JM, van Maris AJ, Pronk JT, Dickinson JR (2008) The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl Environ Microbiol 74(8):2259–2266
Hottiger T, Kuhla J, Pohlig G, Furst P, Spielmann A, Garn M, Haemmerli S, Heim J (1995) 2-Micron vectors containing the Saccharomyces cerevisiae metallothionein gene as a selectable marker: excellent stability in complex media, and high-level expression of a recombinant protein from a CUP1-promoter-controlled expression cassette in cis. Yeast 11(1):1–14
Idicula AM, Blatch GL, Cooper TG, Dorrington RA (2002) Binding and activation by the zinc cluster transcription factors of Saccharomyces cerevisiae. Redefining the UASGABA and its interaction with Uga3p. J Biol Chem 277(48):45977–45983
Iraqui I, Vissers S, Andre B, Urrestarazu A (1999) Transcriptional induction by aromatic amino acids in Saccharomyces cerevisiae. Mol Cell Biol 19(5):3360–3371
Johnston M (1987) A model fungal gene regulatory mechanism: the GAL genes of Saccharomyces cerevisiae. Microbiol Rev 51(4):458–476
Keasling JD (2010) Manufacturing molecules through metabolic engineering. Science 330(6009):1355–1358
Lee KM, DaSilva NA (2005) Evaluation of the Saccharomyces cerevisiae ADH2 promoter for protein synthesis. Yeast 22(6):431–440
Lee K, Hahn JS (2013) Interplay of Aro80 and GATA activators in regulation of genes for catabolism of aromatic amino acids in Saccharomyces cerevisiae. Mol Microbiol 88(6):1120–1134
Liang J, Ning JC, Zhao H (2013) Coordinated induction of multi-gene pathways in Saccharomyces cerevisiae. Nucleic Acids Res 41(4):e54
Lohr D, Venkov P, Zlatanova J (1995) Transcriptional regulation in the yeast GAL gene family: a complex genetic network. FASEB J 9(9):777–787
MacPherson S, Larochelle M, Turcotte B (2006) A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70(3):583–604
Macreadie IG, Horaitis O, Verkuylen AJ, Savin KW (1991) Improved shuttle vectors for cloning and high-level Cu2+-mediated expression of foreign genes in yeast. Gene 104(1):107–111
McIsaac RS, Oakes BL, Wang X, Dummit KA, Botstein D, Noyes MB (2013) Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast. Nucleic Acids Res 41(4):e57
Mumberg D, Muller R, Funk M (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22(25):5767–5768
Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156(1):119–122
Nevoigt E (2008) Progress in metabolic engineering of Saccharomyces cerevisiae. Microbiol Mol Biol Rev 72(3):379–412
Nevoigt E, Fischer C, Mucha O, Matthaus F, Stahl U, Stephanopoulos G (2007) Engineering promoter regulation. Biotechnol Bioeng 96(3):550–558
Nielsen J, Larsson C, van Maris A, Pronk J (2013) Metabolic engineering of yeast for production of fuels and chemicals. Curr Opin Biotechnol 24(3):398–404
Price VL, Taylor WE, Clevenger W, Worthington M, Young ET (1990) Expression of heterologous proteins in Saccharomyces cerevisiae using the ADH2 promoter. Methods Enzymol 185:308–318
Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, Zeevi D, Sharon E, Weinberger A, Segal E (2012) Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 44(7):743–750
Rudolph H, Hinnen A (1987) The yeast PHO5 promoter: phosphate-control elements and sequences mediating mRNA start-site selection. Proc Natl Acad Sci U S A 84(5):1340–1344
Sun J, Shao Z, Zhao H, Nair N, Wen F, Xu JH, Zhao H (2012) Cloning and characterization of a panel of constitutive promoters for applications in pathway engineering in Saccharomyces cerevisiae. Biotechnol Bioeng 109(8):2082–2092
Sze JY, Woontner M, Jaehning JA, Kohlhaw GB (1992) In vitro transcriptional activation by a metabolic intermediate: activation by Leu3 depends on alpha-isopropylmalate. Science 258(5085):1143–1145
Talibi D, Grenson M, Andre B (1995) Cis- and trans-acting elements determining induction of the genes of the gamma-aminobutyrate (GABA) utilization pathway in Saccharomyces cerevisiae. Nucleic Acids Res 23(4):550–557
Todd RB, Andrianopoulos A (1997) Evolution of a fungal regulatory gene family: the Zn(II)2Cys6 binuclear cluster DNA binding motif. Fungal Genet Biol 21(3):388–405
Xiao Z, Lu JR (2014) Strategies for enhancing fermentative production of acetoin: a review. Biotechnol Adv 32(2):492–503
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (2012-R1A1A-3011963).
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Kim and K. Lee contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kim, S., Lee, K., Bae, SJ. et al. Promoters inducible by aromatic amino acids and γ-aminobutyrate (GABA) for metabolic engineering applications in Saccharomyces cerevisiae . Appl Microbiol Biotechnol 99, 2705–2714 (2015). https://doi.org/10.1007/s00253-014-6303-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6303-5