
Abstract
Marine lakes are small bodies of landlocked seawater that are isolated from the open sea and have been shown to house numerous rare and unique taxa. The environmental conditions of the lakes are also characterised by lower pH and salinity and higher temperatures than generally found in the open sea. In the present study, we used a 16S rRNA gene barcoded pyrosequencing approach and a predictive metagenomic approach (PICRUSt) to examine bacterial composition and function in three distinct biotopes (sediment, water and the sponge species Biemna fortis) in three habitats (two marine lakes and the open sea) of the Berau reef system, Indonesia. Both biotope and habitat were significant predictors of higher taxon abundance and compositional variation. Most of the variation in operational taxonomic unit (OTU) composition was related to the biotope (42% for biotope alone versus 9% for habitat alone and 15% combined). Most OTUs were also restricted to a single biotope (1047 for B. fortis, 6120 for sediment and 471 for water). Only 98 OTUs were shared across all three biotopes. Bacterial communities from B. fortis, sediment and water samples were, however, also distinct in marine lake and open sea habitats. This was evident in the abundance of higher bacterial taxa. For example, the phylum Cyanobacteria was significantly more abundant in samples from marine lakes than from the open sea. This difference was most pronounced in the sponge B. fortis. In line with the compositional differences, there were pronounced differences in predicted relative gene count abundance among biotopes and habitats. Of particular interest was the predicted enrichment in B. fortis from the marine lakes for pathways including DNA replication and repair and the glutathione metabolism. This may facilitate adaptation of host and microbes to life in ‘stressful’ low pH, low salinity and/or high temperature environments such as those encountered in marine lakes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Marine lakes are landlocked bodies of water that maintain their marine character through submarine connections to the sea [1]. There are only an estimated 200 marine lakes in the world, most of which are found in Indonesia, Vietnam and Palau [2,3,4]. The environmental parameters of marine lakes are strongly influenced by the degree of connection to the surrounding sea. Studies carried out in 12 marine lakes in East Kalimantan and West Papua and the adjacent open sea habitats have shown that marine lakes generally have lower pH, lower salinity and higher temperature than the surrounding open sea marine environment [3]. Marine lakes may thus provide interesting insights into how future estimates of climate change will affect microbial assemblages and provide a clearly defined spatial setting to study the influence of environmental parameters such as pH and salinity on the composition of marine communities.
In the marine lakes of Berau, Cleary et al. [5,6,7] and Cleary and Polónia [8] previously described host-associated bacterial communities of sponges (Cinachyrella spp.), molluscs (Brachidontes spp.) and jellyfish (Mastigias cf. papua and Tripedalia cf. cystophora). These taxa tended to contain diverse, often taxon-specific bacterial communities that are compositionally very different to bacterial communities in the surrounding water. Bacterial symbionts are a topic of major recent interest due to their ecological and biotechnological importance, particularly in association with marine sponges [9]. Sponges (phylum: Porifera) are sedentary, filter-feeding metazoans that utilise a single layer of flagellated cells to pump water currents through their bodies and are one of the most important benthic invertebrates in marine lakes in terms of abundance, cover and diversity [1]. In the last few decades, sponges have evoked particular interest due to the pharmaceutical potential and biotechnological applications of their secondary metabolites [10,11,12,13,14]. Sponges host diverse microbial populations and, in certain species, these communities make up more than 40% of the total biomass [15,16,17]. The role of sponge symbionts has not been completely elucidated yet; however, it is believed that they play an indispensable role in the trophic link between dissolved organic carbon (DOC) and the autotrophic grazer food chain [18, 19] and play important roles in nutrition [20, 21], immunity [22], defence [23], reproduction [24] and the elimination of toxic metabolic end products [20, 25,26,27,28]. Symbiotic cyanobacteria, for example, benefit sponges via atmospheric nitrogen fixation [29,30,31,32].
In the present study, we compared the composition of bacteria inhabiting the sponge species Biemna fortis (order Biemnida, family Biemnidae), sediment and water (bacterioplankton). The current study focused on three distinct habitats, namely, two marine lakes and the surrounding open sea environment in the Berau region of East Kalimantan, Indonesia. The marine lakes in question are located within the islands of Kakaban and Maratua and were formed approximately 7000–12,000 years before present [3]. The marine lakes of Berau are known to contain highly distinct faunas with a high degree of endemism [33,34,35,36].
Specific aims of the present study were to (1) compare higher taxon relative abundance among habitats and biotopes, (2) identify the most abundant bacterial operational taxonomic units (OTUs) and their closest known relatives using the Basic Local Alignment Search Tool (BLAST), (3) assess to what extent the biotope and habitat structure bacterial composition and (4) assess to what extent the different biotopes and habitats are predicted to harbour functionally distinct bacterial communities.
Material and Methods
Study Site
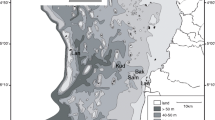
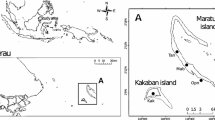
Sampling took place in marine lakes of the islands Kakaban and Maratua and the surrounding marine environment in the Berau region, East Kalimantan, Indonesia (Fig. 1). Yearly rainfall over the period 1987–2007 in Tanjung Redeb, Berau, ranged from 1700 to 3350 mm year−1 (average 2084 mm); monthly precipitation in Berau ranges from 110 to 250 mm with lowest rainfall in August (average 117 mm) and highest from November–January (average 223 mm) (http://www.bmkg.go.id/BMKG_Pusat/, [1]). Becking et al. [3] provided a description of the marine lakes of Kakaban and Maratua. Kakaban is a large island, the centre of which holds a large marine lake (the ca. 4-km2 lake Kakaban hereafter referred to as Kakaban) with southern, western and eastern coasts of the lake fringed by mangroves. The northern shore is predominantly rocky. Tidal amplitude in Kakaban is dampened to 11% of the surrounding sea and the tidal phase has a 3-h and 30-min delay. This indicates limited connection with the surrounding environment [3]. Maratua is a large horseshoe-shaped island further offshore from the main island of Borneo than Kakaban; it encircles a very large semi-enclosed lagoon with depth ranging from 0.5–5 m at low tide and directly connected to the open sea. Maratua contains a number (at least nine) of small anchialine systems including Haji Buang. Haji Buang is an elongated lake of 0.14-km2 surface area located on the western arm of Maratua. Most of the coastline of Haji Buang consists of limestone rock with a small area of mangrove fringing the southern coast. Tidal amplitude of Haji Buang is 48% of the adjacent sea with a tidal delay of 2 h and 30 min indicating a limited connection to the sea but higher than Kakaban. In addition to sampling in lakes, we also sampled the bacterial community from B. fortis, sediment and water (bacterioplankton) in the large lagoon encircled by the island of Maratua. The salinity in Lake Kakaban is only 23–24 ppt, in Lake Haji Buang 26–28 ppt and in the open sea 33–34 ppt. The pH range of the lakes is also lower (7–7.8) than in the open sea (8–8.2) [3].

Map of the study area showing the location of the study sites and Indonesia in the upper left inset. Bottom left and right insets show location of sample areas: K: lake Kakaban; M: lake Haji Buang; Maratua; W: open sea
Sampling
Water, sediment and specimens of the sponge B. fortis were collected from Kakaban, Haji Buang and the open sea using snorkelling from the 17th to 25th of August 2012 (Fig. 1). Biemna fortis Topsent, 1897 (order Biemnida, family Biemnidae) is a drab brown-greyish black sponge that lives partially buried in sediment with irregular processes that are raised above the substratum. This sponge species mostly occurs in very shallow, muddy, sandy environments where it is able to survive exposure to air during very low tides. This species has been reported from East Africa to Indonesia. Sponges belonging to the family Biemnidae can cause dermatitis and have antibacterial, antimalarial and anticancer properties [37,38,39]. This is the first detailed study to assess the bacterial community of a species belonging to the order Biemnida, although Ilan and Abelson [40] showed in a TEM image that the sister species from the Red Sea harboured baceteriocytes containing numerous species of bacteria. The sponge genus Biemna until recently belonged to the order Poecilosclerida, which consists exclusively of low microbial abundance species [41, 42].
Two to three samples of each biotope (sponge, sediment and water) were collected in each habitat (Kakaban, Haji Buang and the open sea of Maratua). Fragments of sponges were collected including the surface and interior of each sponge in order to sample as much as possible of the whole bacterial community [5]. Voucher samples have been deposited in the sponge collection of Naturalis Biodiversity Center (RMNH POR. 10693, 10698, 10699 (Lake Kakaban), RMNH POR. 10700, 10701, 10723 (Lake Haji Buang, Maratua), RMNH POR. 10724, 10725, 10726 (open sea)). Sediment was collected from the upper 5-cm surface layer using a plastic disposable syringe from which the end had been cut in order to facilitate sampling. Water was collected between the depths of 1–2 m with a 1.5-L bottle and subsequently 1 L (± 50 ml) of water was filtered [43] through a Millipore® White Isopore Membrane Filter (0.22-μm pore size) to obtain the bacterioplankton. The filter was subsequently preserved in 96% EtOH. All samples were kept cool (< 4 °C) immediately after collection and during transport. In the laboratory, samples were stored at − 80 °C until DNA extraction.
DNA Extraction and Pyrosequencing
We isolated PCR-ready total community DNA (TC-DNA) from sediment, seawater and sponge samples using the FastDNA® SPIN Kit (MP Biomedicals) following the manufacturer’s instructions. Briefly, we prepared sediment samples by centrifuging each one for 30 min at 4400 rpm and 4 °C; the membrane filter (seawater sample) and sponge samples were each cut into small pieces. The whole membrane filter and 500 mg of sediment and sponge were transferred to lysing matrix E tubes containing a mixture of ceramic and silica particles. The microbial cell lysis was performed in the FastPrep® Instrument (Q Biogene) for 80 s at the speed of 6.0. Extracted DNA was eluted into DNase/pyrogen-free water to a final volume of 50 μl and stored at − 20 °C until use. For the 16S rRNA gene amplification, the first PCR amplification was performed from DNA using the F-27 and R-1494 primers [44]. After a denaturation step at 94 °C for 5 min, 25 thermal cycles of 45 s at 94 °C, 45 s at 56 °C and 1:30 min at 72 °C were carried out followed by an extension step at 72 °C for 10 min. Using the amplicons of the bacterial 16S rRNA gene as template, the V3–V4 region was amplified, with the barcoded fusion forward (V3: 5′-ACTCCTACGGGAGGCAG-3′; [45] and reverse primers (V4: 5′-TACNVRRGTHTCTAATYC-3′; [46]) containing the Roche-454 A and B titanium sequencing adapters, an eight-base barcode sequence in adaptor A and specific sequences for the ribosomal region. After a denaturation step at 94 °C during 4 min, 25 thermal cycles of 30 s at 94 °C, 45 s at 44 °C and 1 min at 68 °C and a final extension at 68 °C for 10 min [5] were carried out with GS 454 FLX titanium chemistry, according to manufacturer’s instructions (Roche, 454 Life Sciences, Brandford, CT, USA).
Following previous studies [47, 48], barcoded pyrosequencing libraries were analysed using the Quantitative Insights into Microbial Ecology (QIIME) software package ([49]; http://www.qiime.org/; last checked 20 January 2014). In QIIME, separate fasta and qual files were used as input for the split_libraries.py script. Default arguments were used except for the minimum sequence length, which was set at 218 bps after removal of forward primers and barcodes; backward primers were removed using the ‘truncate only’ argument and a sliding window test of quality scores was enabled with a value of 50 as suggested in the QIIME description for the script. In addition to user-defined cut-offs, the split_libraries.py script performs several quality filtering steps (http://qiime.org/scripts/split_libraries.html). OTUs were selected using UPARSE with usearch7 using a sequence similarity threshold of 97% [50]. The UPARSE sequence analysis tool [50] provides clustering, chimera checking and quality filtering on de-multiplexed sequences. Chimera checking was performed using the UCHIME algorithm [51]. The quality filtering as implemented in usearch7 filters noisy reads and preliminary results suggest it gives results comparable to other denoisers such as AmpliconNoise, but is much less computationally expensive (http://drive5.com/usearch/features.html; last checked 20 January 2014). First, reads were filtered with the -fastq_filter command and the following arguments -fastq_trunclen 250, -fastq_maxee 0.5, -fastq_truncqual 15. Sequences were then dereplicated and sorted using the -derep_fulllength and -sortbysize commands. OTU clustering was performed using the -cluster_otus command. An additional chimera check was subsequently applied using the -uchime_ref command with the gold.fa database (http://drive5.com/uchime/gold.fa). AWK scripts were then used to convert the OTU files to QIIME format. In QIIME, representative sequences were selected using the pick_rep_set.py script in QIIME using the ‘most_abundant’ method. Taxonomy was assigned to reference sequences of OTUs using default arguments in the assign_taxonomy.py script in QIIME with the ribosomal database project (rdp) method [52]. In the assign_taxonomy.py function, we used a fasta file containing reference sequences from the Greengenes 13_8 release and the rdp classifier method. We used a modified version of the taxonomy file supplied with the Greengenes 13_8 release to map sequences to the assigned taxonomy. Finally, we used the make_otu_table.py script in QIIME to generate a square matrix of OTUs × samples. This was subsequently used as input for further analyses using the R package [53]. Sequence identifiers of closely related taxa of numerically dominant OTUs (≥ 400 sequences) were downloaded using the NCBI Basic Local Alignment Search Tool (BLAST) command line ‘blastn’ tool with the -db argument set to nt [54]. BLAST identifies locally similar regions between sequences, compares sequences to extant databases and assesses the significance of matches; functional and evolutionary relationships can subsequently be inferred. Each run produces a list of hits based on significant similarity between pairs of sequences, i.e. the target sequence and taxa present in the database (or no hits if no significantly similar sequences are found). A discussion of how significance is determined can be found at http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html. The DNA sequences generated in this study can be downloaded from the NCBI SRA: SRP081069 and SRP068454; sample accession numbers are given in Online Resource 1.
Predictive Metagenome Analysis
In the present study, we used PICRUSt [55, 56] to predict the metagenome of each sample with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Output of PICRUSt consists of a table of functional counts, i.e. KEGG pathway counts by sample. Note that because of functional overlap, some KEGG orthologs (KOs) can be represented in multiple pathways. Since KOs can belong to several pathways, we used the categorize_by_function.py script in PICRUSt to collapse the PICRUSt predictions at the level of the individual pathways. Note that the PICRUSt results as presented are predictive and thus provide information on potential enrichment and putative function as opposed to measuring actual gene presence/expression and function.
Higher Taxon Abundance
We tested for significant differences in the relative abundance of selected higher taxa (phyla, classes and orders) and dominance (the relative abundance of the most abundant OTU in each sample) among biotopes and habitats with an analysis of deviance using the glm() function in R ([53]; Online Resource 2). Because the data was proportional, we first applied a generalised linear model (glm) with the family argument set to binomial. The ratio, however, of residual deviance to residual d.f. in the models substantially exceeded 1 so we set family to ‘quasibinomial’. In the ‘quasibinomial’ family the dispersion parameter is not fixed at one so that it can model overdispersion. Using the glm model, we tested for significant variation among biotopes using the anova() function in R with the F test, which is most appropriate when dispersion is estimated by moments as is the case with quasibinomial fits. Ad hoc comparisons were made with the glht() function in the multcomp package (https://cran.r-project.org/web/packages/multcomp/multcomp.pdf; last checked 17 November 2017). Detailed descriptions of the functions used here can be found in R (e.g. ?cmdscale) and online in reference manuals (http://cran.r-project.org/web/packages/vegan/index.html; accessed 27 February 2015).
Composition
For the OTU table (Online Resource 3), OTUs not classified as bacteria or classified as chloroplasts and mitochondria were removed prior to statistical analysis. This table was then loge (x + 1) transformed and distance matrices constructed using the Bray-Curtis index with the vegdist() function in the vegan package [57] in R. The Bray-Curtis index is one of the most frequently applied (dis)similarity indices used in ecology [58, 59]. Variation in OTU composition among biotopes (B. fortis, sediment and water) and habitats (Kakaban, Haji Buang and open sea) was assessed with principal coordinates analysis (PCO) using the cmdscale() function in R with the Bray-Curtis distance matrix as input. Variation among biotopes and habitats was tested for significance using the adonis() function in vegan. In the adonis analysis, the Bray-Curtis distance matrix of species composition was the response variable with biotope and habitat as independent variables; the strata (block) argument was set to habitat so that randomisations were constrained to occur within each habitat and not across all habitats. Detailed descriptions of the functions used here can be found in R (e.g. ?cmdscale) and online in the reference manuals (e.g. http://cran.r-project.org/web/packages/vegan/index.html; checked 2014 September 21).
Results
Sequencing in the present study yielded 80,771 sequences, assigned to 8657 bacterial OTUs (97% sequence similarity threshold) after quality control, OTU picking and removal of chimera, chloroplasts and mitochondria. Of these, 6120 were only recorded in the sediment biotope, 1047 in B. fortis and 471 in water. Ninety-eight OTUs were recorded in all three biotopes while B. fortis shared 106 OTUs with water and 717 with sediment. Water and sediment shared 98 OTUs (Online Resource 4). The total number of bacterial phyla recorded per biotope also varied considerably from 20 in water to 33 in B. fortis and 47 in sediment. The number of classes varied from 47 in water to 82 in B. fortis and 128 in sediment and the number of orders varied from 72 in water to 111 in B. fortis and 157 in sediment. OTU richness followed this general pattern. OTU richness was highest in the sediment biotope and higher in open water than marine lake sediment. OTU richness appeared to be similar in bacterioplankton and B. fortis, although there was a greater range in B. fortis samples with much higher and lower richness than water samples. As in sediment, richness was greater for the B. fortis bacterial community in the open sea than both lake habitats (Fig. 2). Most bacterial sequences belonged to OTUs assigned to Proteobacteria (61571) followed by Cyanobacteria (14656), Acidobacteria (4974), Chloroflexi (4868), Bacteroidetes (4688) and Actinobacteria (3137). The percentage of Proteobacteria varied from 49.5 ± 1.5% for sediment in Haji Buang to 66.1 ± 12.3% for water in the open sea. The percentage of Cyanobacteria, in turn, varied from 0.9 ± 1.0% for sediment in Haji Buang to 34.6 ± 3.9% for water in Kakaban (Fig. 3). The percentage of Cyanobacteria was also much higher in B. fortis from Kakaban (12.1 ± 6.6%) and Haji Buang (23.8 ± 19.2%) than from the open sea (1.1 ± 0.2%).

Rarefied OTU richness for samples of B. fortis from Kakaban (BmK), Haji Buang (BmM) and the open sea (BmW), of sediment from Kakaban (SdK), Haji Buang (SdM) and the open sea (SdW) and of water from Kakaban (WtK), Haji Buang (WtM) and the open sea (WtW)

Mean (error bars represent a single standard deviation) relative abundance of the most abundant bacterial phyla for samples of B. fortis from Kakaban (BmK), Haji Buang (BmM) and the open sea (BmW), of sediment from Kakaban (SdK), Haji Buang (SdM) and the open sea (SdW) and of water from Kakaban (WtK), Haji Buang (WtM) and the open sea (WtW). a Proteobacteria, b Cyanobacteria, c Acidobacteria, d Chloroflexi, e Bacteroidetes, f Actinobacteria, g Firmicutes, h Gemmatimonadetes, i Nitrospirae, j PAUC34f, k SBR1093 and l GN02. Results of the GLM analyses for each taxon are presented in the top right of each subfigure
There were significant differences in the relative abundance of higher bacterial taxa among biotopes and habitats (Fig. 3). Acidobacteria, Chloroflexi, Gemmatimonadetes, PAUC34f and SBR1093 were more abundant in B. fortis or in B. fortis and sediment biotopes than in water samples. Firmicutes were more abundant in sediment and Bacteroidetes and GN02 were more abundant in water samples. Among habitats, Cyanobacteria were significantly more abundant in lake habitat, thus in lake sponge, sediment and water samples (the Tukey test: open sea versus Kakaban, z = − 2.72, P = 0.015; open sea versus Haji Buang, z = − 3.65, P < 0.001). Other taxa showed contrasting responses to habitat. Chloroflexi abundance was, for example, higher in samples of B. fortis collected in the open sea than from the lakes, but higher in lake sediment than open sea sediment. Actinobacteria were more abundant in B. fortis and sediment from lakes. Nitrospirae were much more abundant in open sea sediment (Fig. 3).
At a lower taxonomic level, Gammaproteobacteria, Anaerolineae, Solibacteres, Alteromonadales and HTCC2188 were most abundant in B. fortis (Fig. 4). Deltaproteobacteria, Acidimicrobiia and Desulfobacterales were most abundant in sediment while Alphaproteobacteria, Flavobacteriia, Rhodobacterales and Rickettsiales were most abundant in water samples. In addition to differences in relative abundance among biotopes, there were also significant differences among habitats (Fig. 3). Synechococcophycidae were more abundant in both lake habitats than the open sea whereas the reverse was true for Alphaproteobacteria. There were also significant interactions between biotope and habitat. Anaerolineae and HTCC2188 were, for example, more abundant in B. fortis from the open sea than both lakes whereas the reverse was true for Synechococcophycidae. Likewise, the abundance of Desulfobacterales in sediment was higher in lake than open sea habitat (Fig. 4).

Mean (error bars represent a single standard deviation) relative abundance of the most abundant bacterial classes and orders and the most abundant OTU (dominant OTU) for samples of B. fortis from Kakaban (BmK), Haji Buang (BmM) and the open sea (BmW), of sediment from Kakaban (SdK), Haji Buang (SdM) and the open sea (SdW) and of water from Kakaban (WtK), Haji Buang (WtM) and the open sea (WtW). Note that the abundance of the dominant OTU refers to the abundance of the most abundant OTU per sample and thus not the most abundant OTU overall. a Gammaproteobacteria, b Alphaproteobacteria, c Synechococcophycidae, d Deltaproteobacteria, e Anaerolineae, f Acidimicrobiia, g Solibacteres, h Flavobacteriia, i Alteromonadales, j Chromatiales, k Rhodobacterales, l Rickettsiales, m Rhodospirillales, n Desulfobacterales, o HTCC2188 and p dominant OTU. Results of the GLM analyses for each taxon are presented in the top right of each subfigure
There was a significant difference in composition among biotopes (adonis: F2,20 = 12.64, P < 0.001, R2 = 0.418), habitats (adonis: F4,20 = 2.82, P < 0.001, R2 = 0.093) and the interaction of both factors (adonis: F2,20 = 2.38, P < 0.001, R2 = 0.158). The biotope was the main source of variation in composition, but samples from all biotopes also differed in composition among habitats. This difference was, however, more pronounced for B. fortis and sediment than water. The first PCO axis separated sediment and B. fortis samples from water samples; the second axis separated B. fortis samples from sediment and water (Fig. 5). A number of abundant OTUs were found predominantly or exclusively in B. fortis (Online Resource 5 and Table 1). Most of these OTUs were related to organisms obtained from other sponge species, but also included OTUs related to organisms obtained from a coral and estuarine sediment (Table 1). The sequence similarity with the closest known organism was relatively low for a number of OTUs including those housed mainly in B. fortis such as OTU-1 (sequence similarity = 93.96) and OTU-43 (sequence similarity = 92.62). OTUs found predominantly in water included OTUs 15, 19, 34 and 37 assigned to the Alphaproteobacteria and OTU-28 assigned to the Deltaproteobacteria. All of these OTUs had very high sequence similarity (> 99%) to organisms previously obtained from seawater (Table 1).

Ordination showing the first two axes of the PCO analysis. a Symbols represent samples of B. fortis from Kakaban (BmK), Haji Buang (BmM) and the open sea (BmW), of sediment from Kakaban (SdK), Haji Buang (SdM) and the open sea (SdW) and of water from Kakaban (WtK), Haji Buang (WtM) and the open sea (WtW). Numbers represent abundant (≥ 400 sequence reads) OTUs referred to in Table 1. The circle size of OTUs is proportional to the abundance (number of sequences). The first two axes explain 43% of the variation in the data set
In line with the compositional differences among biotopes and habitats, there were pronounced differences in predicted functional attributes of the bacterial communities (Fig. 6). Samples from B. fortis were predicted to be enriched for peptidases and polyketide sugar unit biosynthesis pathways. Biemna fortis and water were predicted to be enriched for a number of pathways in comparison to sediment. These included DNA replication and repair proteins and valine, leucine and isoleucine biosynthesis pathways whereas sediment was enriched for the transporters, methane metabolism and benzoate degradation pathways. A number of pathways were predicted to be enriched in B. fortis samples from both lakes as compared to B. fortis samples from the open sea including the DNA repair and replication protein, photosynthesis proteins, glutathione metabolism, polyketide sugar unit biosynthesis, terpenoid backbone biosynthesis and the metabolism of xenobiotics by cytochrome P450 pathways. Biemna fortis samples from the open sea, in contrast, were predicted to be enriched for the transporters, steroid biosynthesis and peptidases pathways.

Mean predicted relative gene count abundance for selected KEGG pathways for samples of B. fortis from Kakaban (BmK), Haji Buang (BmM) and the open sea (BmW), of sediment from Kakaban (SdK), Haji Buang (SdM) and the open sea (SdW) and of water from Kakaban (WtK), Haji Buang (WtM) and the open sea (WtW). Error bars represent a single standard deviation. The individual pathways shown include the following KEGG pathways: a cytoskeletal proteins (cytoskel. prot.), b transporters, c two-component system, d DNA repair and recombination proteins (DNA repair and repl. prot.), e phenylalanine, tyrosine and tryptophan biosynthesis (phen., tyr. and try. bio.), f valine, leucine and isoleucine biosynthesis (val., leuc. and iso. bio.), g isoquinoline alkaloid biosynthesis (isoqui. alk. bio.), h methane metabolism, i nitrogen metabolism, j photosynthesis prot. (photosynthesis proteins), k sulphur metabolism, l peptidases, m steroid biosynthesis (steroid bio.), n glutathione metabolism (glutathione met.), o biosynthesis of ansamycins (ansamycins bio.), p biosynthesis of siderophore group nonribosomal peptides (nonribo. peptides bio.), q polyketide sugar unit biosynthesis (polyketide sugar bio.), r terpenoid backbone biosynthesis (terpenoid back. bio.), s benzoate degradation (benzoate degrad.) and t metabolism of xenobiotics by cytochrome P450 (xeno. cyto. P450)
Discussion
Biemna fortis, water and sediment samples were collected from three different habitats with very different environmental conditions, namely two marine lakes in the Berau region and the open sea. Lake Kakaban is the largest lake and although the deepest part of the lake is about 12 m, all samples were collected from a maximum depth of 1 m. The salinity and the pH range of the two marine lakes are lower than in the open sea. As mentioned previously, the recorded pH range was 7.0–7.8 for Kakaban, 7.3–7.8 for Haji Buang and 8.2–8.5 for the open sea; the recorded salinity was 23–24 for Kakaban, 26–28.5 for Haji Buang and 33–34 ppt for the open sea [3]. The different environmental conditions might explain the differences in relative abundance and composition of bacterial communities observed in this study.
OTU richness was lower in the lake sediment bacterial community than in the open sea sediment bacterial community and to a lesser extent lower in the lake water bacterial community than the open sea water bacterial community. The bacterial community of lake sediment was characterised by a higher relative abundance of Anaerolineae, Acidimicrobiia and Desulfobacterales whereas the sediment bacterial community of the open sea was characterised by a higher relative abundance of Alphaproteobacteria. The water bacterial community of the open sea also had a higher relative abundance of Alphaproteobacteria, but a markedly lower relative abundance of Synechococcophycidae (Cyanobacteria) than the lake bacterioplankton communities. Coelho et al. [60] observed a lower relative abundance of Alphaproteobacteria at the most active mud volcanoes (low pH environment) and higher relative abundance of Methylococcales. In a microcosm study, Coelho et al. [61] also observed that the low pH treatment had a significant effect on Alphaproteobacteria and Acidimicrobiia with the most abundant acidimicrobiial OTUs clustering near the reduced seawater pH treatments in an ordination. Importantly, the class Acidimicrobiia includes a number of acidophilic, iron-oxidising bacteria [62]. As with Coelho et al. [61], Monier et al. [63] and Taylor et al. [64] also observed an effect of reduced pH on acidimicrobiial composition.
The most pronounced difference between water of the marine lakes and the open sea was a markedly higher relative abundance of Cyanobacteria in the former where the main cyanobacterial OTUs were assigned to the genus Synechococcus. In an aquarium study, Webster et al. [65] also observed a significant interactive effect of pH (elevated pCO2) and temperature, which was largely driven by the much higher abundance of a single Synechococcus OTU in the low pH treatment. Likewise, Fu et al. [66] observed that elevated temperature and pCO2 increased cell division and photosynthetic rates of Synechococcus. In addition to differences in higher taxon abundance and richness, there were also clear compositional differences between the lakes and the open sea for both sediment and water bacterial communities. Our results thus show that bacterial communities inhabiting the sediment and water of marine lakes differ from those in the open sea. This difference may be related to the lower pH and higher temperature conditions found in these marine lakes, although care must be taken in overemphasising the impact of pH due to the possible confounding effect of salinity, which is also lower in the marine lakes than the open sea. However, it should be noted that the same cyanobacterial OTUs assigned to the genus Synechococcus were found inside and outside the lakes, but were much more abundant in the lakes. In a study of Synechococcus across a salinity gradient, Xia et al. [67] found that the abundance and diversity of Synechococcus assemblages was lower in low salinity waters although Synechococcus members have been shown to be tolerant to low salinity conditions [68].
Biemna fortis samples from both lakes had lower relative abundances of Gammaproteobacteria, Anaerolineae and Solibacteres, higher relative abundance of Acidimicrobiia and much higher relative abundance of Synechococcophycidae compared to B. fortis from the open sea. The lower abundance of Gammaproteobacteria in B. fortis from both lakes was partially due to a lower abundance of OTUs assigned to the order HTCC2188 in the lakes. Characterised as oligotrophic [69], OTUs assigned to the HTCC2188 order have been recorded in a number of marine sponges [5, 70,71,72]. They were also an abundant component of the bacterial community of Cinachyrella australiensis outside of the marine lakes in Berau, but were absent in specimens of Cinachyrella inhabiting the marine lakes [5]. Morrow et al. [70] also observed a reduction in the relative abundance of gammaproteobacterial symbionts of the coral species Acropora millepora and Porites cylindrica at a low pH, CO2 seep. Two sponge species at the seep (Coelocarteria singaporensis and Cinachyra sp.) had lower relative abundances of Alpha-, Beta-, Gamma- and Deltaproteobacteria compared to a control site. Coelocarteria singaporensis also had lower abundances of Chloroflexi, Acidobacteria and Nitrospirae at the CO2 seep. Both coral species and both sponge species at the CO2 seep, however, showed twofold increases in the relative abundance of Cyanobacteria at the seep compared to the control site (e.g. 15 to 30% for Cinachyra sp. and 35 to 70% for Coelocarteria singaporensis). As in the present study, the main cyanobacterial OTUs in both corals and sponges were assigned to the genus Synechococcus with sponges and corals housing distinct taxa.
The increased abundance of OTUs assigned to the genus Synechococcus in the present study and in other natural low pH habitats such as CO2 seeps suggests that strains of the genus help their host organisms adapt to living in reduced pH environments and may give their host organisms a competitive advantage in terms of growth compared to organisms with a less flexible symbiotic bacterial community [70]. The shift in bacterial composition from Gammaproteobacterial orders such as HTCC2188 to cyanobacteria suggests a shift in how nutrients are acquired within the sponge host with potential repercussions for nutrient cycling.
Sponges are also found in much greater densities within lakes than in the surrounding sea including coral reefs and mangroves [1]. In the marine lakes, sponges often completely cover the roots of mangrove trees fringing the lakes and can also be found embedded in the sediment as is the case with B. fortis. In addition to reduced predation pressure and the lack of competition from other groups of benthic invertebrates (e.g. corals), the increased density may be at least partially related to the marked increase of Cyanobacteria in marine lake sponges. So far, two distinct genera, Biemna and Cinachyrella, both showed an increase in Synechococcus abundance in lake as opposed to open sea habitat. Future research should assess if this trend is more widespread among lake sponges and try to ascertain if a general shift to symbiotic cyanobacteria in lake sponges can explain the disparity in density between lake sponges and those in the surrounding sea.
In line with the differences in higher taxon abundance and composition, there were clear differences in predicted functional attributes among biotopes and between lake and open sea habitats. Biemna fortis from both lakes were enriched for the DNA repair and replication protein, photosynthesis proteins, glutathione metabolism, polyketide sugar unit biosynthesis and terpenoid backbone biosynthesis KEGG pathways. Morrow et al. [70] observed that dominant KEGG pathways for Synechococcus included photosynthesis and DNA repair and recombination. The primary KEGG level 2-predicted pathways for Synechococcus were the energy metabolism (particularly pathways involved in photosynthesis), carbohydrate metabolism and amino acid metabolism [70].
In addition to being enriched for photosynthesis, lake sponges were also enriched for pathways that are advantageous for life in stressful environments including DNA repair and replication and the glutathione metabolism. Higher replication and repair may also be a sign of faster growth in the bacterial community as a consequence of increasing carbon and nitrogen fixation, high temperatures and more available CO2. The DNA repair and replication pathway is part of the KEGG genetic information processing category, which includes genes that are responsible for growth and proliferation [73]. DNA repair and replication helps to preserve genetic information and its accurate propagation to the next generation. DNA can be damaged by both endogenous and exogenous factors and needs repairing so as not to disrupt nucleic acid replication [74]. Obligate primary symbionts, however, can lose functions including DNA repair and replication [75,76,77], which places them at a disadvantage in stressful environments. Having a flexible symbiont community, in contrast, enables sponges and other host organisms to colonise and survive in these environments.
Lake sponges were also enriched for the glutathione metabolism pathway. Glutathione is a powerful antioxidant that can prevent damage to cells by stressors including free radicals, peroxides, lipid peroxides and heavy metals [78, 79]. This pathway is known to be present in Cyanobacteria, but not in all bacteria. In addition to providing their host organism with photosynthates, Synechococcus spp. may also help their host organisms to flourish in marine lake environments.
Conclusion
This study showed clear differences in higher taxon abundance, composition and predicted functional attributes among biotopes and between marine lake and open sea habitats. Although most of the variation in OTU composition was related to the biotope, significant differences in the relative abundance and composition of bacterial communities were related to the different environmental conditions encountered in lake versus open sea habitat. Taxa detected in higher relative abundances in lake environments (e.g. Cyanobacteria) have been linked to functions that confer advantages for life in stressful environments.
References
Becking LE, Cleary DFR, De Voogd NJ (2013) Sponge species composition, abundance, and cover in marine lakes and coastal mangroves in Berau, Indonesia. Mar. Ecol. Prog. Ser. 481:105–120. https://doi.org/10.3354/meps10155
Dawson MN, Hamner WM (2005) Rapid evolutionary radiation of marine zooplankton in peripheral environments. P Natl Acad Sci USA 102:9235–9240. https://doi.org/10.1073/pnas.0503635102
Becking LE, Renema W, Santodomingo NK, Hoeksema BW, Tuti Y, De Voogd NJ (2011) Recently discovered landlocked basins in Indonesia reveal high habitat diversity in anchialine systems. Hydrobiologia 677:89–105. https://doi.org/10.1007/s10750-011-0742-0
Becking LE, De Leeuw C, Vogler C (2014) Newly discovered “jellyfish lakes” in Misool, Raja Ampat, Papua, Indonesia. Mar. Biodivers. 45:597–598. https://doi.org/10.1007/s12526-014-0268-6
Cleary DFR, Becking LE, Pires ACC, de Voogd NJ, Egas C, Gomes NCM (2013) Habitat and host related variation in sponge bacterial communities in Indonesian coral reefs and marine lakes. FEMS Microbiol. Ecol. 85:465–482. https://doi.org/10.1111/1574-6941.12135
Cleary DFR, Becking LE, Polónia ARM, Freitas RM, Gomes NCM (2015) Composition and predicted functional ecology of mussel-associated bacteria in Indonesian marine lakes. A Van Leeuw J Microb 107:821–834. https://doi.org/10.1007/s10482-014-0375-1
Cleary DFR, Becking LE, Polónia ARM, Freitas R, Gomes NCM (2016) Jellyfish-associated bacterial communities and bacterioplankton in Indonesian Marine lakes. FEMS Microbiol Ecol 92:fiw064. https://doi.org/10.1093/femsec/fiw064
Cleary DFR, Polónia ARM (2017) Bacterial and archaeal communities inhabiting mussels, sediment and water in Indonesian anchialine lakes. A Van Leeuw J Microb. doi: https://doi.org/10.1007/s10482-017-0944-1
Caston CB, Nowlin WH, Gaulke A, Vanni MJ (2009) The relative importance of heterotrophic bacteria to pelagic ecosystem dynamics varies with reservoir trophic state. Limnol. Oceanogr. 54:2143–2156
Blunt JW, Munro MHG (1998) MarinLit. A database of the literature on marine natural products for use on a Macintosh computer prepared and maintained by the Marine Chemistry Group Department of Chemistry. University of Canterbury, Canterbury, New Zealand
Van Soest RWM, Braekman JC (1999) Chemosystematics of Porifera: a review. Mem Queensl Mus 44:569–598
Faulkner DJ, Harper MK, Haygood MG, Salomon CE, Schmidt EW (2000) Symbiotic bacteria in sponges: sources of bioactive substances. In: Fusetani N (ed) Drugs from the sea. Karger, Basel, Switzerland, pp 107–119
Sipkema D, Franssen MCR, Osinga R, Tramper J, Wijffels RH (2005) Marine sponges as pharmacy. Mar. Biotechnol. 7:142–162. https://doi.org/10.1007/s10126-004-0405-5
Taylor MW, Radax R, Steger D, Wagner M (2007) Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol R 71:295–347. https://doi.org/10.1128/MMBR.00040-06
Unson MD, Holland ND, Faulkner DJ (1994) A brominated secondary metabolite synthesized by the cyanobacterial symbiont of a marine sponge and accumulation of a crystalline metabolite in the sponge tissue. Mar Biol 119:1–11. https://doi.org/10.1007/BF00350100
Friedrich AB, Fischer I, Proksch P, Hacker H, Hentschel U (2001) Temporal variation of the microbial community associated with the Mediterranean sponge Aplysina aerophoba. FEMS Microbiol. Ecol. 38:105–113. https://doi.org/10.1111/j.1574-6941.2001.tb00888.x
Sacristán-Soriano O, Banaigs B, Casamayor EO, Becerro MA (2011) Exploring the links between natural products and bacterial assemblages in the sponge Aplysina aerophoba. Appl. Environ. Microbiol. 77:862–870. https://doi.org/10.1128/AEM.00100-10
Currie DJ, Kalff JK (1984) The relative importance of bacterioplankton and phytoplankton in phosphorus uptake in freshwater. Limnol. Oceanogr. 29:311–321
Danger M, Umarou CO, Benest D, Lacroix G (2007) Bacteria can control stoichiometry and nutrient limitation of phytoplankton. Funct. Ecol. 21:202–210. https://doi.org/10.1111/j.1365-2435.2006.01222.x
Reiswig HM (1975) Bacteria as food for temperate-water marine demosponges. Can. J. Zool. 53:582–589
Baumann P (2005) Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 59:155–189. https://doi.org/10.1146/annurev.micro.59.030804.121041
Macdonald TT, Monteleone G (2005) Immunity, inflammation, and allergy in the gut. Science 307:1920–1925. https://doi.org/10.1126/science.1106442
Scarborough CL, Ferrari J, Godfray HC (2005) Aphid protected from pathogen by endosymbiont. Science 310:1781. https://doi.org/10.1126/science.1120180
Hurst GD, Werren JH (2001) The role of selfish genetic elements in eukaryotic evolution. Nat Rev Genet 2:597–606. https://doi.org/10.1038/35084545
Wilkinson CR (1978) Microbial association in sponges. II. Numerical analysis of sponge and water bacterial populations. Mar. Biol. 49:169–176
Hoffmann F, Larsen O, Rapp HT, Osinga R (2005) Oxygen dynamics in choanosomal sponge explants. Mar. Biol. Res. 1:160–163. https://doi.org/10.1080/17451000510019006
Hoffmann F, Radax R, Woebken D, Holtappels M, Lavik G, Rapp HT, Schlaeppy ML, Schleper C, Kuypers MMM (2009) Complex nitrogen cycling in the sponge Geodia barretti. Environ. Microbiol. 11:2228–2243. https://doi.org/10.1111/j.1462-2920.2009.01944.x
Holmes B, Blanch H (2007) Genus-specific associations of marine sponges with group I crenarchaeotes. Mar. Biol. 150:759–772. https://doi.org/10.1007/s00227-006-0361-x
Carpenter EJ, Foster RA (2002) Marine cyanobacterial symbioses. In Cyanobacteria in Symbiosis. Springer, Netherlands, pp 11–17
Wilkinson C, Fay P (1979) Nitrogen fixation in coral reef sponges with symbiotic cyanobacteria. Nature 279:527–529. https://doi.org/10.1038/279527a0
Fiore CL, Jarett JK, Olson ND, Lesser MP (2010) Nitrogen fixation and nitrogen transformations in marine symbioses. Trends in microbiol 18:455–463
Ribes M, Dziallas C, Coma R, Riemann L (2015) Microbial diversity and putative diazotrophy in high- and low-microbial-abundance Mediterranean sponges. Appl. Environ. Microbiol. 81:5683–5693. https://doi.org/10.1128/AEM.01320-15
Holthuis LB (1973) Caridean shrimps found in land-locked saltwater pools at four indo-west pacific localities (Sinai Peninsula, Funafuti Atoll, Maui and Hawaii Islands), with the description of one new genus and four new species. Zool Verhandel 128:1–48
Maciolek J (1983) Distribution and biology of Indo-Pacific insular hypogeal shrimps. B Mar Sci 33:606–618
Tomascik T, Mah AJ (1994) The ecology of ‘Halimeda open sea’: an anchialine open sea of a raised atoll, Kakaban Island, East Kalimantan, Indonesia. Tropical Biodiversity 2:385–399
Massin C, Tomascik T (1996) Two new holothurians (Echinodermata: Holothuroidea) from an anchialine open sea of an uplifted atoll, Kakaban Island, East Kalimantan, Indonesia. Raffles B Zool 44:157–172
Gros E, Martin MT, Sorres J, Moriou C, Vacelet J, Frederich M et al (2015) Netamines O–S, five new tricyclic guanidine alkaloids from the Madagascar sponge Biemna laboutei, and their antimalarial activities. Chem. Biodivers. 12:1725–1733. https://doi.org/10.1002/cbdv.201400350
Moran DAP, Takada K, Ise Y, Bontemps N, Davis RA, Furihata K et al (2015) Two cell differentiation inducing pyridoacridines from a marine sponge Biemna sp. and their chemical conversions. Tetrahedron 71:5013–5018. https://doi.org/10.1016/j.tet.2015.05.070
Youssef DT, Badr JM, Shaala LA, Mohamed GA, Bamanie FH (2015) Ehrenasterol and biemnic acid; new bioactive compounds from the Red Sea sponge Biemna ehrenbergi. Phytochem. Lett. 12:296–301. https://doi.org/10.1016/j.phytol.2015.04.024
Ilan M, Abelson A (1995) The life of a sponge in a sandy open sea. Biol. Bull. 189:363–369. https://doi.org/10.2307/1542154
Gloeckner V, Wehrl M, Moitinho-Silva L, Gernert C, Schupp P, Pawlik JR et al (2014) The HMA-LMA dichotomy revisited: an electron microscopical survey of 56 sponge species. Biol. Bull. 227:78–88. https://doi.org/10.1086/BBLv227n1p78
Morrow C, Cárdenas P (2015) Proposal for a revised classification of the Demospongiae (Porifera). Front in Zool 12:7. https://doi.org/10.1186/s12983-015-0099-810.1086/BBLv227n1p78
Bowen JL, Morrison HG, Hobbie JE, Sogin ML (2012) Salt marsh sediment diversity: a test of the variability of the rare biosphere among environmental replicates. ISME J 6:2014–2023. https://doi.org/10.1038/ismej.2012.47
Gomes NCM, Heuer H, Schönfeld J, Costa R, Mendonca-Hagler L, Smalla K (2001) Bacterial diversity of the rhizosphere of maize (Zea mays) grown in tropical soil studied by temperature gradient gel electrophoresis. Plant Soil 232:167–180. https://doi.org/10.1007/978-94-010-0566-1_17
Yu Y, Lee C, Kim J, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89:670–679
Vaz-Moreira I, Egas C, Nunes OC, Manaia CM (2011) Culture-dependent and culture-independent diversity surveys target different bacteria: a case study in a freshwater sample. Antonie Van Leeuwenhoek 100:245–257
Cleary DFR, de Voogd NJ, Polónia ARM, Freitas R, Gomes NC (2015) Composition and predictive functional analysis of bacterial communities in seawater, sediment and sponges in the Spermonde Archipelago, Indonesia. Microb. Ecol. 70:889–903
de Voogd NJ, Cleary DFR, Polónia ARM, Gomes NCM (2015) Bacterial community composition and predicted functional ecology of sponges, sediment and seawater from the thousand islands reef complex, West Java, Indonesia. FEMS Microbiol Ecol 91:fiv019. https://doi.org/10.1093/femsec/fiv019
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10:996–998. https://doi.org/10.1038/nmeth.2604
Edgar R, Haas B, Clemente J, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Wang Q, Garrity G, Tiedje J, Cole J (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73:5261–5267. https://doi.org/10.1128/AEM.00062-07
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria ISBN 3-900051-07-0. Available from http://www.R-project.org/
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7:203–214. https://doi.org/10.1089/10665270050081478
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31:814–821. https://doi.org/10.1038/nbt.2676
Polónia ARM, Cleary DRF, Duarte LN, de Voogd NJ, Gomes NCM (2013) Composition of Archaea in seawater, sediment and sponges in the Kepulauan Seribu reef system, Indonesia. Microb. Ecol. 67:553–567. https://doi.org/10.1007/s00248-013-0365-2
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Szoecs E, Wagner H (2017) Vegan: community ecology package. R package version 2:4–3 https://CRAN.R-project.org/package=vegan
Legendre P, Gallagher ED (2001) Ecologically meaningful transformations for ordination of species data. Oecologia 129:271–280. https://doi.org/10.1007/s004420100716
Cleary DFR (2003) An examination of scale of assessment, logging and ENSO-induced fires on butterfly diversity in Borneo. Oecologia 135:313–321. https://doi.org/10.1007/s00442-003-1188-5
Coelho FJRC, Louvado A, Domingues P, Cleary DFR, Ferreira M, Almeida A, Cunha MR, Cunha Â, Gomes NCM (2016) Integrated characterization of bacterial and microeukaryotic communities in non-active to active mud volcanoes in the Gulf of Cadiz. Sci. Rep. 6:35272. https://doi.org/10.1038/srep35272
Coelho FJRC, Cleary DFR, Rocha RJM, Calado R, Castanheira J, Silva AMS, Simões MMQ, Oliveira V, Lillebo A, Almeida A, Cunha Â, Lopes I, Ribeiro R, Moreira-Santos M, Marques CR, Costa R, Pereira R, Gomes NCM (2015) Unravelling the interactive effects of climate change and oil contamination on lab-simulated estuarine benthic communities. Glob Change Biol 21:1871–1886. https://doi.org/10.1111/gcb.12801
Stackebrandt E, Rainey FA, Ward-Rainey N (2012) Order I. Acidimicrobiales. In: Whitman W, Goodfellow M, Kämpfer P, Hans-Jürgen B, Trujillo M, Ludwig W et al (eds) Bergey’s manual of systematic bacteriology: volume 5: the Actinobacteria. Springer, New York, p 1750
Monier A, Findlay HS, Charvet S, Lovejoy C (2014) Late winter under ice pelagic microbial communities in the high Arctic Ocean and the impact of short-term exposure to elevated CO2 levels. Front. Microbiol. 5:490. https://doi.org/10.3389/fmicb.2014.00490
Taylor JD, Ellis R, Milazzo M, Hall-Spencer JM, Cunliffe M (2014) Intertidal epilithic bacteria diversity changes along a naturally occurring carbon dioxide and pH gradient. FEMS Microbiol. Ecol. 89:670–678. https://doi.org/10.1111/1574-6941.12368
Webster NS, Negri AP, Botté ES, Laffy PW, Flores F, Noonan S, Schmidt C, Uthicke S (2016) Host-associated coral reef microbes respond to the cumulative pressures of ocean warming and ocean acidification. Sci. Rep. 6:19324. https://doi.org/10.1038/srep19324
Fu FX, Warner ME, Zhang YH, Feng YY, Hutchins DA (2007) Effects of increased temperature and CO2 on photosynthesis, growth, and elemental ratios in marine Synechococcus and Prochlorococcus (Cyanobacteria). J. Phycol. 43:485–496. https://doi.org/10.1111/j.1529-8817.2007.00355.x
Xia X, Guo W, Tan S, Liu H (2017) Synechococcus assemblages across the salinity gradient in a salt wedge estuary. Front. Microbiol. 8:1254. https://doi.org/10.3389/fmicb.2017.01254. ECollection 2017
Liu X, Xiao WP, Landry MR, Chiang KP, Wang L, Huang BQ (2016) Responses of phytoplankton communities to environmental variability in the East China Sea. Ecosystems 19:832–849
Cho JC, Giovannoni SJ (2004) Cultivation and growth characteristics of a diverse group of oligotrophic marine Gammaproteobacteria. Appl. Environ. Microbiol. 70:432–440. https://doi.org/10.1128/aem.70.1.432-440.2004
Morrow KM, Bourne DG, Humphrey C, Botté ES, Laffy P, Zaneveld J, Uthicke S, Fabricius KE, Webster NS (2015) Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J 9:894–908. https://doi.org/10.1038/ismej.2014.188
Ribes M, Calvo E, Movilla J, Logares R, Coma R, Pelejero C (2016) Restructuring of the sponge microbiome favors tolerance to ocean acidification. Environ. Microbiol. Rep. 8:536–544. https://doi.org/10.1111/1758-2229.12430
Cleary DFR, Polónia ARM, Becking LE, de Voogd NJ, Purwanto GH, Gomes NCM (2017) Compositional analysis of bacterial communities in seawater, sediment and high and low microbial abundance sponges in the Misool coral reef system, Indonesia. Mar. Biodivers.:1–13. https://doi.org/10.1007/s12526-017-0697-0
Innocenti F (ed) (2008) Genomics and pharmacogenomics in anticancer drug development and clinical response. Humana Press, New York, USA, p 378. https://doi.org/10.1007/978-1-60327-088-5
Eggleston AK (2007) DNA replication and repair. Nature 447:923–923. https://doi.org/10.1038/447923a
Shigenobu S, Watanabe H, Hattori M, SaKi Y, Ishikawa H (2000) Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407:81–86. https://doi.org/10.1038/35024074
Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M, Aksoy S (2002) Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 32:402–407. https://doi.org/10.1038/ng986
Dale C, Wang B, Moran N, Ochman H (2003) Loss of DNA recombinational repair enzymes in the initial stages of genome degeneration. Mol Biol Evol 20:1188–1194. https://doi.org/10.1093/molbev/msg138
Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF (2003) The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 66:1499–1503. https://doi.org/10.1016/S0006-2952(03)00504-5
Yadav SK (2010) Heavy metals toxicity in plants: an overview on the role of glutathione and phytochelatins in heavy metal stress tolerance of plants. S. Afr. J. Bot. 76:16–179. https://doi.org/10.1016/j.sajb.2009.10.007
Acknowledgements
We are grateful to the Indonesian State Ministry of Research and Technology (RISTEK) for providing research permits. We thank the following people for their help in various ways: Leontine Becking, Rossana Freitas, Suharsono, Y. Tuti, E.Oberhauser, R. Suhr and the staff of Nabucco Island Dive Resort.
Funding
The research was sponsored by the Indonesian Institute of Sciences (LIPI) and funded by the Portuguese Foundation for Science and Technology, FCT, project LESS CORAL, PTDC/AAC-AMB/115304/2009. Ana R.M. Polónia was supported by a postdoctoral scholarship [SFRH/BPD/117563/2016] funded by FCT, Portugal (QREN-POPH Type 4.1—Advanced Training, subsidised by the European Social Fund and national funds MCTES). Thanks are also due, for the financial support to CESAM [UID/AMB/50017], to FCT/MEC through national funds and co-funding by FEDER, within the PT2020 Partnership Agreement and Compete 2020.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cleary, D.F.R., Polónia, A.R.M. & de Voogd, N.J. Bacterial Communities Inhabiting the Sponge Biemna fortis, Sediment and Water in Marine Lakes and the Open Sea. Microb Ecol 76, 610–624 (2018). https://doi.org/10.1007/s00248-018-1156-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1156-6