
Abstract
Different from other fungal species that can be largely cultivated in ‘axenic conditions’ using plant material (e.g., species of Lentinula and Pleurotus in ‘sterile’ straw-based substrate), the commercial Agaricus bisporus cultivation system relies heavily on ecological relationships with a broad range of microorganisms present in the system (compost and casing). Since the A. bisporus cultivation system consists of a microbial manipulation process, it is important to know the microbial community dynamics during the entire cultivation cycle to design further studies and/or crop management strategies to optimize this system. To capture the bacterial community ‘flow’ from compost raw materials to the casing to the formation and maturation of mushroom caps, community snapshots were generated by direct DNA recovery (amplicon sequencing). The ‘bacterial community flow’ revealed that compost, casing and mushrooms represent different niches for bacteria present in the cultivation system, but at the same time, a bacterial exchange between microenvironments can occur for a portion of the community. Within each microenvironment, compost showed intense bacterial populational dynamics, probably due to the environmental changes imposed by composting conditions. In casing, the colonization of A. bisporus appeared, to reshape the native bacterial community which later, with some other members present in compost, becomes the core community in mushroom caps. The current bacterial survey along with previous results provides more cues of specific bacteria groups that can be in association with A. bisporus development and health.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the agricultural sector, cultivation of mushrooms is an important economic income for many regions around the world [1, 2]. Among many mushroom species cultivated on a commercial basis, Agaricus bisporus is the most cultivated in western countries [2, 3]. The USA alone produced 361,000 tonnes of A. bisporus in 2019, valued at US$ 1.09 billion [4]. Like other horticultural commodities, the cultivation of A. bisporus is an intensive food production system carried out in indoor settings (controlled environment) year-round and is highly efficient in terms of production per area cultivated [4].
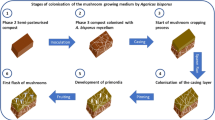
A. bisporus is a poor competitor on the dead organic matter; however, as a secondary decomposer it is specifically adapted to grow on partially decomposed humic-rich plant material [1, 5]. Different from other cultivated fungal species that can be largely cultivated in ‘axenic conditions’ using non-degrade plant material (e.g. species of Lentinula and Pleurotus), the commercial A. bisporus cultivation system relies heavily on ecological relationships with a broad range of microorganisms present in the system [6, 7]. In addition, some of these microorganisms can be harmful to the cultivated fungus, developing parasitic or antagonistic relationships (‘pathogens’). These microorganisms (beneficial or not) coexist with A. bisporus in two heterogeneous microenvironments; the compost and the casing layer, which differ in nature and function. These two microenvironments are merged in a sequential cultivation process that can be divided into three main stages, compost preparation, compost/casing colonization and mushroom formation and harvest (Fig. 1).

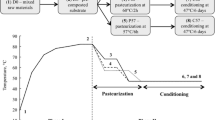
Illustration of the Agaricus bisporus cropping process and sampling time/points. * Samples: D0—mixed compost raw materials; PI—compost sample at the end of phase I; PII—compost sample at the end of phase II; PIII—compost sample at the end of spawn run; CoF1—compost sample during the first mushroom harvest; CoF2—compost sample during the second mushroom harvest; Ca—mixed peat moss; CaF1—casing sample during the first mushroom harvest; CaF2—casing sample during the second mushroom harvest; CapF1—mushroom caps during the first harvest and; CapF2—mushroom caps during the second harvest. On top of the figure is a description (temperature graph) of the temperature during the Agaricus bisporus cultivation process
The compost represents the major nutrient source for the fungus mycelium and is made to produce a selective substrate for A. bisporus. The heterogeneous mix of vegetal and animal raw materials (e.g. wheat straw, horse manure and chicken manure) needs to be partially depolymerized [8] by other decomposer microorganisms during a two-phase composting process [9, 10]. The remaining microbial community will then interact directly with the fungus mycelium in subsequent cropping stages (compost/casing colonization, and mushroom formation and harvest). The second microenvironment is the casing layer (added on top of colonized compost), which provides a suitable environment for mushroom formation. Typically, the casing layer is composed of peat moss with a high capacity to hold water and rich in organic matter. In addition, it has been speculated that the casing materials contain bacteria involved in the transformation of vegetative mycelial growth into fruiting body formation [11, 12]. Additionally, microbial populations found in the casing material may contribute to diseases on mushroom caps [13, 14]. Different than compost, the microbial communities of casing materials are introduced to the system after the fungus mycelium is already well established, i.e. ready to change into reproductive structures (mushrooms).
Indeed, the microbial ecology of the A. bisporus cultivation system has been continually studied since it gained popularity in the second half of the last century [7, 12]. Portions of the cropping process were investigated regarding microbial communities, and their ecological niche, e.g. Vieira and Pecchia [10] and Cao et al. [15], investigated the bacterial community dynamics during the two-phase composting process, McGee et al. [16, 17] investigated the microbial dynamics in compost during a commercial cropping process, Taparia et al. [18] evaluated the microbial community dynamics in different casing materials during an experimental cropping process, Martins et al. [19] investigated the bacterial community associated with blotch disease using asymptomatic and symptomatic mushroom caps collect from an organic farm and Carrasco et al. [20] examined the microbial community dynamics in compost and casing in a commercial cropping setting. These previous studies will be discussed along with the current results when appropriate.
Despite previous studies on the microbial ecology of the system, it is still unclear as to the microbial community pattern during the entire cultivation process, from compost raw materials to the casing to the formation and maturation of mushroom caps. Since the A. bisporus cultivation system consists of a microbial manipulation process, it is important to know the microbial community dynamics during the entire cultivation cycle to design further studies and/or crop management strategies to optimize this system. Thus, an experimental design was set up attempting to capture the bacterial community ‘flow’ (community snapshots generated by direct DNA recovery, amplicon sequencing) within the same microenvironment over time, between microenvironments (compost and casing) and compared with bacterial communities associated with mushroom caps.
Material and Methods
Cropping Process
The A. bisporus cropping trial was carried out using standards procedures at the Mushroom Research Center (https://plantpath.psu.edu/research/centers/mushroom-research-center) at Pennsylvania State University, PA, USA. The crop process consisted of composting phases I and II, compost/casing colonization, mushroom fructification and harvest (Fig. 1 and Fig. S1). The compost formula was based on wheat straw-bedded horse manure, dried poultry manure, dried distiller’s grain and gypsum (Table S1). The raw compost materials (minus dried distiller’s grain) were mixed in a Jaylor feed mixer (model S2545, Ontario, Canada) to reach approximately 70% starting moisture. The mixed ingredients (~ 3.5 tonnes, wet basis) were filled into a bunker with a forced aeration system supplying fresh air to prevent anaerobic conditions. At day 3, the compost matrix was removed from the bunker, mixed in the Jaylor feed mixer with the addition of the dried distiller’s grain, watered and returned to the bunker for an additional 3 days. At the end of phase I (a total of 6 days), the compost matrix was removed from the bunker, mixed and watered (to adjust the moisture to ~ 74%) and transferred into a ‘pasteurization tunnel’ (rectangular-shaped building) for phase II. The pasteurization tunnel is designed with a forced aeration system supplying filtered fresh air to prevent anaerobic conditions and to regulate the temperature for pasteurization and conditioning. The temperature and length for the phase II were set up at 60 °C for 4 h for pasteurization and 47 °C for 5 days for conditioning, totaling 6 days of phase II. A day prior to spawning, the temperature of conditioned compost was adjusted to 25 °C by increasing the intake of filtered fresh air. Subsequently, compost (with moisture of ~ 68%) was spawned at a rate of 3% (commercial strain of A. bisporus on millet grain, Triple X—Amycel, CA, USA) and the addition of 4% of commercial supplement (Promycel gold [54% protein]—Amycel, CA, USA). Spawn and supplement ratios were based on dry weights of compost. For phase III, 22.5 kg of spawned and supplemented compost (wet weight) was transferred to rectangular PVC containers (35-cm height, 70-cm length and 50-cm width) before transferred to growing rooms. The spawn run (compost colonization, PIII) was carried out with temperature of compost adjusted to 24–25 °C and air humidity of 95%, in the dark, until complete compost colonization, 16 days. A commercial casing material (Scotts sphagnum peat moss, Ontario, Canada) was mixed in a mixer with crushed agricultural limestone to raise the pH to near 8.0, CAC (mycelium used for casing, Triple X—Amycel) and water prior to adding to the top of the colonized compost. Approximately 10 kg of peat moss with moisture adjusted to ~ 75% was placed on top of colonized compost, forming a 5-cm layer. From there, casing colonization took place, and after ~ 17 days mushroom started being picked. Mushrooms were picked in two harvest cycles with an interval of 8 days between. Mushrooms were harvested based on their size and maturity (2–4 cm in diameter) and were weighed for yield calculations. The whole cultivation process was completed within 60 days (from composting day 0 to the second mushroom harvest), and biological efficiency for 2 cycles was approximately ~ 60% (g of mushrooms [wet wt.] per g of dry compost from phase II).
Sampling, DNA Extraction and Sequencing
Samples were taken from 11 time/points during the cultivation process including compost, casing and mushroom caps, and each sample represents a ‘community snapshot’ at that moment (Fig. 1 and Fig. S1). Compost samples were collected at the beginning of composting (D0—mixed compost raw materials), at the end of phases I, II and III (PI, PII and PIII) and in the middle of the first and second harvests (CoF1 and CoF2). Compost samples D0 and PI were taken at three different times during unloading of the mixer. The PII compost samples were collected in the same way, three different times during unloading of the Phase II tunnel. During the cropping process, compost samples were taken from destructive experimental units (compost blocks), i.e. three independent compost blocks were destroyed for each time/point (PIII, CoF1 and CoF2). For casing, samples were collected on casing day in three different times during unloading of the mixer (Ca—mixed peat moss) and in the middle of the first and second harvests (CaF1 and CaF2). Mushroom samples were collected during the middle of the first and second harvests (CapF1 and CapF2). For mushrooms, 10 mushroom caps (2–4 cm in diameter) were randomly collected in each compost block. After collecting mushroom caps, 200 g of casing and compost was taken as well. In this way, the mushrooms, casing and compost samples during the cropping process originated from the same experimental unit (compost block). All samples were collected in triplicate (i.e. three samples for each time/point) and were used as individual samples for DNA extraction, PCR amplified, library preparation and sequencing.
All samples were collected and immediately frozen in liquid nitrogen and stored at − 80 °C until genomic DNA (gDNA) extraction. After removing from storage, samples were freeze dried, and 10 g of each sample was homogenized (by making a fine powder); 50 mg of the fine powder was used for gDNA extraction using a FastDNA Spin Kit for Soil (MP Biomedicals, OH, USA) following the manufacturer’s instructions. Positive control samples were included during the library preparation, which consisted of gDNA of a commercial mock community (ZymoBiomics Microbial Community DNA Standard II, CA, USA) with 8 species of bacteria. The gDNAs were checked for concentration using Qubit dsDNA BR Assay Kit and visualized by agarose gel electrophoresis (agarose 1%, voltage 150 V for 40 min). Normalized gDNA samples (30 ng) were amplified using a dual-indexing approach [21] with primers (515F and 806R) targeting the V4 region of the 16S rRNA gene [22]. Subsequently, Agencourt AMPure XP beads (Beckman-Coulter Life Sciences) were used to purify PCR products followed by a qualification using Agilent 2100 bioanalyzer. Purified PCR products were normalized and sequenced using a 250 PE kit in a HiSeq 2500 Illumina platform at the Beijing Genomic Institute.
Sequence Processing and Bioinformatics Analyses
The resulting read files (Fastq files) were processed using the DADA2 R package version 1.16.0 [23] in RStudio version 1.4.1103 [24] and R version 4.02 [25]. Raw reads were checked for ambiguity (zero Ns were allowed), and primer removal (forward and reverse) was performed by cutadapt command-line tool version 2.8 [26] as a plugin in DADA2. Subsequently, reads were inspected for their quality score, and reads < Q 30 were removed from the dataset. High quality reads (220 bp and 220 bp for forward and reverse reads, respectively) were then merged, with a read average length of 253 bp (length threshold chosen for the V4 region was 250–260 bp and sequences shorter or longer were removed). After chimera removal, the remaining sequences were aligned using a naïve Bayesian classifier method [27] against Silva 16S rRNA database version 138.1 [28]. The DADA2 output files (taxonomic assignment and abundance of sequences) were manipulated by phyloseq R package version 1.34.0 [29]. The sequencing and data processing ‘evaluation of accuracy’ was carried out by simply checking the presence and absence of sequences in the positive samples (commercial mock community). All 8 sequences from the mock community were recovered in positive control samples. The dataset was pruned, and classified sequences only at the bacteria domain (i.e. no taxonomic classification at phylum level and beyond, 225 sequences) were removed prior to some downstream analyses. Most of the analyses were carried out using unrarefied and rarefied data for comparison and were discussed if appropriated. To facilitate the display of results, candidate phyla were removed (NB1-j, MBNT15, SAR324 clade, WPS2, WS1 and WS4, 138 sequences in total). Datasets rarefied or not (to the sample with the fewest number of reads) and pruned (exclusion of candidate phyla) or not can be accessed in the supplemental material (Tables S2–S4).
Taxa distribution, community profile, core community and taxa prevalence calculations and visualization were carried out using R packages, MiscMetabar version 0.20 (http://github.com/adrientaudiere/Miscmetabar), microbiome version 1.10.0 [30] and phyloseq. Beta diversity indexes were calculated using Bray–Curtis [31] between microenvironments and unweighted Unifrac [32] within the same microenvironment and for samples from different microenvironments. Both ecological metrics were visualized using Non-metric Multi-dimensional Scaling (NMDS) plots with phyloseq and ggplot2 R package version 3.3.3 [33]. Analysis of variance for beta diversity indices was performed by Adonis and pairwise PERMANOVAs with vegan R package version 2.5–7 [34].
Alpha diversity measures were calculated and visualized using richness, Shannon and Simpson indices with phyloseq. Normality test (Shapiro–Wilk) and Q-Q plots of the residuals were performed using stats R package version 4.0.2 [25] which showed a non-normal distribution for the dataset, and a non-parametric test (Kruskal–Wallis) was used. Post hoc test using Dunn’s test was performed using FSA R package version 0.8.32 [35] with adjustment for p values using Bonferroni correction.
Results
The bacterial community succession displayed below was based on community snapshots generated by direct DNA recovery of samples from major crop stages (Fig. 1, Fig. S1). After sequence processing, more than half of the raw sequences (~ 62%) remained for taxonomic assignment (Table S5). Sequencing depth showed high coverage trend to casing and mushroom samples (Fig. S2). In the mock community, seven of eight SVs (sequence variants) were recovered with a matching score of 100% with reference sequences, and one SV had an alignment of 97% similarity (data not shown). Based on unrarefied and unpruned data, 3776 SVs were detected, and 33 of them were singletons (SVs present only in one sample) (Table S3). The percentage of sequences classified below the phylum level decreased as follows: Class 99%, Order 97%, Family 90% and Genus 70%. At the species level, 245 SVs compressing ~ 17% of total sequences were classified with a matching score of 100% with the reference sequences (silva database version 138.1).
Without any data treatment associated with taxonomy assignments, 35 bacterial phyla were identified globally (all samples, Table S3). From there, 6 phyla with the status of ‘candidate phyla’ (Table S4) and SVs classified only at Bacteria domain were removed from the dataset for some visualization analyses (a total of 15 SVs compressing 353 sequences). Among the remaining 29 phyla, Proteobacteria, Firmicutes, Actinobacteriota and Bacteroidota were the most abundant, comprising 90% of total sequences (Fig. 2a and Table S4). Some phyla appear to have specificity distribution patterns regarding the microenvironment (Fig. 2b and Fig. S3), e.g. Firmicutes in compost (Fig. S4, samples D0, PI, PII, PIII, CoF1 and CoF2) and Bacteroidota in mushroom caps (Fig. S5, samples CapF1 and CafF2). On the other hand, some phyla were more uniformly distributed in all microenvironments, e.g. Proteobacteria (Fig. 2b and Fig. S6), while others were more uniformly distributed between microenvironments adjacent to each other, e.g. Actinobacteriota in compost and casing (Fig. S7). Between crop stages, phyla distribution tended to be consistent within the same microenvironment (Fig. 3a), e.g. Firmicutes in compost (D0, PI, PII, PIII, CoF1 and CoF2) and Bacteroidota in mushroom caps (CapF1 and CafF2).

a Absolute number of bacterial sequences at the phylum level. b Absolute number of bacterial sequences at the phylum level distributed by microenvironments (compost, casing and mushrooms)


a Relative abundance of sequences at the phylum level distributed by crop stages. b Absolute number of sequences at the species level distributed by microenvironments
At a low taxonomic level, approximately 630 genera and 245 species were identified, and 14 SVs/species had a relative abundance higher than 1% within species level, Fig. 3b. As in higher taxonomic level (e.g. phylum), the distribution of species displayed specificity patterns related to the microenvironment. Beta diversity indices were used to compare community composition between microenvironments, within the same microenvironment over time or for different time/points between microenvironments (Fig. 4a–b, Table S6). Between microenvironments, community composition was significantly different (p < 0.05) between compost and mushrooms. Within the same microenvironment, over time, community composition in compost was significantly different (p < 0.05) between early and late crop stages. Mixed compost raw materials (D0) and post thermophilic stage (PI) were the most different (p < 0.05) compared with all other compost samples, while mature compost (PII) was significantly different than compost samples at harvest (CoF1 and CoF2). Colonized compost (PIII) was significantly different than compost samples from second harvest (CoF2). In casing and mushroom microenvironments over time, community composition did not differ (p > 0.05). For different time/points between microenvironments, compost samples from early crop stages (D0, PI and PII) were significantly different (p < 0.05) than casing samples (Ca, CaF1 and CaF2). Colonized compost (PIII, CoF1 and CoF2) and colonized casing (CaF1 and CaF2) did not differ (p > 0.05). Mushroom caps (CapF1 and CapF2) were significantly different (p < 0.05) compared to all other samples.

a Beta diversity indices calculated by microenvironment. b Beta diversity indices calculated between crop stages. Samples: D0—mixed compost raw materials; PI—compost sample at the end of phase I; PII—compost sample at the end of phase II; PIII—compost sample at the end of spawn run; CoF1—compost sample during the first mushroom harvest; CoF2—compost sample during the second mushroom harvest; Ca—mixed peat moss; CaF1—casing sample during the first mushroom harvest; CaF2—casing sample during the second mushroom harvest; CapF1—mushroom caps during the first harvest and; CapF2—mushroom caps during the second harvest
Alpha diversity measures (richness, Shannon and Simpson) were compared as beta diversity, i.e. between microenvironments, or within the same microenvironment over time or for different time/points between microenvironments (Fig. 5a–b and Table S7). Community diversity tends to be higher in compost comparing to casing and mushroom samples. Independently of the alpha metric tested with or without rarefied data, significant differences (p < 0.05) were observed between the compost and mushrooms. For some alpha diversity metrics, compost was significantly different (p < 0.05) than casing. Within the same microenvironment, community diversity over time in compost was not significantly different (p > 0.05) as well as compared with casing samples (p > 0.05). In mushrooms, alpha diversity values were not significant (p > 0.05) between the first and second harvest (CapF1 and CapF2). Significant differences (p < 0.05) were observed between samples from different microenvironments, mixed compost raw materials (D0) and mushroom (CapF1 and CapF2), mixed compost raw materials and casing raw materials (Ca) and compost PI and mushrooms (CapF1).


a Alpha diversity metrics calculated between crop stages. b Alpha diversity metrics calculated by microenvironments. Samples: D0—mixed compost raw materials; PI—compost sample at the end of phase I; PII—compost sample at the end of phase II; PIII—compost sample at the end of spawn run; CoF1—compost sample during the first mushroom harvest; CoF2—compost sample during the second mushroom harvest; Ca—mixed peat moss; CaF1—casing sample during the first mushroom harvest; CaF2—casing sample during the second mushroom harvest; CapF1—mushroom caps during the first harvest and; CapF2—mushroom caps during the second harvest
The core community (SV/species level) was calculated globally (all samples), within the same microenvironment and between microenvironments (Table S8). Arbitrarily, a threshold of 1% detection (taxa with at least 1% relative abundance) and 50% prevalence (taxa present in at least 50% of samples) was used for the analyses. The global core community was represented by 1 SV. Within the same microenvironment, the core community in compost was represented by 11 SVs while in casing 13 and mushrooms 13. Between microenvironments, compost and casing shared 3 SVs and compost and mushroom 6 SVs while casing and mushroom shared 10 SVs. A second threshold (1% detection and 100% prevalence) was used to calculate the core community within the same microenvironment and compost had 1 SV while casing 2 and mushrooms 4. Comparing compost-casing-mushroom samples at the same crop stage using a threshold of 1% detection and 100% prevalence did not detect any SV among these samples (i.e. CoF1-CaF1-CapF1 or CoF2-CaF2-CapF2).
Discussion
Microorganisms present in compost and casing microenvironments cohabit and interact with A. bisporus, and such ecological relationships are described as important driver factors of mycelial growth and fruiting bodies development as detailed in previous reviews [6, 7, 12, 16]. The results of the current work combined with previous findings provide more cues of microbial community patterns in the A. bisporus cultivation system which perhaps can contribute to crop management programs focusing on microbial manipulation. Our contribution is an assessment of the bacterial community dynamics over time in different microenvironments of the A. bisporus cultivation system (compost, casing and mushrooms).
The cultivation process starts with the substrate preparation (i.e. composting) which is the major event contributing to bacterial community shifts in the compost microenvironment (Figs. 4a and 5a). The bacterial communities present in the heterogeneous mix of vegetal and animal sources were reshaped by environmental conditions imposed by the composting process (carbon and nitrogen ratios, water content, piling up effect, temperature, etc.), and such environmental conditions affect the rise or decline of certain bacterial populations in a short period of time. For example, sequences of Thermus sp. (SV9, Deinococcota, Table S3) were recovered in residual levels in mixed compost raw materials (D0) but increased to ~ 36,000 sequences after PI. After PII, sequences of Thermus sp. dropped to residual levels. However, sequences of Pseudomonas formosensis (SV22, Proteobacteria, Table S3) decreased from ~ 16,000 in mixed compost raw materials (D0) to residual levels after PI and subsequent crop stages. Despite intense population shifts between pre- or post-thermophilic periods (D0 to PI and to PII), the decreasing trend in community diversity was not significantly different (p > 0.05). On the other hand, community composition shifted significantly between pre- and post-thermophilic periods, D0, PI and PII (Fig. 4b and Table S6). After the composting process, the presence of A. bisporus mycelium and supplement did not significantly affect community diversity and composition in compost over time.
The core community in compost was formed by a few members of Firmicutes, Proteobacteria and Actinobacteriota (11 SVs compressing ~ 15% of total sequences, Table S8), and previous studies have reported a large number of sequences that belong to these phyla in compost during preparation [10, 15] or in colonized compost [20]. Sequences of Firmicutes were the most abundant, especially during the thermophilic stage (PI). This trend may indicate the ability of Firmicute members to grow and/or survive in environmental conditions imposed during PI (e.g. high temperature and high levels of ammonia [10]). Almost half of the total sequences at the end of PI belong to the family Bacillaceae (Firmicutes, Fig. S8), which is composed of rod-shaped bacteria known to form endospores and survive adverse physical and chemical conditions [36]. Within the Bacillaceae family, Bacilli and Clostridia were the most abundant members, and Bacilli have been speculated to play a role in compost selectivity. Some species of Bacillus (Bacilli) are present in compost raw materials and can act as a biological control agent against certain pathogens, e.g. Trichoderma (green mold disease) [37, 38]. In addition, inoculants of Bacillus spp. are commercially available for application in matured compost (PII) and/or casing to control green mold. On the other hand, the impact of native populations of Bacillus spp. on compost fitness against diseases occurrence is still unknown. In the current survey, 16 SVs that belong to Bacillus were identified in compost, comprising just a few sequences (~ 2500 sequences), and the majority of them were recovered in colonized compost (PIII, CoF1 and CoF2, Tables S3–S4). In casing, 9 of these SVs (Bacillus sp.) were detected in residual levels, and in mushrooms 1 SV was detected in residual level. Besides Firmicutes, sequences of Proteobacteria were detected in large amounts, and Rickettsiales comprised ~ 40% of sequences within the phylum. The majority of Rickettsiales sequences were recovered during compost conditioning (PII) and colonized compost (PIII, CoF1 and CoF2). Previous studies investigating the microbial changes in different composting settings (aerobic and vermicomposting of green waste) reported the presence of sequences of Rickettsiales [39, 40]. Some of their members are pathogenic to eukaryotes including humans [41]. Sequences of Leitomonas sp. and Chelativorans sp. were the second and third most abundant genera found within Proteobacteria. These two genera are known to play an important role in biomass degradation in composting processes [42]. Actinobacteriota was the third most abundant phylum in compost, and sequences of Thermobifida sp. were recovered in large amounts within the phylum. In addition, sequences of Thermobifida sp. were found in all microenvironments. Zhang et al. [43] reported Thermobifida sp. as a common isolate in mature compost, and it appeared that they are involved with organic matter cycling.
Similar to compost, casing materials carry bacterial communities from adjacent environments (typically peat moss), and when it is incorporated into the system, environmental changes reshape the community (e.g. inoculation of A. bisporus, water content, application of supplements and/or pesticides, etc.). The casing layer is not thought to be a nutritive source for the mushroom mycelia like the compost, but it provides a suitable environment for the transformation of vegetative mycelium into fruiting bodies [9, 12]. As observed in compost, population shifts occur in the casing microenvironment when the raw casing (Ca) is incorporated to the system. For example, sequences of Flavobacterium arsenitoxidans (SV1, Bacteroidota, Table S3) increased from residual levels in raw casing (Ca) to ~ 17,000 in colonized casing (CaF1). In mushrooms (CapF1 and CapF2), sequences of F. arsenitoxidans were recovered in large numbers (~ 56,000 sequences). On the other hand, sequences of Massilia sp. (SV16, Proteobacteria, Table S3) decreased from raw casing (~ 10,000 sequences) to residual level in colonized casing (CaF1 and CaF2). In mushrooms, sequences of Massilia sp. (SV16) were recovered in residual levels.
Even with a certain populational dynamics between uncolonized (Ca) and colonized casing (CaF1 and CaF2), the increasing trend in community diversity, as well as shifts in community composition, was not significantly different. The casing microenvironment was less diverse than compost, and Proteobacteria and Bacteroidota (former Bacteroidetes) phyla comprised the majority of sequences, forming the core community in casing (Table S8). Previous studies have reported a large number of sequences that belong to Proteobacteria and Bacteroidota in the casing microenvironment during the mushroom cropping process [18, 20]. Proteobacteria appear to be the most abundant phylum in the casing microenvironments, and among several members, sequences of Pseudomonadaceae were recovered in large numbers in the casing and mushrooms. In some cases (using dependent-culturable methods, i.e. isolation of bacteria from casing), members of Pseudomonadaceae represented up to 80% of the bacterial community recovered in casing microenvironments [11].
Compared with compost and casing materials in which the bacterial community loads come from adjacent environments, the bacterial community that resides on mushroom caps is formed by recruiting microorganisms already present in casing and/or compost. Unfortunately, samples of water, air, tools and other potential sources of bacteria at the production facility were not collected. Casing materials appear to be the major source of microorganisms that reside on mushroom caps, and three genera (Pseudomonas, Pedobacter and Flavobacterium) comprised 2/3 of the total sequences on mushroom caps (Table S4). Among them, Pseudomonas (Proteobacteria, Pseudomonadaceae) probably are the most well-known regarding ecological relationships with A. bisporus [12, 13, 19, 44]. Globally, 33 SVs (~ 0.8% of total SVs) were classified as members of Pseudomonas, and 7 of them were classified at the species level (P. formosensis, P. pertucinoger, P. mendocina, P. oleovorans, P. saudimassiliensis, P. flexibilis and P. alcaligenes, Table S3). In raw compost (D0), 21 SVs (Pseudomonas sp.) were detected, and half of them were detected at the end of the composting process (PII). Most of Pseudomonas sequences in compost were detected in residual levels, except SV22 P. formosensis in raw compost (D0), which dropped to residual levels at the end of the composting process. In raw casing, 10 SVs (Pseudomonas sp.) were detected, and two of them comprised a large number of sequences (SV3 and SV4). Searching in a broader database (NCBI nucleotide collection), SV3 and SV4 had a match score of 100% (253/253 bp) with P. putida (GenBank: MT641244.1 and MT065816.1, respectively), a bacterium known to stimulate mushroom fruit body formation [12, 44]. In mushroom caps, 15 SVs (~ 1.5% of total SVs) were classified as members of Pseudomonas, comprising ~ 33% of the total sequences in mushroom caps (CapF1 and CapF2). The major sequences of Pseudomonas found in mushroom caps were also detected in large number in the casing microenvironment, e.g. SV3 and SV4 (Table S3). Besides Pseudomonas, two other genera comprised a large number of sequencing in mushroom caps, Flavobacterium and Pedobacter (Table S3), and such genera have been less studied regarding their potential relationships with A. bisporus. It is worth noting that together, these three genera (71 SVs) comprised ~ 2/3 of the total sequences found in mushroom caps in the current study.
Comparing the bacterial community between microenvironments, the compost microenvironment tends to be more diverse than the casing and mushrooms. However, previous studies have reported opposite trends. Carrasco et al. [20] evaluated the bacterial community dynamics in compost and casing over time in commercial settings and found that compost samples were less diverse than the casing, leading to the hypothesis that the application of casing on top of colonized compost (PIII) promoted bacterial translocation from casing to compost which increased the community diversity in the compost microenvironment over time. In the current survey, the community diversity in raw casing tended to increase after contact with compost, and the ‘bacterial motility’ appeared to occur from compost to casing (even watering the casing during the crop process). Besides bacterial mobility, Taparia et al. [18] investigated the microbial dynamics in different casing materials, and threefold higher diversity index values were reported compared with the current results and with the study of Carrasco et al. [20]. It is unclear if these variances in community diversity between studies are results of biological differences (casing materials, crop management, etc.) or differences of methodological strategies (DNA extraction, primers, bioinformatics analyses pipelines, etc.). Perhaps, a metanalyses (i.e. large collection of datasets) using different datasets can help elucidate some of this differences. In addition, the current analysis of the bacterial communities based on amplicon sequencing as well as others presented here only represents snapshots of the communities in the A. bisporus cultivation system, and future research may look into the functionality of this communities with hopes to understand the association of A. bisporus and bacteria in a community level.
This attempt to capture the ‘bacterial community flow’ (from raw materials to the mushroom caps) in the A. bisporus cultivation system revealed that compost, casing and mushrooms represent different niches for bacteria present in the cultivation system, but at the same time, a bacterial exchange between microenvironments can occur for a portion of the community. Looking within each microenvironment, compost showed intense bacterial populational dynamics, probably due to the environmental changes imposed by composting conditions. In casing, the colonization of A. bisporus appeared, to reshape the native bacterial community which later, with some other members present in compost, becomes the core community in mushroom caps.
Data Availability
The data presented in this study are available in the article (generated sequences are in the supplementary material).
Code Availability
Not applicable
References
Chang ST, Miles PG (2004) Mushrooms: cultivation, nutritional value, medicinal effect, and environmental impact, 2nd edn. CRC Press Taylor & Francis Group, Boca Raton
Chang ST, Wasser SP (2017) The cultivation and environmental impact of mushrooms, Oxford research encyclopedia of environmental science (agriculture and environment). Oxford University Press, New York
Royse DJ, Baars J, Tan Q (2017) Current overview of mushroom production in the world. In: Zied DC, Pardo-Giménez A (eds) Edible and medicinal mushrooms technology and applications. John Wiley & Sons Ltd., Chichester, pp 5–12
USDA National Agricultural Statistics Service (NASS) (2020) Agricultural Statistics Board, United States Department Agriculture (USDA), ISSN: 1949–1530. https://www.nass.usda.gov/Statistics_by_State/Wisconsin/Publications/Crops/2020/US-Mushrooms-08-20.pdf. Accessed 20 Jan 2021
Kerrigan RW, Challen MP, Burton KS (2013) Agaricus bisporus genome sequence: a commentary. Fungal Genet Biol 55:2–5
McGee CF (2018) Microbial ecology of the Agaricus bisporus mushroom cropping process. Appl Microbiol Biotechnol 102:1075–1083
Kertesz MA, Thai M (2018) Compost bacteria and fungi that influence growth and development of Agaricus bisporus and other commercial mushrooms. Appl Microbiol Biotechnol 102:1639–1650
Kabel MA, Jurak E, Mäkelä MR, de Vries RP (2017) Occurrence and function of enzymes for lignocellulose degradation in commercial Agaricus bisporus cultivation. Appl Microbiol Biotechnol 101:4363–4369
Van Griensven LJLD (1988) The cultivation of mushrooms, 1st ed. Rustington - Darlington Mushroom Laboratories, Sussex, United Kingdom
Vieira FR, Pecchia JA (2018) An exploration into the bacterial community under different pasteurization conditions during substrate preparation (composting—Phase II) for Agaricus bisporus cultivation. Microb Ecol 75:318–330
Colauto NB, Fermor TR, Eira AF, Linde GA (2016) Pseudomonas putida stimulates primordia on Agaricus bitorquis. Curr Microbiol 72:482–488
Baars JJP, Scholtmeijer K, Sonnenberg ASM, van Peer A (2020) Critical factors involved in primordia building in Agaricus bisporus: a review. Molecules 25:e2984
Osdaghi E, Martins S, Ramos-Sepulveda L, Vieira FR, Pecchia JA, Beyer DM (2019) 100 years since Tollas: bacterial blotch of mushrooms in the 21st century. Plant Dis 103:2714–2731
Taparia T, Krijger M, Haynes E, Elphinstone JG, Noble R, van der Wolf J (2020) Molecular characterization of Pseudomonas from Agaricus bisporus caps reveal novel blotch pathogens in western Europe. BMC Genomics 21:5050
Cao G, Song T, Shen Y, Jin Q, Feng W, Fan L, Cai W (2019) Diversity of bacterial and fungal communities in wheat straw compost for Agaricus bisporus cultivation. HortScience 54:100–109
McGee CF, Byrne H, Irvine A, Wilson J (2017) Diversity and dynamics of the DNA- and cDNA-derived compost fungal communities throughout the commercial cultivation process for Agaricus bisporus. Mycologia 109:475–484
McGee CF, Byrne H, Irvine A, Wilson J (2017) Diversity and dynamics of the DNA and cDNA-derived bacterial compost communities throughout the Agaricus bisporus mushroom cropping process. Ann Microbiol 67:751–761
Taparia T, Hendrix E, Hendricks M, Nijhuis E, de Boer W, van der Wolf J (2021) Casing soil microbiome mediates suppression of bacterial blotch of mushrooms during consecutive cultivation cycles. Soil Biol Biochem 155:108161
Martins SJ, Trexler RV, Vieira FR, Pecchia JA, Kandel PP, Hockett KL, Bell TH, Bull CT (2020) Comparing approaches for capturing bacterial assemblages associated with symptomatic (bacterial blotch) and asymptomatic mushroom (Agaricus bisporus) caps. Phytobiomes 4:90–99
Carrasco J, García-Delgado C, Lavega R, Tello ML, Toro M, Barba-Vicente V, Rodríguez-Cruz MS, Sánchez-Martín MJ, Pérez M, Preston GM (2020) Holistic assessment of the microbiome dynamics in the substrates used for commercial champignon (Agaricus bisporus) cultivation. Microb Biotechnol 13:1933–1947
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J (2014) An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6
Caparoso GJ, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 108:4516–4522
Callaran BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583
RStudio Team (2020) RStudio: Integrated Development for R. RStudio, PBC, Boston. http://www.rstudio.com/
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The silva ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:590–596
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8:e61217
Lathi L, Shetty S (2017) Tools for microbiome analysis in R. http://microbiome.github.com/microbiome
Bray JR, Curtis JT (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27:325–349
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Oksanen J, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2020) vegan: community ecology package. R package version 2.5–7. https://CRAN.R-project.org/package=vegan
Ogle DH, Wheeler P, Dinno A (2020) FSA: Fisheries Stock Analysis. https://github.com/droglenc/FSA
Mandic-Mulec I, Stefanic P, van Elsas JD (2015) Ecology of Bacillaceae. Microbiol Spectr 3:TBS-0017-2013. https://doi.org/10.1128/microbiolspec.TBS-0017-2013
Pandin C, Védie R, Rousseau T, Le Coq D, Aymerich S, Briandet R (2018) Dynamics of compost microbiota during the cultivation of Agaricus bisporus in the presence of Bacillus velezensis QST713 as biocontrol agent against Trichoderma aggressivum. Biol Control 127:39–54
Kosanovic D, Potocnik I, Duduk B, Vukojevic J, Stajic M, Rekanovic E, Milijasevic-Marcic S (2013) Trichoderma species on Agaricus bisporus farms in Serbia and their biocontrol. Ann Appl Biol 163:218–230
Cai L, Gong X, Sun X, Li S, Yu X (2018) Comparing of chemical and microbiological changes during the aerobic composting and vermicomposting of green waste. PloS One 13:e0207494
Galitskaya P, Biktasheva L, Saveliev A, Grigoryeva T, Boulygina E, Selivanovskaya S (2017) Fungal and bacterial successions on the process of composting of organic wastes as revealed by 454 pyrosequencing. PloS One 12:e0186051
Salje J (2021) Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle. Nat Rev Microbiol. https://doi.org/10.1038/s41579-020-00507-2
Székely AJ, Sipos R, Berta B, Vajna B, Hajdú C, Márialiget K (2009) DGGE and T-RFLP analysis of bacterial succession during mushroom compost production and sequences-aided T-RFLP prolife of mature compost. Microb Ecol 57:522–533
Zhang HL, Wei JK, Wang QH, Yang R, Gao XJ, Sang YX, Cai PP, Zhang GQ, Chen QJ (2019) Lignocellulose utilization and bacterial communities of millet straw based mushroom (Agaricus bisporus) production. Sci Rep 9:1151. https://doi.org/10.1038/s41598-018-37681-6
Soler-Rivas C, Jolivet S, Arpin N, Olivier JM, Wichers HJ (1999) Biochemical and physiological aspects of brown blotch disease of Agaricus bisporus. FEMS Microbiol Rev 23:591–614
Carrasco J, Preston GM (2020) Growing edible mushrooms: a conversation between bacteria and fungi. Environ Microbiol 22:858–872
Liu C, Sheng J, Chen L, Zheng Y, Lee DYW, Yang Y, Xu M, Shen L (2015) Biocontrol activity of Bacillus subtilis isolated from Agaricus bisporus mushroom compost against pathogenic fungi. J Agrc Food Chem 63:6009–6018
Acknowledgements
We thank all the previous authors mentioned in the current manuscript for the relevant contribution to the field as well as the reviewers and the editors of the journal.
Funding
This research was funded by the United States Department of Agriculture (USDA/NIFA—National Institute of Food and Agriculture) and Hatch Appropriations under Project #PEN04741 and Accession #1023198, John B. Swayne Mushroom Endowment (College of Agricultural Sciences, The Pennsylvania State University) and Giorgi Mushroom Competitive Grant (College of Agricultural Sciences, The Pennsylvania State University).
Author information
Authors and Affiliations
Contributions
F.R.V. and J.A.P. designed the experiments. F.R.V. performed research. F.R.V. analyzed data. F.R.V. drafted the manuscript. F.R.V. and J.A.P. revised and edited the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
All authors agree with the final version of the manuscript as well as the submission to Microbial Ecology Journal.
Conflict of Interest
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Vieira, F.R., Pecchia, J.A. Bacterial Community Patterns in the Agaricus bisporus Cultivation System, from Compost Raw Materials to Mushroom Caps. Microb Ecol 84, 20–32 (2022). https://doi.org/10.1007/s00248-021-01833-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-021-01833-5