
Abstract
Both varied and strong promoters are essential for metabolic and pathway engineering applications in any host organism. To enable this capacity, here we demonstrate a generalizable method for the de novo construction of strong, synthetic hybrid promoter libraries. Specifically, we demonstrate how promoter truncation and fragment dissection analysis can be utilized to identify both novel upstream activating sequences (UAS) and core promoters—the two components required to generate hybrid promoters. As a base case, the native TEF promoter in Yarrowia lipolytica was examined to identify putative UAS elements that serve as modular synthetic transcriptional activators. Resulting synthetic promoters containing a core promoter region activated by between one and twelve tandem repeats of the newly isolated, 230 nucleotide UASTEF#2 element showed promoter strengths 3- to 4.5-fold times the native TEF promoter. Further analysis through transcription factor binding site abrogation revealed the GCR1p binding site to be necessary for complete UASTEF#2 function. These various promoters were tested for function in a variety of carbon sources. Finally, by combining disparate UAS elements (in this case, UASTEF and UAS1B), we developed a high-strength promoter with for Y. lipolytica with an expression level of nearly sevenfold higher than that of the strong, constitutive TEF promoter. Thus, the general strategy described here enables the efficient, de novo construction of synthetic promoters to both increase native expression capacity and to produce libraries for tunable gene expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High, tunable levels of gene expression are necessary for metabolic and pathway engineering applications in model host organisms. However, newly isolated or poorly understood organisms often lack well-characterized promoters required for high transcription rates and tunable gene expression. Prior attempts to modulate gene expression include altering endogenous promoter strength through point mutations (Alper et al. 2005; Nevoigt et al. 2006), tuning intergenic regions in operons (Pfleger et al. 2006), and saturation mutagenesis of non-conserved promoter spacer regions (Jensen and Hammer 1998; Jeppsson et al. 2003; Rud et al. 2006). While successful at generating large ranges in relative expression, these methods are quite limited by their tendency to decrease gene expression levels below the original starting point and thus rely on a strong, well-defined starting promoter. To overcome these limitations, we have recently demonstrated the utility of a synthetic hybrid promoter engineering approach in the model yeast Saccharomyces cerevisiae and in the oleaginous yeast Yarrowia lipolytica (Blazeck et al. 2011, 2012). Here we seek to generalize this approach through the de novo isolation and evaluation of a novel UAS element and, in doing so, create one of the strongest known promoters for Y. lipolytica.
Y. lipolytica is an excellent example of a unique host with intriguing capabilities whose applications, such as native or heterologous protein excretion, were until recently limited by a lack of strong characterized promoters (Barth and Gaillardin 1997; Beopoulos et al. 2009; Madzak et al. 2004; Muller et al. 1998; Nicaud et al. 2002). Y. lipolytica's transcriptional machinery is amenable to a hybrid promoter approach (Blazeck et al. 2011; Madzak et al. 2000), and simple genetic tools are readily available (Chen et al. 1992; Davidow et al. 1985; Fournier et al. 1993; Juretzek et al. 2001; Ledall et al. 1994; Matsuoka et al. 1993; Vanheerikhuizen et al. 1985; Vernis et al. 1997, 2001).
Synthetic hybrid promoters consist of a core promoter region fused to a single upstream activating sequence (UAS) or multiple tandem UAS repeats that augment and modulate promoter function (Blazeck et al. 2011; Deboer et al. 1983; Madzak et al. 2000; Mukai et al. 1992; Rosenberg and Tekamp-olson 1992). These upstream activation regions help localize trans-acting regulatory elements (transcription factors) to increase the transcriptional activity of the core promoter region. Thus, the UAS and core promoter regions act as modular synthetic parts that can be combined to produce a strong, functional UAS-core promoter chimera. In this manner, promoter strength can be elevated by either utilizing a stronger core promoter or by increasing the number of UAS repeats (Blazeck et al. 2011, 2012). All prior hybrid promoter engineering efforts in the oleaginous yeast Y. lipolytica have utilized the same 105 basepair upstream activating sequence named UAS1B as the sole UAS element. UAS1B was originally isolated through a functional dissection of the strong, tightly regulated XPR2 native promoter and showed promising constitutive transcriptional activation abilities (Blanchin-roland et al. 1994; Madzak et al. 1999, 2000). Multiple tandem repeats of this UAS element adjacent to a core minimal LEU2 promoter or core TEF-based promoters resulted in significant transcriptional activation (Blazeck et al. 2011; Madzak et al. 2000).
The full flexibility of hybrid promoter generation has been limited by the reliance on only well-established upstream activating regions—a building block likely unidentified in poorly understood hosts. To circumvent this limitation, we describe here the methodology for the isolation, characterization, and incorporation of novel UAS elements into strong, tunable hybrid promoter libraries in Y. lipolytica. In particular, we demonstrate the modular transcriptional activation capacity of a 257-basepair upstream region in the Y. lipolytica TEF promoter (dubbed UASTEF). A full dissection of UASTEF element yielded a fully active, 230 bp UAS, element, dubbed UASTEF#2. Only six tandem repeats are necessary for saturation of promoter enhancement using this UAS, and a mutational analysis implicates the GCR1p transcription factor binding site to be essential for full UAS function. Kinetic time course experiments reveal that TEF and UASTEF#2-based promoters are fully active after 2 days growth before beginning to lose transcriptional activity. Furthermore, characterization in alternative carbon sources demonstrates that these promoters are constitutively active in a variety of conditions. Thus, this work establishes a generalizable approach of hybrid promoter engineering through de novo isolation of novel UAS elements and incorporation into strong, tunable promoter libraries.
Materials and methods
Strains and media
Escherichia coli strain DH10B was used for all cloning and plasmid propagation. DH10B was grown at 37 °C with constant shaking in LB Media Broth (Teknova) supplemented with 50 μg/ml ampicillin for plasmid propagation. Y. lipolytica strain PO1f (ATCC # MYA-2613), a leucine and uracil auxotroph devoid of any secreted protease activity (Madzak et al. 2000), was used for all experiments. Y. lipolytica PO1f containing plasmid was routinely cultivated at 30 °C with constant agitation in YSC-LEU media consisting of 20 g/L glucose (Fisher Scientific), 0.69 g/L CSM-Leucine (MP Biomedicals), and 0.67 g/L Yeast Nitrogen Base (Becton, Dickinson, and Company). When testing on alternative carbon sources, glucose was replaced by 20 g/L sucrose (Acros Organics), 20 g/L glycerol (Fisher Scientific), or 30 g/L oleic acid (Sigma Aldrich). Solid media for E. coli and Y. lipolytica was prepared by adding 15 g/L agar (Teknova) to liquid media.
Cloning procedures
All restriction enzymes were purchased from New England Biolabs, and all digestions were performed according to standard protocols. PCR reactions were set up with recommended conditions using Phusion high fidelity DNA polymerase (Finnzymes). Ligation reactions were performed overnight at 22 °C using T4 DNA Ligase (Fermentas). Gel extractions were performed using the Fermentas GeneJET extraction kit purchased from Fisher ThermoScientific. E. coli minipreps were performed using the Zyppy Plasmid Miniprep Kit (Zymo Research Corporation). Transformation of E. coli strains was performed using standard electroporator protocols (Sambrook and Russell 2001). Transformation of Y. lipolytica was performed using the Zymogen Frozen EZ Yeast Transformation Kit II (Zymo Research Corporation). Genomic DNA was extracted from Y. lipolytica using the Wizard Genomic DNA Purification kit (Promega).
Plasmid construction
Primer sequences can be found in Table S1. All Y. lipolytica plasmids were centromeric, replicative vectors derived from plasmid pSl16-Cen1-1(227) (Yamane et al. 2008), which was modified to include a multicloning site, a hrGFP green fluorescent reporter gene (pIRES-hrGFP, Agilent), and a cyc1 terminator (Mumberg et al. 1995) to create plasmid pMCS-hrGFP. A TEF promoter (Damude et al. 2006; Muller et al. 1998) was added to pMCS-hrGFP to form pMCS-TEF-hrGFP. Vectors pMCS-HrGFP, pMCS-TEF-HrGFP, pUC-UAS1B16-TEF, pUC-UAS1B8-TEF, pMCS-TEF(136)-hrGFP, pMCS-TEF-LacZ, and pUC-UAS1B2-Leum have been described previously (Blazeck et al. 2011). Unless stated otherwise, all PCRs utilized pMCS-TEF-hrGFP as template. All plasmids were sequenced confirmed.
Construction of UASTEF(n)-TEF and UASTEF(n)-Leum hrGFP expression cassettes
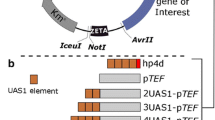
A UASTEF fragment amplified by primers JB438/439 was SphI/HindIII digested and inserted in place of the UAS1B16 fragment in pUC-UAS1B16-TEF to form pUC-UASTEF(1)-TEF. A second UASTEF fragment (primers JB440/437) was inserted into pUC-UASTEF(1)-TEF with BstBI/SphI to form pUC-UASTEF(2)-TEF. UASTEF(1)-TEF and UASTEF(2)-TEF promoters were gel extracted and inserted into pMCS-hrGFP with BstBI/AscI to create pMCS-UASTEF(1)-TEF-hrGFP and pMCS-UASTEF(2)-TEF-hrGFP, respectively. A final UASTEF fragment (primers JB442/443) was inserted into pMCS-UASTEF(2)-TEF-hrGFP with XmaI/BstBI to form pMCS-UASTEF(3)-TEF-hrGFP, completing UASTEF(n)-TEF expression cassette construction (Fig. 1a).


Construction of plasmids for this study. a A schematic picture is provided detailing the construction of UASTEF(n)-TEF and UASTEF(n)-Leum promoter hrGFP fluorescence cassettes, b the construction of the UASTEF(2)-UAS1B8-TEF-hrGFP expression cassette, c and the construction of a library of UASTEF#2 hrGFP expression cassettes. Restriction enzymes utilized are abbreviated as follows: A—AscI, Ba—BamHI, B—BstBI, E—EcoRI, H—HindIII, Sa—SalI, S—SphI, Xb—XbaI, X—XmaI
An EcoRI/SphI digested UASTEF fragment amplified by primers JB436/437 was inserted in place of the UAS1B2 fragment in plasmid pUC-UAS1B2-Leum to create plasmid pUC-UASTEF(1)-Leum. A BstB1/EcoRI digested UASTEF fragment (primers JB440/441) was inserted into pUC-UASTEF(1)-Leum to create pUC-UASTEF(2)-Leum. UASTEF(1)-Leum and UASTEF(2)-Leum promoters were inserted into pMCS-hrGFP with BstB1/AscI to create pMCS-UASTEF(1)-Leum-hrGFP and pMCS-UASTEF(2)-Leum-hrGFP, respectively. A final UASTEF fragment (primers JB442/443) was inserted into pMCS-UASTEF(2)-Leum-hrGFP with XmaI/BstBI to form pMCS-UASTEF(3)-Leum-hrGFP, completing UASTEF(n)-Leum expression cassette construction.
Construction of the UASTEF(2)-UAS1B8-TEF-hrGFP expression cassette
A BstBI/EcoRI digested UASTEF fragment amplified with primers JB440/441 was inserted 5′ of the UAS1B8 region of pUC-UAS1B8-TEF to form pUC-UASTEF-UAS1B8-TEF. Promoter UASTEF-UAS1B8-TEF was extracted with BstBI/AscI and inserted into pMCS-hrGFP, and a final 5′ UASTEF fragment was added with XmaI/BstBI to create pMCS-UASTEF(2)-UAS1B8-TEF-hrGFP (Fig. 1b).
Dissection of the TEF upstream region
Twenty-two overlapping fragments spanning the UASTEF region were inserted 5′ of the TEF(136) minimal promoter in plasmid pMCS-TEF(136)-hrGFP with XmaI/BstBI digests to form plasmids pMCS-UASTEF#1-TEF(136)-hrGFP through pMCS-UASTEF#22-TEF(136)-hrGFP. Primer pairs JB442/443, JB442/508, JB442/509, JB442/510, and JB442/511 amplified fragments UASTEF#1through UASTEF#5. JB503/443, JB503/507, JB503/508, JB503/510, and JB503/511 amplified fragments UASTEF#6 through UASTEF#10. JB504/443, JB504/509, and JB504/511 amplified fragments UASTEF#11 through UASTEF#13. JB505/507, JB505/508, JB505/510, and JB505/511 amplified fragments UASTEF#14 through UASTEF#17. Finally, JB506/443, JB506/507, JB506/508, JB506/510, and JB506/511 amplified fragments UASTEF#18 through UASTEF#22 (Fig. 4a).
Construction of library of UASTEF#2(n)-TEF(136) hrGFP expression cassettes
An initial UASTEF#2 region amplified by primers JB544/537 was inserted into plasmid pUC-UAS1B8-TEF(136) in place of UAS1B8 with EcoRI/HindIII to form plasmid pUC-UASTEF#2(1)-TEF(136). Second (primers JB546/539) and third UASTEF#2 (JB548/541) elements were inserted into pUC-UASTEF#2(1)-TEF(136) with EcoRI/BamHI and then EcoRI/XbaI digests to create pUC-UASTEF#2(2)-TEF(136) and then pUC-UASTEF#2(3)-TEF(136), respectively. To enable plasmid construction, a multicloning site annealed together from JB630/631 was inserted into p416-MCS-yECitrine (Blazeck et al. 2012) to created plasmid pTMCS. A 737-bp fragment containing three tandem UASTEF#2 elements in series amplified from pUC-UASTEF#2(3)-TEF(136) plasmid DNA with primers JB620/621 was inserted twice sequentially into pTMCS, first using SalI-HF/HindIII then SphI-HF/EcoRI-HF to create plasmids pTMCS-UASTEF#2(3) and pTMCS-UASTEF#2(6). This same 737-bp fragment, digested with SphI/EcoRI, was inserted into the three pUC-UASTEF#2(1, 2, and 3)-TEF(136) plasmids to create plasmids pUC-UASTEF#2(4, 5, and 6)-TEF(136). The six promoters—UASTEF#2(1)-TEF(136) through UASTEF#2(6)-TEF(136)—were BstBI/AscI-extracted and inserted into pMCS-hrGFP to create pMCS-UASTEF#2(1)-TEF(136)-hrGFP through pMCS-UASTEF#2(6)-TEF(136)-hrGFP. Three or six UASTEF#2 tandem repeats were BstBI/XmaI extracted from pTMCS-UASTEF#2(3) or pTMCS-UASTEF#2(6) and inserted into pMCS-UASTEF#2(6)-hrGFP to create pMCS-UASTEF#2(9)-TEF(136)-hrGFP and pMCS-UASTEF#2(12)-TEF(136)-hrGFP, completing UASTEF#2(n)-TEF(136)-hrGFP library construction (Fig. 1c).
Construction of the lacZ expression cassettes
The β-galactosidase gene encoded by E. coli lacZ (Kalnins et al. 1983) was gel-extracted from pMCS-TEF-lacZ with AscI/PacI and inserted in place of hrGFP in certain pMCS- hrGFP-based plasmid series to generate lacZ expression cassettes.
Construction of mutant UASTEF#2 elements
Utilizing plasmid pUC-UASTEF#2(1)-TEF(136) as DNA template for the Stratagene Quikchange mutagenesis kit, three putative transcription factor binding sites (TFBSs) were removed from UASTEF#2. Primers JB682/683 deleted a “tgtgt” motif to abrogate a putative NDT80p TFBS. Primers JB680/681 deleted a “ttaag” motif to remove a putative MCM1 TFBS, and primers JB684/685 deleted a “gccatc” motif to remove a GCRp putative TFBS. Sequential mutagenesis reactions created all combinations of NDT80p, MCM1p, and GCR1p TFBS deletion mutants in the pUC-UASTEF#2(1)-TEF(136) background. These seven mutants were BstB1/AscI-extracted and inserted into pMCS-hrGFP to complete mutant UASTEF#2 expression cassette construction (Fig. 4c).
Promoter characterization by flow cytometry
The hrGFP green fluorescent protein has been validated as an ideal tool to assess promoter strength in Y. lipolytica at the single cell level (Blazeck et al. 2011), and thus was employed to assess relative promoter activity for the majority of results generated in this study. Y. lipolytica PO1f strains, transformed with different plasmids, were inoculated directly from glycerol stock (in biological triplicate) in YSC-LEU media for 48 h at 30 °C with shaking, and then normalized to an OD600 of 0.01 in 2 ml fresh YSC-LEU and incubated for another 48 h (unless otherwise stated) at 30 °C in a rotary drum (CT-7, New Brunswick Scientific) at speed seven. A time course of fluorescence value showed 48 h to be an optimal incubation time for high expression levels from native and hybrid promoters (Blazeck et al. 2011). To harvest, the cultures were spun down at 4 °C at 1,000×g for 5 min, washed, and resuspended in 1 ml ice cold water before testing with a FACS Fortessa (BD Biosciences) using the GFP fluorochrome, a voltage of 319, and a 10,000 cell count for hrGFP detection. Samples were kept on ice during the test, and the data was analyzed using FlowJo software (Tree Star Inc., Ashland, OR) to compute mean fluorescence values. Day-to-day variability was mitigated by analyzing all comparable strains on the same day. An average fluorescence and standard deviation were calculated from the mean values of biological replicates.
Promoter characterization through β-galactosidase assay
Y. lipolytica PO1f strains, transformed with different plasmids, were inoculated directly from glycerol stock (in biological triplicate) in YSC-LEU media for 48 h at 30 °C in a rotary drum (CT-7, New Brunswick Scientific) at speed seven, and then normalized to an OD600 of 0.01 in 2 ml fresh YSC-LEU and incubated for another 48 h in the same conditions. The cultures were washed twice and resuspended in 1 ml Z buffer, and their OD600 readings were recorded (Gaillardin and Ribet 1987; Miller 1972). β-galactosidase assays were performed as described by Miller (1972) using 25 μl of chloroform-permeabilized cells, with a reaction time of 17 min.
Kinetic analysis of promoters
Y. lipolytica PO1f strains, transformed with different plasmids, were inoculated directly from glycerol stock (in biological triplicate) in YSC-LEU media for 48 h at 30 °C in a rotary drum (CT-7, New Brunswick Scientific) at speed seven and then normalized to an OD600 of 0.01 in 2 ml fresh YSC-LEU and incubated for either 24, 48, 72, or 96 h in the same conditions. Cultures were inoculated such that all cultures (for the 24-, 48-, 72-, and 96-h time points) were harvested and tested via flow cytometry at the same time.
Results
Isolation of a Y. lipolytica TEF promoter region UAS
Prior analysis of the 406 basepair full length TEF promoter revealed that the 136 basepair proximal to the ATG start codon, dubbed TEF(136), was unable to drive the expression of an hrGFP reporter gene (Blazeck et al. 2011). However, when this core promoter fragment was used in conjunction with a known UAS element, resulting TEF(136)-based hybrid promoters generated high fluorescence levels. These results established TEF(136) as a functional, low-strength core promoter that had been apparently stripped of its native UAS elements (Blazeck et al. 2011). Analysis of the TEF promoter revealed that a drastic change in GC content coincided with the end of all putative TATA elements at 149 basepairs upstream of the ATG start codon. Consequently, we postulated that a strong upstream activating sequence responsible for the majority of the TEF promoter's transcriptional activation ability must be located further upstream in the remaining 257 basepairs. We tested this hypothesis by constructing two distinct series of hybrid promoters in which between one and three tandem repeats of a putative 257 basepair TEF upstream activating sequence, dubbed UASTEF, were fused to a core promoter. Hybrid promoter series UASTEF(n)-TEF employed the 406 basepair native TEF promoter (TEF) as a core promoter, while series UASTEF(n)-Leum employed a minimal core LEU2 promoter (Leum). Flow cytometry analysis of the UASTEF(n)-TEF and UASTEF(n)-Leum hybrid promoters revealed strong expression enhancement by tandem UASTEF elements (Fig. 2). The enhancement of fluorescence levels were strongly linearly correlated to the number of UASTEF copies (r 2 = 0.9899 for TEF core and r 2 = 0.9983 for Leum core—r 2 values calculated using a linear regression in Microsoft Excel), demonstrating the modularity and functional additivity of this novel UAS. Net promoter strength strongly depended on core promoter choice, as hybrid promoters employing the TEF core were more than threefold stronger than Leum-based counterparts. The maximum expression amplification imparted to each core promoter differed as well. In this vain, three UASTEF tandem repeats generated a 6.45-fold amplification of expression when fused to the Leum core promoter (compared to basal Leum), while only a 3.44-fold increase emerged in the corresponding TEF-based constructs. These results echo our previous findings that amplification levels were highest with more minimal core promoter constructs.

Functional testing of the novel UASTEF element. Relative fluorescence values indicate that the UASTEF element functions as a strong modular upstream activating sequence, linearly increasing expression capacity of both the TEF native promoter (r 2 = 0.9899) and the Leum minimal promoter (r 2 = 0.9983). Three tandem repeats of the UASTEF element elevated protein expression to more than threefold base levels for the TEF promoter and sixfold for the Leum promoter. Final expression capacity of the UASTEF(3)-TEF promoter approached UAS1B16-TEF and UAS1B16-Leum levels, two of the strongest previously characterized hybrid promoter in Y. lipolytica. Error bars represent the standard deviation of measurements between biological triplicates
Investigating the effect of incorporating disparate UAS sequences
We next investigated the impact of combining disparate UAS elements. The UASTEF element proximal to the core promoter in promoter UASTEF(3)-TEF was replaced by an UAS1B8 fragment (Blazeck et al. 2011) to form hybrid promoter UASTEF(2)-UAS1B8-TEF. The mean fluorescence levels generated by UASTEF(2)-UAS1B8-TEF are at least as high as the strongest promoters yet described in Y. lipolytica (Blazeck et al. 2011), advocating future work assessing potential benefits of incorporating multiple, distinct UAS elements into hybrid promoters (Fig. 3).

Combining the novel UASTEF with the UAS1B element. Relative fluorescence values indicate that the UASTEF (2)-UAS1B8-TEF promoter surpasses in strength the previously characterized UAS1B16-TEF and UAS1B32-Leum promoters, the strongest promoters constructed in Y. lipolytica. Error bars represent the standard deviation of measurements between biological triplicates
Functional dissection analysis of the UASTEF element through promoter truncation and transcription factor binding site removal
With the 257-bp UASTEF established as a modular, synthetic transcriptional amplifier, we sought to dissect its genetic sequence in search of a more minimal, compact UAS element. Twenty-two unique but overlapping fragments spanning the UASTEF sequence were PCR-amplified to create a UASTEF-based truncation library (Fig. 4a). Putative UAS library elements were individually fused to the minimal TEF(136) core promoter to form promoters UASTEF#1-TEF(136) through UASTEF#22-TEF(136). The minimal TEF(136) core promoter was selected for hybrid promoter construction to allow for the highest sensitivity in measuring UAS activity. The newly constructed hybrid promoter series was tested with hrGFP-based flow cytometry analysis. The fluorescence data reveal that the majority of truncated UASTEF fragments continue to retain at least modest UAS activity (Fig. 4a, b). This result suggests that nearly all of these fragments can serve to create functional, synthetic hybrid promoters. However, hybrid promoter strength showed a pronounced correlation to UASTEF fragment length, providing evidence that the majority of the original 257 bp UASTEF sequence is required for full activity (Fig. 4b). UASTEF#2 displayed the highest activating capacity of the UASTEF truncations tested, elevating the strength of the TEF(136) core promoter to levels 1.5-fold times the native TEF promoter (Fig. 4a). Thus, we selected UASTEF#2 for further hybrid promoter library construction and for a more rigorous examination.


Dissection of the UASTEF element. a Twenty-two overlapping fragments spanning the UASTEF sequence were inserted upstream of a TEF(136)-hrGFP expression cassette and tested for transcriptional amplification activity. Relative fluorescence values are shown. b Relative fluorescence values are plotted as a function of putative UAS length, with a noticeable correlation between decreasing putative UAS strength and decreasing UAS length. c A simplified schematic picture is provided detailing the location of specific consensus transcription factor binding sites within the UASTEF#2 element and the TEF native promoter. d Three putative transcription factor binding sites (NDT80p, MCM1p, and GCR1p) were removed individually and combinatorially from the UASTEF#2 element with at UASTEF#2(1)-TEF(136)-hrGFP expression cassette. Relative fluorescence values are shown. Error bars represent the standard deviation of measurements between biological triplicates
The majority of the UASTEF#2 element is necessary for full UAS activity, precluding a more thorough characterization through truncation analyses. Thus, we characterized UASTEF#2 through the systematic removal of three putative transcription factor binding sites predicted within this sequence. In our truncation analysis, we observed that removing 27 basepair at the 5′ end of UASTEF (−406 to −386 deletion) tended to decrease UAS strength, but further truncation had little effect. We also observed that the removal of 27 basepairs from the 3′ end of the original UASTEF (−149 to −176 deletion) often increased UAS strength, but further 3′ truncation (−176 to −196/−216/−236 deletion) always decreased UAS strength (Fig. 4a, c). Thus, we concluded the existence of an upstream repressive element (URS) between basepairs −149 to −176 and enhancing UAS elements in the remaining truncated regions (−406 to −386 and −176 to −196/−216/−236 deletion).
An analysis of the native TEF and the UASTEF#2-TEF(136) promoters at the sequence level in the yeast promoter database (SCPD) (Zhu and Zhang 1999) revealed that consensus binding sites for yeast transcription factors MCM1p and PUT3p are lost in the 27 basepair truncation. Further sequence analysis of the UASTEF#2 element also highlighted consensus binding sites for transcription factors NDT80p at position −386, for GCR1p at position −186, and for a separate MCM1p at position −257 (Fig. 4c). We hypothesized that the GCR1p and NDT80p binding sites grant UAS activity, while this second native MCM1p binding site reduces UAS activity. To test this hypothesis, we employed site-directed mutagenesis to delete the NDT80p, GCR1p, and MCM1p binding sites from the UASTEF#2-TEF(136) promoter (Fig. 4c), and tested and compared binding site deletion mutants with hrGFP-based flow cytometry. As expected, removal of the NDT80p and GCR1p bindings sites reduced UASTEF#2-TEF(136) promoter strength, by 15 % and 31 %, respectively (Fig. 4d). Combinatorial deletion of NDT80p and GCR1p binding sites only slightly further decreased promoter strength, revealing the GCR1p binding site as the predominate site responsible for UASTEF #2 function. Deletion of the MCM1p binding site had no effect on UASTEF#2-TEF(136) promoter strength (Fig. 4d). Thus, the GCR1p binding site motif is essential for UAS capability, while URS activity is not conferred by the MCM1p binding site.
Utilizing the hybrid promoter approach to construct a novel tandem UAS series
Above, we have demonstrated how UAS elements can be deduced and mechanistically studied through promoter truncation analysis. To complete the generic hybrid promoter construction process, we utilized one, two, three, four, five, six, nine, or twelve tandem repeats of the newly isolated UASTEF#2 sequence to enhance expression of the minimal TEF(136) core promoter. These new UASTEF#2 based promoters were tested and compared to the native TEF promoter, amongst other controls, via hrGFP fluorescence by flow cytometry. Multiple copies of UASTEF#2 increased hybrid promoter strength for up to six tandem repeats, at which point expression enhancement from added UASTEF #2 elements had been saturated (Fig. 5a). Ultimately, UASTEF #2-TEF(136) hybrids yielded expression levels 3.5-fold higher than the native, full length TEF promoter starting point (Fig. 5a). Expression cassettes containing tandem UASTEF #2 copies without the TEF(136) promoter generated no expression above background levels. To further demonstrate the utility of the UASTEF#2-TEF(136) series, we utilized the β-galactosidase gene as alternate reporter protein. Once again, UASTEF#2-mediated enhancement increased promoter activity, producing a final expression enhancement to nearly twice that of the native TEF using this marker (Fig. 5b).

Functional testing of the novel UASTEF#2 element to complete de novo hybrid promoter construction. a Relative fluorescence values indicate that the UASTEF#2 element functions as an upstream activating sequence, increasing expression capacity of the TEF(136) promoter to levels 3.5-fold higher than the native TEF promoter. Final UASTEF#2-based promoter strength remained below UASTEF-based and previously characterized promoters, UAS1B32-Leum and UAS1B16-TEF. b Hybrid promoters were tested with a β-galactosidase reporter gene, yielding similar results to the hrGFP assay. Error bars represent the standard deviation of measurements between biological triplicates
Kinetic analysis of hybrid promoters and effect of media formulation
Previous analysis of the UAS1B element (the only other known upstream activating sequence in Y. lipolytica) revealed expression profiles dependent on both time course and media formulation (Madzak et al. 2000, 2004). Thus, we sought to further characterize our novel UASTEF-based promoters and compare them to several previously constructed hybrid promoters utilizing a thorough time course kinetic analysis and analyzing the effects of alternate carbon sources on promoter activity.
We analyzed the UASTEF#2(n)-TEF(136) series, several high expression UASTEF-truncation hybrids, the novel UASTEF(2)-UAS1B8-TEF promoter, and our previously constructed strong hybrids promoters UAS1B8-TEF(406), UAS1B16-TEF(406), UAS1B16-Leum, and UAS1B32-Leum (Blazeck et al. 2011) via hrGFP flow cytometry after 1, 2, 3, and 4 days of growth to discern effects of cell phase on promoter activity. UASTEF#2-TEF(136) promoters demonstrated fairly constitutive activity, peaking in expression levels after two days before decreasing (Fig. 6a). Interestingly, the UASTEF#2(6)-TEF(136) and UASTEF#2(12)-TEF(136) promoter attain nearly identical peak expression level, but the UASTEF#2(12)-TEF(136) promoter retains full activity much longer. Hybrid promoters based on the UASTEF truncations displayed similar constitutive expression profiles, with the UASTEF#2-TEF(136) promoting the highest expression levels (Fig. 6b). While stronger than the UASTEF-based hybrids, the UAS1B16-Leum and UAS1B32-Leum promoters were not highly activated until the third day of growth, confirming prior observation that the UAS1B element is most active in early stationary phase (Madzak et al. 1999, 2000). The UAS1B8-TEF(406) and UAS1B16-TEF(406) promoters displayed very high fluorescence levels and were fully expressed after only 2 days. The UASTEF(2)-UAS1B8-TEF promoter reached maximal expression capacity quicker than any other tested promoter, further demonstrating the benefit of incorporating multiple, distinct UAS elements into the same hybrid promoter (Fig. 6c).


Kinetic analysis of hybrid promoters. a Relative fluorescence values indicate that tandem copies of UASTEF#2 showed progressive increased in transcription capacity. b Various UASTEF truncation hybrid promoters (from those described in Fig. 3a) displayed similar expression profiles, with the UASTEF#2-TEF(136) promoting the highest expression levels. c UAS1B16-Leum and UAS1B32-Leum promoters were not highly activated until the third day of growth. UAS1B8-TEF(406) and UAS1B16-TEF(406) promoters displayed very high fluorescence levels and were fully expressed after only 2 days, and the UASTEF(2)-UAS1B8-TEF promoter exhibited the high expression capacity quicker than any other tested promoter. Error bars represent the standard deviation of measurements between biological triplicates
To analyze the effects of media on promoter expression capacity, we tested many of the hybrid constructs for protein expression when grown in media utilizing glucose, sucrose, glycerol, or oleic acid as the sole carbon source. UASTEF#2-TEF(136) promoters were minimally impacted by carbon source, with sucrose enabling the highest expression, and glycerol the lowest expression capacity (Fig. 7a). In contrast, promoters containing UAS1B elements were very strongly dependent on media composition, with highest expression on sucrose or oleic acid (Fig. 7b). Thus, hybrid promoters constructed with the newly isolated TEF UAS were more constitutive than previous Y. lipolytica hybrid promoters.

Effect of media formulation on hybrid promoter expression. a Hybrid constructs were tested in medium containing glucose, sucrose, glycerol, or oleic acid as the sole carbon source. UASTEF#2-TEF(136) promoters did not significantly vary with carbon source. b UAS1B promoters were further activated when grown on sucrose or oleic acid and had lower levels when grown on glycerol. Error bars represent the standard deviation of measurements between biological triplicates
Discussion
This study generalizes a synthetic hybrid promoter approach for generating promoter libraries of increasing strength, especially in organisms lacking strong, characterized promoters. Specifically, this study illustrated the isolation and characterization of a novel upstream activating sequence. In this regard, this approach expands the quantity and quality of parts required for synthetic biology research (Liu et al. 2011; Young and Alper 2010).
In this work, we validated the existence of a strong UAS element upstream of the minimal core TEF(136) sequence, and demonstrated its capacity as a synthetic amplifier when fused to either the minimal leucine promoter or the full native TEF promoter. In this regard, the novel UASTEF enhancer acts as a modular element that relieves enhancer-limited transcription at core promoter elements. Hybrid promoters using the TEF core were much stronger (at least threefold) than their Leum-based counterparts, as expected from the difference in their basal core strength. We further demonstrated a rigid linear correlation between promoter strength and the number of UASTEF enhancer elements. This relationship is in contrast to the Hill-curve dynamics seen with the UAS1B enhancement (Blazeck et al. 2011). This difference suggests that UAS elements have distinct transfer functions of activity. Moreover, both UAS enhancer element and core promoter elements contribute to overall hybrid promoter strength in a logical manner, raising the possibility of rationally designing synthetic hybrid promoters with specified expression strength for a desired metabolic engineering application.
The UASTEF enhancer element represents a newly isolated upstream activating sequence in Y. lipolytica. We conducted a dissection analysis of this UASTEF to eliminate extraneous (or potentially interfering) DNA sequence as a means of demonstrating a generic methodology for identifying useful UAS elements. While the majority of this 257 bp UASTEF element was confirmed to be necessary to enable full transcriptional capacity of TEF, the removal of 27 bp from 3′ UASTEF resulted in increased activation potential. Removing this specific portion of DNA alleviates latent repressive activity in the otherwise highly constitutive TEF promoter sequence. A comparison of the native TEF and the UASTEF#2-TEF(136) revealed that MCM1p and PUT3p binding sites are lost in this truncation, and a deletion analysis of putative transcription factor binding sites revealed that MCM1p binding sites have no effect on TEF promoter activity, implicating the PUT3p binding site as a repressive element. Further mutational analysis highlighted the GCR1p binding site as the predominant site responsible for the full transcriptional capacity associated with the TEFUAS element. Thus, by combining a truncation analysis with targeted binding site mutagenesis, we were able to propose transcription factors localized by the TEFUAS region that both activate and repress transcription and attain a greater understanding of Y. lipolytica native promoters. Following this dissection, we fused the strongest UASTEF truncation identified, UASTEF#2 (which contained the 3′ deletion), in tandem copies to the TEF(136) core promoter. Only six UASTEF#2 elements were necessary to saturate gains in promoter expression capacity from tandem UAS addition, potentially because of a shortage of essential transcription factors utilized by this enhancer element, a metabolic burden observed in high-strength heterologous promoter systems in S. cerevisiae (Gorgens et al. 2001). Even so, the maximal fluorescence yielded by the UASTEF #2 library reached levels 3.5-fold that of the native TEF promoter. Thus, UAS elements isolated from native promoters can be utilized to increase available promoter strength within an organism.
As described above, all prior hybrid promoter attempts in Y. lipolytica have been limited to only one UAS element. By combining disparate UAS elements, we obtain surprisingly high levels of gene expression, as high as the strongest promoters previously described in Y. lipolytica (Blazeck et al. 2011) in a much shorter total promoter size (1,850 bp vs. up to 3,700 bp). We hypothesize that the distinct UAS elements localize different transcription activating factors with unexpected cooperative effects and thus more efficiently enhance transcription. Prior evidence has demonstrated that the combination of GCR1 and RAP1 regulatory sequences constitute one of the strongest activating sequences known in S. cerevisiae (Drazinic et al. 1996). The UAS1B sequence was initially identified through a deletion analysis of the XPR2 promoter and was shown to be necessary for XPR2 promoter function (Blanchin-roland et al. 1994). The UAS1B sequence contains a TUF/RAP1 transcription factor binding site that by itself rescues native promoter activity when inserted to replace a UAS1B deletion (Blanchin-roland et al. 1994). Our transcription factor deletion analysis of the consensus GCR1 binding site found in the UASTEF sequence confirmed its importance towards transcriptional activation. Hence, the unison of GCR1 and RAP1 transcription factor binding sites within the same promoter could drastically increase transcriptional capacity, and it is likely that the localization of these regulatory sequences serves as the mechanism behind the synergy seen when combining the disparate UASTEF and UAS1B8 elements to form hybrid promoters. These types of synergies will form the basis of developing predictive models for designing de novo promoters.
UASTEF#2-based hybrid promoters retained the constitutive expression properties found in the native TEF promoter and exhibited consistent expression levels regardless of media formulation or cell phase. As the UASTEF#2(12)-TEF(136) promoter retained full activity much longer than other library members, increasing UASTEF#2 copy number may serve to delay the onset of reduced promoter activity. In contrast, hybrid promoters containing only UAS1B elements (UAS1B16 –Leum and UAS1B32–Leum) displayed a cell-phase-dependent time course expression pattern and remained only marginally active until later cell phases. The addition of a TEFUAS element in the form of a TEF core promoter (UAS1B8-TEF and UAS1B16-TEF) shortened this lag period and facilitated quicker promoter activation. This phenomena was seen more prominently with the UASTEF(2)-UAS1B8-TEF promoter. Thus, we have shown that both hybrid promoter potency and regulation can be manipulated through the choice of UAS element and core element.
Sucrose-activated UAS1B hybrid promoters yielded surprisingly high expression values compared to glucose, presenting it as an alternative carbon source for protein expression. However, maximum cell density was significantly decreased in sucrose-grown cultures. As sucrose is a disaccharide comprised of glucose and fructose, fructose-grown cultures were also analyzed for protein expression capability but generated less expression than glucose-grown cultures. Thus, further research is necessary to determine the mechanism for such high heterologous protein expression in sucrose-grown cultures.
Collectively, these results illustrate a detailed example of hybrid promoter library construction to increase promoter strength in an organism. The basic architecture of hybrid promoters informs the need for both upstream activation sequences and core promoter regions. Dissection analyses of native promoters can mediate the efficient isolation of novel UAS elements. Tandem UAS elements help bypass the enhancer limited nature of promoters by serving as transcriptional amplifiers, and fusion of UAS repeats to native promoters elevates their basal strength, and therefore cell-wide transcriptional capacity. We have demonstrated that this amplification is generic for all promoters (regardless of length) and thus not restricted to minimal promoters. Nevertheless, minimal promoters can be isolated through a general truncation and UAS replacement analysis. Minimal promoters increase the ultimate dynamic range of the hybrid promoter approach and allow for fine-tuned gene expression starting at a lower level. Exploitation of both native and minimal promoters under the control of tandem UAS elements permits an otherwise unattainable range of gene expression. Moreover, combining unrelated UAS sequences offers tantalizing potential for ever higher levels of gene expression and controllable regulation. In conclusion, the generic approach for hybrid promoter engineering advanced here is an important synthetic biology method enabling the construction of high-level and fine-tuned promoters.
References
Alper H, Fischer C, Nevoigt E, Stephanopoulos G (2005) Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA 102:12678–12683
Barth G, Gaillardin C (1997) Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS Microbiol Rev 19:219–237
Beopoulos A, Chardot T, Nicaud JM (2009) Yarrowia lipolytica: a model and a tool to understand the mechanisms implicated in lipid accumulation. Biochimie 91:692–696
Blanchin-roland S, Otero RRC, Gaillardin C (1994) Two upstream activation sequences control the expression of the XPR2 gene in the yeast Yarrowia lipolytica. Mol Cell Biol 14:327–338
Blazeck J, Liu L, Redden H, Alper H (2011) Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl Environ Microbiol 77:7905–7914
Blazeck J, Garg R, Reed B, Alper H (2012) Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol Bioeng. doi:10.1002/bit.24552
Chen DC, Yang BC, Kuo TT (1992) One step transformation of yeast in stationary phase. Curr Genet 21:83–84
Damude H.G.H., Gillies, Peter John, Macool, Daniel Joseph, Picataggio, Stephen K., Pollak, Dana Walters M., Ragghianti, James John, Xue, Zhixiong, Yadav, Narendra S., Zhang, Hongxiang, Zhu, Quinn Qun. (2006) High eicosapentaenoic acid producing strains of Yarrowia lipolytica, United States
Davidow LS, Apostolakos D, Odonnell MM, Proctor AR, Ogrydziak DM, Wing RA, Stasko I, Dezeeuw JR (1985) Integrative transformation of the yeast Yarrowia lipolytica. Curr Genet 10:39–48
Deboer HA, Comstock LJ, Vasser M (1983) The Tac promoter—a functional hybrid derived from the Trp and Lac promoters. Proc Natl Acad Sci USA 80:21–25
Drazinic CM, Smerage JB, Lopez MC, Baker HV (1996) Activation mechanism of the multifunctional transcription factor repressor-activator protein 1 (Rap1p). Mol Cell Biol 16:3187–3196
Fournier P, Abbas A, Chasles M, Kudla B, Ogrydziak DM, Yaver D, Xuan JW, Peito A, Ribet AM, Feynerol C, He F, Gaillardin C (1993) Colocalization of centromeric and replicative functions on autonomously replicating sequences isolated from the yeast Yarrowia lipolytica. Proc Natl Acad Sci USA 90:4912–4916
Gaillardin C, Ribet AM (1987) LEU2 directed expression of beta-galactosidase activity and phleomycin resistance in Yarrowia lipolytica. Curr Genet 11:369–375
Gorgens JF, van Zyl WH, Knoetze JH, Hahn-Hagerdal B (2001) The metabolic burden of the PGK1 and ADH2 promoter systems for heterologous xylanase production by Saccharomyces cerevisiae in defined medium. Biotechnol Bioeng 73:238–245
Jensen PR, Hammer K (1998) The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl Environ Microbiol 64:82–87
Jeppsson M, Johansson B, Jensen PR, Hahn-Hagerdal B, Gorwa-Grauslund MF (2003) The level of glucose-6-phosphate dehydrogenase activity strongly influences xylose fermentation and inhibitor sensitivity in recombinant Saccharomyces cerevisiae strains. Yeast 20:1263–1272
Juretzek T, Le Dall MT, Mauersberger S, Gaillardin C, Barth G, Nicaud JM (2001) Vectors for gene expression and amplification in the yeast Yarrowia lipolytica. Yeast 18:97–113
Kalnins A, Otto K, Ruther U, Mullerhill B (1983) Sequence of the lacZ gene of Escherichia coli. EMBO J 2:593–597
Ledall MT, Nicaud JM, Gaillardin C (1994) Multiple-copy integration in the yeast Yarrowia lipolytica. Curr Genet 26:38–44
Liu L, Reed B, Alper H (2011) From pathways to genomes and beyond: the metabolic engineering toolbox and its place in biofuels production. Green 1:81–95
Madzak C, Blanchin-Roland S, Otero RRC, Gaillardin C (1999) Functional analysis of upstream regulating regions from the Yarrowia lipolytica XPR2 promoter. Microbiology 145:75–87
Madzak C, Treton B, Blanchin-Roland S (2000) Strong hybrid promoters and integrative expression/secretion vectors for quasi-constitutive expression of heterologous proteins in the yeast Yarrowia lipolytica. J Mol Microbiol Biotechnol 2:207–216
Madzak C, Gaillardin C, Beckerich JM (2004) Heterologous protein expression and secretion in the non-conventional yeast Yarrowia lipolytica: a review. J Biotechnol 109:63–81
Matsuoka M, Matsubara M, Daidoh H, Imanaka T, Uchida K, Aiba S (1993) Analysis of regions essential for the function of chromosomal replicator sequences from Yarrowia lipolytica. Mol Gen Genet 237:327–333
Miller J.H. (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory, [Cold Spring Harbor, N.Y.]
Mukai H., Horii H., Tsujikawa M., Kawabe H., Arimura H., Suyama T. (1992) Yeast promoter and process for preparing heterologous protein, GREEN CROSS CORPORATION (3-3, Imabashi 1-chome Chuo-ku, Osaka-shi, Osaka, JP), Europe
Muller S, Sandal T, Kamp-Hansen P, Dalboge H (1998) Comparison of expression systems in the yeasts Saccharomyces cerevisiae, Hansenula polymorpha, Klyveromyces lactis. Schizosaccharomyces pombe and Yarrowia lipolytica. Cloning of two novel promoters from Yarrowia lipolytica. Yeast 14:1267–1283
Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122
Nevoigt E, Kohnke J, Fischer CR, Alper H, Stahl U, Stephanopoulos G (2006) Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl Environ Microbiol 72:5266–5273
Nicaud JM, Madzak C, van den Broek P, Gysler C, Duboc P, Niederberger P, Gaillardin C (2002) Protein expression and secretion in the yeast Yarrowia lipolytica. Fems Yeast Res 2:371–379
Pfleger BF, Pitera DJD, Smolke C, Keasling JD (2006) Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol 24:1027–1032
Rosenberg S., Tekamp-olson P. (1992) Enhanced yeast transcription employing hybrid GAPDH promoter region constructs, Chiron Corporation (Emeryville, CA), United States
Rud I, Jensen PR, Naterstad K, Axelsson L (2006) A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum. Microbiology 152:1011–1019
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y
Vanheerikhuizen H, Ykema A, Klootwijk J, Gaillardin C, Ballas C, Fournier P (1985) Heterogeneity in the ribosomal RNA genes of the yeast Yarrowia lipolytica—cloning and analysis of two size classes of repeats. Gene 39:213–222
Vernis L, Abbas A, Chasles M, Gaillardin CM, Brun C, Huberman JA, Fournier P (1997) An origin of replication and a centromere are both needed to establish a replicative plasmid in the yeast Yarrowia lipolytica. Mol Cell Biol 17:1995–2004
Vernis L, Poljak L, Chasles M, Uchida K, Casaregola S, Kas E, Matsuoka M, Gaillardin C, Fournier P (2001) Only centromeres can supply the partition system required for ARS function in the yeast Yarrowia lipolytica. J Mol Biol 305:203–217
Yamane T, Sakai H, Nagahama K, Ogawa T, Matsuoka M (2008) Dissection of centromeric DNA from yeast Yarrowia lipolytica and identification of protein-binding site required for plasmid transmission. J Biosci Bioeng 105:571–578
Young E, Alper H (2010) Synthetic biology: tools to design, build, and optimize cellular processes. J Biomed Biotechnol 2010:130781
Zhu J, Zhang MQ (1999) SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics 15:607–611
Acknowledgements
This work was funded by the Office of Naval Research Young Investigator Program and DuPont Young Professor Grant for funding. We would like to thank Masayoshi Matsuoka for the generous gift of plasmid pSl16-Cen1-1(227).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 25 kb)
Rights and permissions
About this article
Cite this article
Blazeck, J., Reed, B., Garg, R. et al. Generalizing a hybrid synthetic promoter approach in Yarrowia lipolytica . Appl Microbiol Biotechnol 97, 3037–3052 (2013). https://doi.org/10.1007/s00253-012-4421-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4421-5