
Abstract
In this study, we used a 16S rRNA gene barcoded pyrosequencing approach to sample bacterial communities from six biotopes, namely, seawater, sediment and four sponge species (Stylissa carteri, Stylissa massa, Xestospongia testudinaria and Hyrtios erectus) inhabiting coral reefs of the Spermonde Archipelago, South Sulawesi, Indonesia. Samples were collected along a pronounced onshore to offshore environmental gradient. Our goals were to (1) compare higher taxon abundance among biotopes, (2) test to what extent variation in bacterial composition can be explained by the biotope versus environment, (3) identify dominant (>300 sequences) bacterial operational taxonomic units (OTUs) and their closest known relatives and (4) assign putative functions to the sponge bacterial communities using a recently developed predictive metagenomic approach. We observed marked differences in bacterial composition and the relative abundance of the most abundant phyla, classes and orders among sponge species, seawater and sediment. Although all biotopes housed compositionally distinct bacterial communities, there were three prominent clusters. These included (1) both Stylissa species and seawater, (2) X. testudinaria and H. erectus and (3) sediment. Bacterial communities sampled from the same biotope, but different environments (based on proximity to the coast) were much more similar than bacterial communities from different biotopes in the same environment. The biotope thus appears to be a much more important structuring force than the surrounding environment. There were concomitant differences in the predicted counts of KEGG orthologs (KOs) suggesting that bacterial communities housed in different sponge species, sediment and seawater perform distinct functions. In particular, the bacterial communities of both Stylissa species were predicted to be enriched for KOs related to chemotaxis, nitrification and denitrification whereas bacterial communities in X. testudinaria and H. erectus were predicted to be enriched for KOs related to the toxin–antitoxin (TA) system, nutrient starvation and heavy metal export.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coastal marine ecosystems influence climate, nutrient cycling and primary productivity on a global scale [1]. Despite the acknowledged importance of these ecosystems, they have been severely affected by anthropogenic disturbances. This is particularly the case with coral reef ecosystems that have been adversely affected by a number of disturbances including local perturbations such as overfishing, eutrophication and heavy metal pollution [2–4] and global disturbances related to warming such as coral bleaching [3–6]. The intensity of these disturbances is predicted to increase over the coming decades [7, 8].
Microbes play key roles in the functioning of coral reef ecosystems [9]. Relatively little research has, however, focused on microbial communities in coral reefs when compared to other taxa such as corals or fish. In coral reef ecosystems, microbes can be found in the plankton and sediment but are also important symbionts in higher taxa such as corals and sponges. Here, we studied communities of bacteria in six coral reef biotopes in the Spermonde Archipelago, a coral reef system off the coast of Makassar, Indonesia, and located in an area known as the coral triangle. These included two non-host biotopes namely sediment and seawater and four host biotopes namely the sponge species Stylissa carteri and Stylissa massa (order Halichondrida: family Dictyonellidae), Xestospongia testudinaria (order Haplosclerida: family Petrosiidae) and Hyrtios erectus (order Dictyoceratida: family Thorectidae). Sponges are both abundant and ecologically important in coral reef ecosystems [10]. They also harbour very high microbial densities; high microbial abundance (HMA) sponges can contain 1010 bacterial cells per gram wet weight of sponge. This is orders of magnitude higher than the surrounding seawater [11–15]. In most cases, bacteria make up the lion’s share of prokaryotic diversity [15–17]. There has been a recent surge in studies of bacteria and their functions in a number of biotopes including sponges [18–20]. At present, however, relatively little is known about the functions of sponges and their bacterial symbionts in the reefs of the coral triangle, which contains the most diverse coral reefs in the world [21]. It is important, however, to have some idea of how sponges may affect the coral reef environment given that they are predicted to increase in abundance in the future [22, 23].
Unfortunately, very few bacterial symbionts of sponges have been cultured. It is, therefore, difficult to identify the functions of the majority of sponge-associated symbionts [24]. Recent advances in ‘omics’ techniques such as metatranscriptomics [18] and proxy techniques including predictive analysis using marker genes, however, now enable predictions of metagenomic functional content. In the present paper, we use a recently developed bioinformatic tool, PICRUSt, that enables us to both predict gene enrichment and identify the taxonomy of bacteria carrying these genes [25].
Few studies have assessed the composition and functions of bacteria in multiple coral reef biotopes, particularly in the coral triangle. Our main goals with this study were to (1) compare higher taxon abundance among biotopes, (2) assess to what extent the biotope and environment (sampling zone) influence composition, (3) assess if different biotopes harbour functionally distinct bacterial communities and (4) assign putative functions to the bacterial communities of different sponge species.
Material and Methods
Study Site
All sampling took place in the Spermonde Archipelago, South Sulawesi, Indonesia, which consists of 160 fringing, barrier and patch reefs [26]. The Spermonde is situated adjacent to the city of Makassar, a city with more than two million inhabitants [27]. Previous studies have shown a pronounced onshore to offshore gradient in environmental conditions related to anthropogenic disturbances and river discharge including sedimentation, agricultural runoff, oil spills, destructive fisheries, tourism and coral mining [28–30].
Sampling
Sediment, seawater and four sponge species were collected from reefs in the Spermonde Archipelago (Lae Lae, Samalona, Kudingkareng Keke, Bone Baku and Langkai) using SCUBA in August 2012 (Fig. 1). The Spermonde is a well-documented carbonate coastal shelf subject to several environmental influences along an onshore to offshore gradient. The environmental influences include sewage and other forms of pollution from the city of Makassar and fluvial discharge and erosion products from the Jene Berang River that transverses Makassar [31]. Previous studies have divided the Spermonde into four zones that run parallel to the coast. These zones were based on geomorphology, reef development, geology, shelf depth and offshore distance [32, 33]. The innermost zone, zone 1, is approximately bounded by the 20-m isobath and mainly consists of cay-crowned reefs. Visibility is limited in zone 1, and salinity is lower and nutrient, silt and sand content higher than the other zones. This zone is most under influence of land-based pollution. The sample site Lae Lae was sampled in zone 1. Nutrient levels in the other zones are comparable and exhibit minor fluctuations throughout the year [34]. Zone 2 begins >4 km offshore, mainly consists of reefs crowned with islets, and maximum depth is approximately 30 m. The sites Samalona and Bone Baku were sampled in zone 2. Zone 3 begins 12.5 km offshore, mainly consists of submarine shoals with few emerging cays, and maximum depth ranges from 30 to 50 m depending on the reef. The site Kudingareng Keke was sampled in zone 3. Zone 4 consists of the outer rim of the reef system and starts approximately 30 km offshore. Maximum depth ranges from 40 to 50 m on the eastern side and beyond 100 m on the westward drop. The reefs of zone 4 form a barrier-type reef crowned by some islets. The site Langkai was sampled in zone 4.

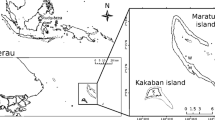
Bathymetric map of the Spermonde Coral Reef System showing the location of the sample sites: Lae (Lae Lae), Sam (Samalona), Bak (Bone Baku), Kud (Kudingareng Keke) and Lan (Langkai). The inset in the upper left corner shows the location of the Spermonde Archipelago in Southeast Asia
At each site in each zone (Lae Lae, Samalona, Kudingareng Keke), one sample of each biotope (sediment, seawater, S. carteri, S. massa, X. testudinaria, H. erectus) was taken. Sediment, seawater, S. massa, X. testudinaria and H. erectus were sampled from Lae Lae, Samalona, Kudingkareng Keke and Langkai. Unfortunately, S. carteri was not present in two of these sites (Lae Lae and Langkai). We, therefore, sampled additional specimens of S. massa from Kudingareng Keke and another site, Bone Baku. Sediment was sampled using the mini core method as previously described [35, 36]. Seawater was sampled by filtering approximately 1 l [37, 38] of seawater (collected between 1 and 3 m depth) through a Millipore® White Isopore Membrane Filter (0.22-μm pore size). Sponges were sampled including fragments of surface and interior following previously described methods [36]. All samples were stored in 96 % EtOH [39, 40] and kept cool (<4 °C) after collection and during transport. In the laboratory, samples were stored at −20 °C until DNA extraction.
DNA Extraction and Pyrosequencing
PCR-ready genomic DNA was isolated from seawater, sediment and sponge samples with FastDNA® SPIN Kit (MP Biomedicals) following the manufacturer’s instructions. This is an extraction method frequently used for this purpose [36, 41, 42]. Briefly, the whole membrane filter and 500 mg of sediment or sponge were transferred to Lysing Matrix E tubes containing a mixture of ceramic and silica particles. The microbial cell lysis was performed in the FastPrep® Instrument (Q Biogene) for 80 s at speed 6.0. Extracted DNA was eluted into DNase/pyrogen-free water to a final volume of 50 μl and stored at −20 °C until use. Prior to pyrosequencing, the amplicons of the bacterial 16S ribosomal RNA (rRNA) gene were obtained using bacterial specific primers 27F and 1494R [43]. After a denaturation step at 94 °C for 5 min, 25 thermal cycles of 1 min at 94 °C, 1 min at 56 °C and 2 min at 68 °C were carried out followed by an extension step at 68 °C for 10 min. Using the amplicons of the bacterial 16S rRNA gene as template, the V3V4 region was amplified, using barcoded fusion primers with the Roche-454 A Titanium sequencing adapters, a six-base barcode sequence, forward V3 primer 5′-ACTCCTACGGGAGGCAG-3′ [44] and V4 reverse degenerate primer 5′-TACNVRRGTHTCTAATYC-3′ (Ribosomal Database Project [RDP], Release 10, Update 20, http://rdp.cme.msu.edu/; last checked 06 April 2015). Sequence analysis was performed using previously described methods ([36, 39, 45]; see Online Resource 1 for a detailed description). Briefly, barcoded pyrosequencing libraries were analysed using the Quantitative Insights Into Microbial Ecology (QIIME) software package ([46]; http://www.qiime.org/; last checked 20 Jan 2014) on a computer running the BioLinux 7 operating system (http://nebc.nerc.ac.uk/; checked 02 June 2015). In QIIME, fasta and qual files were used as input for the split_libraries.py script. OTUs were selected using UPARSE with usearch7 [47]. Chimera checking was performed using the UCHIME algorithm, which is the fastest and most sensitive chimera checking algorithm currently available [48]. OTU clustering was performed using the -cluster_otus command (cut-off threshold at 97 %). The DNA sequences generated in this study can be downloaded from the NCBI SRA: SRP047468.
BLAST, Phylogenetic and Predictive Metagenome Analysis
Closely related organisms to numerically dominant OTUs (>300 sequences) were identified using the NCBI Basic Local Alignment Search Tool (BLAST) command line ‘blastn’ tool with the -db argument set to nt [49]. We used PICRUSt, a bioinformatics tool that uses marker genes, in this case 16S rRNA, to predict metagenome gene functional content. A detailed description of these methods has been published previously [25, 36] and can be found in the supplementary methods (Online Resource 1). In the present study, we used the KEGG database and focused on a selected set of KEGG orthologs (KOs). In the KEGG database, KOs are sets of homologous sequences, from a large array of organisms, that have been assigned a specific molecular function. KOs are in turn arranged hierarchically and grouped into biological pathways. Note that because of functional overlap, some KOs can be represented in more than one pathway. We used R to generate bar graphs showing the estimated number of genes for selected KOs (K00087, K00575, K00673, K00991, K01076, K01426, K01770, K03409, K03696, K04517, K04561, K05522, K05982, K06200, K07239, K07334, K07658, K07665, K10535 and K12339; the selection of KOs was based on a preliminary analysis of KO variation among biotopes) for each sample and the contribution of selected taxonomic orders; the latter was obtained using the metagenome_contributions.py script in PICRUSt. Note that the PICRUSt results as presented are predictive and thus provide information on potential enrichment and putative function as opposed to measuring actual gene presence/expression and function.
Higher Taxon Abundance
We tested for significant differences in the relative abundance of selected higher taxa (classes and orders) and dominance (the relative abundance of the most abundant OTU in each sample) among biotopes with an analysis of deviance using the glm() function in R [50]. Because the data was proportional, we first applied a glm with the family argument set to binomial. The ratio, however, of residual deviance to residual d.f. in the models substantially exceeded 1, so we set family to ‘quasibinomial’. In the quasibinomial family, the dispersion parameter is not fixed at one so that it can model overdispersion. Using the glm model, we tested for significant variation among biotopes using the anova() function in R with the F test, which is most appropriate when dispersion is estimated by moments as is the case with quasibinomial fits. Detailed descriptions of the functions used here can be found in R (e.g., ?cmdscale) and online in reference manuals (http://cran.r-project.org/web/packages/vegan/index.html; accessed 27 Feb 2015).
Composition
Two tables containing (1) the presence and abundance of all OTUs per sample and (2) a table of predicted KO counts were imported into R using the read.Table() function. For the OTU table, OTUs with <20 sequences, not classified as bacteria or classified as chloroplasts and mitochondria, were removed prior to statistical analysis. Both tables were log10 (x + 1) transformed (in order to normalise the distribution of data) and distance matrices constructed using the Bray–Curtis index with the vegdist() function in the vegan package [51] in R. The Bray–Curtis index is one of the most frequently applied (dis)similarity indices used in ecology [52, 53]. Variation in OTU and KO composition among biotopes (sediment, seawater, S. massa, S. carteri, X. testudinaria and H. erectus) was assessed with principal coordinate analysis (PCO) using the cmdscale() function in R with the Bray–Curtis distance matrix as input. Variations among biotopes and reef zones (pooling samples from the same zone but different biotopes) were tested separately for significance using the adonis() function in vegan. In the Adonis analysis, the Bray–Curtis distance matrix of species composition was the response variable with biotope as independent variable. The number of permutations was set at 999; all other arguments used the default values set in the function. Weighted averages scores were computed for OTUs and KOs on the first two PCO axes using the wascores() function in the vegan package.
Results
Sequencing yielded 76,510 sequences assigned to 4141 OTUs after quality control, OTU picking and removal of chimera. All OTUs were assigned to 44 phyla, 101 classes and 124 orders. Most (57,409) sequences were assigned to the Proteobacteria followed by the Bacteroidetes (3318 sequences) and Nitrospirae (3115 sequences) (Online Resource 2).
Higher Taxon Abundance
Proteobacteria was the dominant abundant phylum in all biotopes but was particularly abundant in both Stylissa species (Online Resource 2). There were highly significant differences in the relative abundance of selected classes and orders among biotopes (Fig. 2). OTUs assiged to the Entotheonellales, for example, were mainly restricted to X. testudinaria and H. erectus whereas OTUs assiged to the NB1-j were most abundant in both Stylissa species. Dominance was most pronounced in both Stylissa species, particularly in S. carteri, and was least pronounced in sediment. At the phylum level, the main effect was a marked increase in the abundance of Bacteroidetes in seawater and H. erectus in the inshore site (Lae Lae). This effect, however, was not apparent in sediment or other sponge taxa (Online Resource 2).

Mean relative abundance of the most abundant bacterial classes, orders and the dominant OTU for samples from S. carteri (Sc), S. massa (Sm), X. testudinaria (Xt), H. erectus (He), sediment (Sd) and seawater (Wt). Error bars represent a single standard deviation. The dominant OTU represents the mean abundance for the single most abundant OTU in each sample, thus not necessarily the same OTU. Results of glm are shown in the top-right corner of each graph
OTU Composition
The two, by far, most abundant OTUs (OTUs 1 and 2) were related to organisms previously obtained from S. carteri sampled in Saudi Arabia (Table 1). Both of these OTUs (and the alphaproteobacterium OTU-11) were restricted to both Stylissa hosts and were absent in all other biotopes. In addition to the above, OTU-12 was restricted to S. massa. A large number of OTUs were restricted to X. testudinaria and H. erectus (e.g., OTUs 4, 8, 9, 13 and 16) including OTUs such as OTU-17 that was restricted to X. testudinaria and OTU-14 that was restricted to H. erectus. A number of OTUs were more abundant in seawater (e.g., OTUs 3, 15, 51, 58) or sediment (e.g., OTUs 44 and 741), but these organisms were also found in sponges albeit in low abundances. Most of the abundant OTUs were closely related to organisms previously isolated from other sponges (e.g., OTUs 1, 2, 4, 5, 7, 8, 9 and 10; Table 1 and Online Resource 3). Phylogenetic trees of the most numerically dominant OTUs and selected cultured organisms are presented in Online Resources 4 and 5.
There was a highly significant difference in composition among biotopes (all biotopes: F 5,18 = 25.64, P < 0.001, R 2 = 0.877; excluding S. massa: F 4,15 = 29.97, P < 0.001, R 2 = 0.889). Variation among biotopes thus explained >87 % of the variation in bacterial composition. In contrast, there was no significant difference among zones when pooling samples according to zone (F 3,16 = 0.12, P = 0.999, R 2 = 0.022). A PCO ordination (Fig. 3) of the first two axes revealed three distinct clusters representing samples from the six biotopes. One cluster consisted of samples from seawater and both Stylissa hosts, another cluster consisted of samples from sediment, and the last cluster consisted of samples from X. testudinaria and H. erectus. The first PCO axis separated samples from seawater and both Stylissa hosts from samples of X. testudinaria and H. erectus. The second PCO axis separated sediment samples from all other samples. Most OTUs were restricted to or showed a pronounced preference for specific biotopes as evidenced by the distribution of OTUs in Fig. 3. Including all OTUs (thus, also OTUs <20 sequences), only 1 OTU (OTU-45, family Rhodobacteraceae) out of 4141 OTUs was found in all six biotopes and only 6 were found in five biotopes. In contrast, more than 90 % of OTUs (3749) were only found in a single biotope.

Ordination showing the first two axes of the PCO analysis for OTU composition. Symbols represent samples from S. carteri (Sc), S. massa (Sm), X. testudinaria (Xt), H. erectus (He), sediment (Sd) and seawater (Wt). Numbers refer to OTU numbers in Table 1. The small light grey circles represent OTUs with <300 sequences while the larger light grey circles represent OTUs ≥ 300 sequences
Predictive Metagenome Analysis
Mean (and standard deviation) Nearest Sequenced Taxon Index (NSTI) values for the biotopes sampled in Makassar were 0.220 (0.025) for Sc, 0.195 (0.012) for Sm, 0.197 (0.025) for Xt, 0.198 (0.020) for He, 0.145 (0.004) for Sd and 0.145 (0.010) for Wt. There was a significant difference among biotopes in KO composition (F 5,18 = 18.23, P < 0.001, R 2 = 0.835). Variation among biotopes thus explained almost 84 % of the variation in KO composition. The first axis was primarily related to variation between samples from seawater versus samples from X. testudinaria and H. erectus with samples from sediment and both Stylissa species intermediate. The second axis was primarily related to variation between sediment samples and samples from both Stylissa species (Fig. 4).

Ordination showing the first two axes of the PCO analysis for KO composition. Symbols represent samples from S. carteri (Sc), S. massa (Sm), X. testudinaria (Xt), H. erectus (He), sediment (Sd) and seawater (Wt). Codes refer to KO codes in Table 2
KOs predicted to be enriched in both Stylissa species and X. testudinaria and H. erectus are indicated by their KO identifiers (Fig. 4 and Table 2). KOs predicted to be enriched in both Stylissa species included K00087 (benzoate and aminobenzoate degradation), K00575 (chemotaxis protein methyltransferase CheR), K00673 (arginine and proline metabolism), K01076 (limonene and pinene degradation), K03409 (chemotaxis protein CheX), K04561 (denitrification, nitrate => nitrogen) and K10535 (nitrification, ammonia => nitrite). KOs predicted to be enriched in X. testudinaria and H. erectus included K00991 (terpenoid backbone biosynthesis), K01426 (styrene and aminobenzoate degradation; arginine and proline metabolism, phenylalanine metabolism, and tryptophan metabolism), K01770 (terpenoid backbone biosynthesis), K03696 (heat shock protein), K04517 (phenylalanine, tyrosine and tryptophan biosynthesis; novobiocin biosynthesis), K05522 (replication and repair), K05982 (DNA repair and recombination protein), K06200 (carbon starvation protein), K07239 (heavy metal exporter), K07334 (proteic killer suppression protein), K07658 (PhoR-PhoB phosphate starvation response two-component regulatory system), K07665 (copper resistance phosphate regulon response regulator CusR two-component regulatory system) and K12339 (sulphur metabolism; cysteine and methionine metabolism).
The contributions of selected orders to KO enrichment are presented in Online Resources 6 and 7. In both Stylissa species, OTUs (primarily OTU-1) belonging to the Deltaproteobacteria class contributed strongly to enrichment of K00087, K03409, K04561 and K10535. In X. testudinaria and H. erectus, Solibacteres contributed strongly to enrichment of K05522, K07239, K07334 and K07665. Other important classes that contributed to enrichment were Acidimicrobiia and Gammaproteobacteria (primarily OTU-4) for K05522, Gammaproteobacteria and Nitrospira for K07239, Gammaproteobacteria and Nitrospira for K07334 and Solibacteres and Nitrospira and ‘other’ for K07665.
Discussion
Higher Taxon Abundance
We recorded highly significant differences in the relative abundances of a number of classes and orders. The Rhodospirillales order and Gemm-2 class, for example, were largely restricted to sediment, H. erectus and X. testudinaria, whereas the Rickettsiales were largely restricted to seawater and both Stylissa species. Entotheonellales and HTCC2188 also were most abundant in H. erectus and X. testudinaria. At the phylum level, the relative abundance of Proteobacteria was most pronounced in the bacterial communities of both Stylissa species. Bacterial communities belonging to the other biotopes, while hosting a majority of OTUs assigned to Proteobacteria, had more phylum-level diversity. The relative abundance of the most dominant OTU in each sample was highest in both Stylissa species and lowest in sediment. This result reflects similar findings of these biotopes in coral reefs of Jakarta, Indonesia [54].
Bacterial Composition: Biotope Versus Environment
Biotope proved a significant predictor of variation in composition as opposed to the sampling zone. This suggests that much of the variation in bacterial composition in coral reef habitat is due to differences among distinct biotopes, i.e., seawater, sediment, host organisms and possibly other microhabitats such as crevices and biofilms, the latter of which were not investigated in the present study. It remains to be investigated how bacterial communities from different biotopes respond to environmental gradients. It is probable that bacterioplankton and possibly sediment bacteria respond more strongly than bacteria residing in host organisms such as sponges. A number of previous studies, for example, have found that sponge bacterial communities remain remarkably stable across pronounced geographic and environmental gradients [11, 17, 55, 56]. Reveillaud et al. [56] sampled Hexadella species over a very large bathymetric gradient (15–960 m) and observed ‘remarkably specific and stable sponge–bacteria associations’. Likewise, Lee et al. [17] suggested that sponge microbial communities appear to resist environmental change. This apparent resistance to environmental change of host-related microbes extends beyond sponges. Hawlena et al. [57] collected bacterial communities of fleas and ticks over a range of environmental conditions and sites but found that none of those conditions significantly affected bacterial community composition. Composition was, however, strongly related to the type of host. Bacterioplankton communities while probably more sensitive to changes in environmental conditions are also subject to living in a highly dynamic environment. Bacterioplankton composition though has been shown to vary along pronounced environmental gradients of carbon, temperature and salinity [58, 59].
There is a debate about the degree to which sponges host sponge-specific or sponge species-specific microbial communities [11, 15, 60]. In the present study, three of the most abundant OTUs were only found in Stylissa species (OTUs 1, 2 and 11). These three OTUs were closely related (with a similarity ≥99 %) to organisms found in S. carteri sampled in Saudi Arabia ([61]; OTU-1: GI: 407912992, OTU-2: 407913000; OTU-11: 407913009), Stylissa sp. sampled in Australia ([62] OTU-1: GI:400269236), Axinella sp. sampled in China (Liu unpublished; OTU-1: GI: 597437720; OTU-2: 597437717; OTU-11: GI: 597437738), Axinella corrugata sampled in the Caribbean (Lopez et al. unpublished, Holmes and Blanch unpublished; OTU-1: GI: 209364706; OTU-2: GI: 127692655; OTU-11: GI: 127692617 and GI: 209364724), Axinella verrucosa sampled in the Mediterranean (Steffens unpublished; OTU-2: GI: 34368515) and Phakellia fusca sampled in China ([63]; OTU-11: GI: 340764414). Interestingly, all these sponge hosts (including the Stylissa species) belong to the same taxonomic order (Halichondrida). These results confirm previous findings of Polónia et al. [64] where they found that both Stylissa spp. hosted a single very abundant crenarchaeote assigned to the species Cenarchaeum symbiosum. This crenarchaeote was also found in other sponges including Axinella and Phakellia leading Polónia et al. [64] to suggest the presence of a possibly order-specific symbiosis between Halichondrida and C. symbiosum. C. symbiosum was itself originally isolated from the sponge Dragmacidon mexicanum (previously known as Axinella mexicana) off the California coast [65]. We expand on this and suggest the existence of a small core community of possibly sponge order-specific microbes including one crenarchaeote and three bacteria belonging to the orders NB1-j, Chromatiales and an unclassified alphaproteobacterium. As with C. symbiosum, organisms closely related to these bacterial OTUs were isolated from other halichondrid sponges across a very large geographical range including the Indo-Pacific, Caribbean and Mediterranean. As noted by Polónia et al. [64], this would appear to suggest that this core group is spatially stable and possibly vertically transmitted, i.e., from parent to offspring. This result contrasts with Schmitt et al. [66] who found that bacterial communities of sponges in the same order were not more similar to one another than bacterial communities of sponges in different orders.
The bacterial communities of the HMA sponges X. testudinaria (order: Haplosclerida) and H. erectus (order: Dictyoceratida) were compositionally similar and shared a large number of OTUs. In contrast to the low microbial abundance (LMA) Stylissa species, both HMA sponges were enriched with OTUs closely related to Nitrospira marina (GI: 530902; Online Resource. 3), a well-known lithoautotrophic nitrite-oxidising bacteria previously found in other marine sponges [11]. Very few OTUs found in H. erectus were shared with sediment, seawater and both Stylissa species despite the fact that phylogenetically, the Haplosclerida is more closely related to the Halichondrida than to the Dictyoceratida [67].
Sponge host phylogeny has been shown to have a weak effect on microbial composition [68], but the structure of the sponge tissue matrix may play a more important role in structuring the sponge bacterial community. H. erectus is a small black digitate sponge that lives embedded in sediment and sand. The skeletons of Hyrtios species lack silicious spicules and have a crust of exogenous material, and the choanosome consists of dense spongin fibres, extraneous detritus, sediment grains, foreign sponge spicules and broken shells. X. testudinaria, in turn, is a very long-lived and slow-growing species, the skeleton of which consists of a dense network of silicious spicules [69]. In contrast to the previous species, Stylissa spp. are probably fast growers with a loose collagen-rich skeleton containing relatively large spicules [70]. Like a bath sponge, the loose skeletal structure of Stylissa spp. has the capacity to retain much higher amounts of water in their tissue.
In addition to the above, X. testudinaria is a confirmed high microbial abundance (HMA) sponge while H. erectus is a presumed HMA sponge [71]. Our results would appear to confirm H. erectus as a HMA sponge given the similarity of its bacterial community with X. testudinaria. Stylissa spp., in contrast, are confirmed low microbial abundance (LMA) sponges [72]. LMA sponges typically have limited phylum-level diversity dominated by Proteobacteria and are known to filter larger water volumes than HMA sponges, thereby increasing similarity with bacterioplankton communities [14, 61, 72–75]. This fits well with our results and results from archaeal communities inhabiting Stylissa spp. in Makassar, Indonesia [64], but not Jakarta [36]. In addition to the above, the sponge metabolism is believed to be only influenced by microbes in HMA sponges, which has led to a focus on HMA sponges [76]. Importantly, our data confirms that both LMA Stylissa species maintain a bacterial community that is similar to, but still distinct from, the surrounding seawater and includes highly abundant OTUs that were absent in all other biotopes including seawater. This result is in line with de Voogd et al. [54] who found the same for S. massa in Jakarta and Moitinho-Silva et al. [72] who found the same for S. carteri in the Red Sea.
Predictive Functional Analysis
As mentioned previously, PICRUSt provides a prediction of microbiome function but not an actual measurement of such function. There are, however, methods of quality control that test the reliability of PICRUSt predictions including the weighted NSTI scores. NSTI, which was developed to evaluate the predictive accuracy of PICRUSt, calculates dissimilarity between reference genomes and the metagenome under study. In poorly characterised environments, there are relatively few reference genome sequences available; thus, the PICRUSt predictions of these genomes tend to be less accurate than for well-known microbial environments. In the present study, NSTI scores were relatively high, most notably for sponges, a reflection of the relative novelty of the bacterial communities of the coral reef sponges studied here. Mean scores for three of the four sponge species were below 0.20, but the highest value was obtained for S. carteri at 0.220. Langille et al. [25] showed that the accuracy of PICRUSt decreased with increasing NSTI scores but still produced reliable results for a dataset of soil samples with a mean NSTI score of 0.17. Accuracy was, however, lower for a dataset from the Guerrero Negro microbial mat with a mean NSTI score of 0.23. Langille et al. [25] noted, however, that this was also related to shallow sequencing at a depth that was insufficient to fully sample the community’s genomic composition. The relatively high NSTI scores obtained here indicate that the PICRUSt predictions must be treated with caution. The results, however, still provide some interesting insights into potential bacterial community functioning that, in the future, should be tested with studies that measure actual gene presence or expression.
One notable difference between OTU and KO composition was the similarity in bacterial composition between seawater and Stylissa samples, but the distinct difference in KO composition. Despite the abundance of symbionts shared between seawater and both Stylissa species and the lower number of sponge-specific symbionts found, sponge-specific symbionts exhibited the most pronounced dominance in both Stylissa species and contributed strongly to certain predicted metabolic functions. In particular OTU-1, assigned to the Deltaproteobacteria, was largely responsible for the pronounced enrichment of both Stylissa species for K00087 (Benzoate and Aminobenzoate degradation), K03409 (chemotaxis protein CheX), K04561 (denitrification, nitrate => nitrogen) and K10535 (nitrification, ammonia => nitrite). Moitinho-Silva et al. [18] found that S. carteri from the Red Sea exhibited high expression of functions related to stress response and membrane transporters. In both Stylissa species, we observed predicted enrichment of KOs related to bacterial chemotaxis (K00575 and K03409) and xenobiotics degradation (K00087, K01076).
The predicted contribution of Deltaproteobacteria to both nitrification and denitrification is in line with similar findings for S. massa in Jakarta [54] and highlights the potential importance of this class and OTU-1 in particular to nitrogen cycling with Stylissa species. In other marine environments, Deltaproteobacteria have also been shown to play a key role in the nitrogen cycle. In the Eastern South Pacific, for example, Nitrospina-like bacteria (order Desulfobacterales) were identified as the main drivers of nitrite oxidation in a seasonal upwelling area [77].
The contrast in predicted metabolic enrichment of both Stylissa species with X. testudinaria and H. erectus is interesting. KOs enriched in X. testudinaria and H. erectus included KOs involved in terpenoid backbone biosynthesis (K00991 and K01770), DNA repair (K05522, K05982), heavy metal efflux (K07239), copper resistance (K07665), carbon starvation (K06200) and proteic killer suppression (K07334) proteins. Two KOs (K07658, K07665) involved in copper resistance and phosphate starvation enriched in X. testudinaria and H. erectus are part of the signal transduction system known as the two-component regulatory system [78]. Signal transductors belonging to the two-component regulatory system enable bacteria to respond to a very wide range of nutrients, stressors (including antibiotics) and environmental conditions [79].
In addition to the previously mentioned KOs related to stress management (nutrient starvation, heat proteins and heavy metal exporters), X. testudinaria and H. erectus were also enriched for the proteic killer suppression protein higA. The higA (host inhibition of growth) protein is required for cloning of the killer protein HigB, part of the toxin–antitoxin (TA) system. TA systems consist of sets of two or more genes that include a toxin (e.g., higB) and anti-toxin (e.g., higA) and are believed to confer an advantage on the fitness of plasmids that carry them [80]. They are key regulators of cellular processes that influence survival under stressful conditions, are involved in essential cellular processes like replication, gene expression and cell wall synthesis and play a role in persistence, biofilm formation, antibiotic resistance and bacterial virulence [81, 82]. Interestingly, in a survey of TA loci, Pandey and Gerdes [83] found that TA loci were highly abundant in free living prokaryotes but absent from obligate intracellular organisms. They suggested that is a reflection of the beneficial role that TA loci play for free living prokaryotes in coping with stress.
The type of predicted functional enrichment displayed by the bacterial communities of X. testudinaria and H. erectus would appear to suggest adaptations to surviving and indeed persisting (in the case of X. testudinaria for very long periods of time) in stressful environments. Many sponges including X. testudinaria and sponge symbionts are known to produce antibacterial compounds, so host symbionts need to have mechanisms such as TA loci to cope with these compounds [84, 85]. X. testudinaria is also often found in highly perturbed environments and can even be extremely abundant in such environments [86–88]. The specific bacterial community of X. testudinaria may play a role in enabling the sponge to persist and survive in stressful environments.
In addition to the above, KOs related to the phenylalanine metabolism (K01426) and phenylalanine tyrosine and tryptophan biosynthesis (K04517) were predicted to be enriched in X. testudinaria and H. erectus. Phenylalanine is an essential amino acid, which is converted to tyrosine and is produced for a variety of medicinal and nutritional applications. Tyrosine is an amino acid that occurs in proteins belonging to signal transduction processes, plays a role in photosynthesis and is a precursor to alkaloids and phenols [89, 90]. Enrichment in the tyrosine and phenylalanine metabolic pathways and the importance of these pathways for the biosynthesis of alkaloids and phenols are in line with the numerous bioactive compounds that have been isolated from X. testudinaria and H. erectus [91–95]. Numerous bioactive compounds have also been isolated from Stylissa species including dimeric alkaloids (e.g., dibromophakellin and sceptrin), brominated pyrrole alkaloids and other brominated alkaloids. These compounds are of particular interest due, among other things, to their ability to inhibit protein kinases. Both Stylissa species, X. testudinaria and H. erectus, also produce a large range of highly selective antibiotic compounds [96–99].
Here, we have provided a detailed analysis of the bacterial communities inhabiting distinct coral reef biotopes. More than 87 % of the variation in the composition of these communities could be attributed to differences among biotopes. Despite sampling along a pronounced environmental gradient, the sampling zone proved a poor predictor of bacterial composition. Future research should focus on how bacterial communities from different biotopes respond to environmental variation. Bacterioplankton, for example, may show more of a response than bacterial communities housed within host organisms such as sponges. Although LMA sponges belonging to the genus Stylissa contained communities that were similar to seawater, they also contained highly abundant OTUs that were absent in all other biotopes. One of these OTUs, assigned to the class Deltaproteobacteria, contributed substantially to the predicted enrichment of genes related to chemotaxis, denitrification and nitrification in both Stylissa species. X. testudinaria and H. erectus displayed diverse microbial communities that differed strongly from seawater. The bacterial communities of X. testudinaria and H. erectus were predicted to be enriched for genes related to the toxin–antitoxin (TA) system and genes that convey tolerance to heavy metal pollution and nutrient starvation suggesting adaptation to stressful environmental conditions.
References
Solan M, Cardinale BJ, Downing AL, Engelhardt KAM, Ruesink JL, Srivastava DS (2004) Extinction and ecosystem function in the marine benthos. Science 306:1177–1180
Carpenter KE, Arbar M, Aeby G, Aronson RB et al (2008) One-third of reef-building corals face elevated extinction risk from climate change and local impacts. Science 321:560–563
Jackson JBC, Kirby MX, Berger WH, Bjorndal KA, Botsford LW, Bourque BJ, Bradbury RH, Cooke R, Erlandson J, Estes JA, Hughes TP, Kidwell S, Lange CB, Lenihan HS, Pandolfi JM, Peterson CH, Steneck RS, Tegner MJ, Warner RR (2001) Historical overfishing and the recent collapse of coastal ecosystems. Science 293:629–638
Pandolfi JM, Bradbury RH, Sala E, Hughes TP, Bjorndal KA, Cooke RG, McArdle D, McClenachan L, Newman MJH, Paredes G, Warner RR, Jackson JBC (2003) Global trajectories of the long-term decline of coral reef ecosystems. Science 301:955–958
Bruno JF, Selig ER (2007) Regional decline of coral cover in the Indo-Pacific: timing, extent, and subregional comparisons. PLoS One 2:e711
De’ath G, Fabricius KE, Sweatman H, Puotinen M (2012) The 27-year decline of coral cover on the Great Barrier Reef and its causes. Proc Natl Acad Sci U S A 109:17995–17999
Done TJ, DeVantier LM, Turak E, Fisk DA, Wakeford M, van Woesik R (2010) Coral growth on three reefs: development of recovery benchmarks using a space for time approach. Coral Reefs 29:815–833
Hughes TP, Graham NAJ, Jackson JBC, Mumby PJ, Steneck RS (2010) Rising to the challenge of sustaining coral reef resilience. Trends Ecol Evol 25:633–642
Garren M, Azam F (2012) New directions in coral reef microbial ecology. Environ Microbiol 14:833–844. doi:10.1111/j.1462-2920.2011.02597.x
Diaz MC, Rutzler K (2001) Sponges: an essential component of Caribbean coral reefs. B Mar Sci 69:535–546
Hentschel U, Hopke J, Horn M, Friedrich AB, Wagner M, Hacker J, Moore BS (2002) Molecular evidence for a uniform microbial community in sponges from different oceans. Appl Environ Microbiol 68:4431–4440
Hentschel U, Piel J, Degnan SM, Taylor MW (2012) Genomic insights into the marine sponge microbiome. Nat Rev Microbiol 10:641–654
Hentschel U, Usher KM, Taylor MW (2006) Marine sponges as microbial fermenters. FEMS Microbiol Ecol 55:167–177
Kamke J, Taylor M, Schmitt S (2010) Activity profiles for marine sponge-associated bacteria obtained by 16S rRNA vs 16S rRNA gene comparisons. ISME J 4:498–508
Taylor MW, Radax R, Steger D, Wagner M (2007) Sponge associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol R 71:295–347
Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, Thomas T (2012) Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci U S A 109:E1878–E1887
Lee OO, Wang Y, Yang J, Lafi FF, Al-Suwailem A, Qian PY (2011) Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J 5:650–664. doi:10.1038/ismej.2010.165
Moitinho-Silva L, Seridi L, Ryu T, Voolstra CR, Ravasi T, Hentschel U (2014) Revealing microbial functional activities in the Red Sea sponge Stylissa carteri by metatranscriptomics. Environ Microbiol. doi:10.1111/1462-2920.12533
Radax R, Rattei T, Lanzen A, Bayer C, Rapp HT, Urich T, Schleper C (2012) Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Environ Microbiol 14:1308–1324
Sanders JG, Beinart RA, Stewart FJ, Delong EF, Girguis PR (2013) Metatranscriptomics reveal differences in in situ energy and nitrogen metabolism among hydrothermal vent snail symbionts. ISME J 7:1556–1567
McLeod E, Timmermann A, Salm R et al (2010) Warming seas in the Coral Triangle: coral reef vulnerability and management implications. Coast Manag 38:518–539
Bell JJ, Davy SK, Jones T , Taylor MW, Webster NS (2013) Could some coral reefs become sponge reefs as our climate changes? Glob Chang Biol 19:2613–2624
McMurray SE, Henkel TP, Pawlik JR (2010) Demographics of increasing populations of the giant barrel sponge Xestospongia muta in the Florida Keys. Ecology 91:560–570
Montalvo NF, Hill RT (2011) Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl Environ Microbiol 77:7207–7216
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. doi:10.1038/nbt.2676
de Voogd NJ, Cleary DFR, Hoeksema B, Noor A, van Soest R (2006) Sponge beta diversity in the Spermonde Archipelago, SW Sulawesi, Indonesia. Mar Ecol-Prog Ser 309:131–142
Renema W (2010) Is increased calcarinid (foraminifera) abundance indicating a larger role for macro-algae in Indonesian Plio-Pleistocene coral reefs? Coral Reefs 29:165–173
Cleary DFR, Becking LE, Voogd NJ, Pires ACC, Polónia ARM, Egas C, Gomes NCM (2013) Habitat-and host-related variation in sponge bacterial symbiont communities in Indonesian waters. FEMS Microbiol Ecol 85:465–482
Cleary DFR, Renema W (2007) Relating species traits of foraminifera to environmental parameters in the Spermonde Archipelago, Indonesia. Mar Ecol-Prog Ser 334:73–82
de Voogd NJ, Cleary DFR (2007) Relating species traits to environmental variables in Indonesian coral reef sponge assemblages. Mar Freshw Res 58:240–249
Renema W, Troelstra SR (2001) Larger foraminifera distribution on a mesotrophic carbonate shelf in SW Sulawesi (Indonesia). Palaeogeogr, Palaeoclim, Palaeoecol 175:125–146
de Klerk LG (1983) Zeespiegels, riffen en kustvlakten in Zuidwest Sulawesi, Indonesië; een morphogenetisch-bodemkundige studie. Utrecht, the Netherlands, Pp. 174
Hoeksema BW, Moka W (1989) Species assemblages and phenotypes of mushroom corals (Fungiidae) related to coral reef habitats in the Flores Sea. Neth J Sea Res 23:149–160
Erftemeijer PLA (1994) Differences in nutrient concentrations and resources between seagrass communities on carbonate and terrigenous sediments in South Sulawesi, Indonesia. Bull Mar Sci 54:403–419
Capone DG, Dunham SE, Horrigan SG, Duguay LE (1992) Microbial nitrogen transformations in unconsolidated coral reef sediments. Mar Ecol-Prog Ser 80:75–88
Polónia ARM, Cleary DRF, Duarte LN, de Voogd NJ, Gomes NCM (2013) Composition of Archaea in seawater, sediment and sponges in the Kepulauan Seribu reef system, Indonesia. Microb Ecol 67:553–567
Bowen JL, Morrison HG, Hobbie JE, Sogin ML (2012) Salt marsh sediment diversity: a test of the variability of the rare biosphere among environmental replicates. ISME J 6:2014–2023
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci U S A 103:12115–12120
Cleary DFR, Becking LE, de Voogd NJ, Renema W, de Beer M, van Soest RWM, Hoeksema BW (2005) Variation in the diversity and composition of benthic taxa as a function of distance offshore, depth and exposure in the Spermonde Archipelago, Indonesia. Estuar Coast Shelf Sci 65:557–570
Previsic A, Walton C, Kucinic M, Mitrikeski PT, Kerovec M (2009) Pleistocene divergence of Dinaric Drusus endemics (Trichoptera, Limnephilidae) in multiple microrefugia within the Balkan Peninsula. Mol Ecol 18:634–647
Costa R, Keller-Costa T, Gomes NCM, da Rocha, Ulisses N, van Overbeek L, van Elsas JD (2013) Evidence for selective bacterial community structuring in the freshwater sponge Ephydatia fluviatilis. Microb Ecol 65:232–244
Urakawa H, Martens-Habbena W, Stahl DA (2010) High abundance of ammonia-oxidizing Archaea in coastal waters, determined using a modified DNA extraction method. App Environ Microb 76:2129–2135
Gomes NCM, Heuer H, Schönfeld J, Costa RS, Mendonça-Hagler LCS et al (2001) Bacterial diversity of the rhizosphere of maize (Zea mays) grown in tropical soil studied by temperature gradient gel electrophoresis. Plant Soil 232:167–180. doi:10.1023/A:1010350406708
Wang Y, Qian P (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4:e7401
Pires ACC, Cleary DFR, Almeida A, Cunha Â, Dealtry S, Mendonça-Hagler LCS, Smalla K, Gomes NCM (2012) Denaturing gradient gel electrophoresis and barcoded pyrosequencing reveal unprecedented archaeal diversity in mangrove sediment and rhizosphere samples. App Environ Microb 78:5520–5528
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughp community sequencing data. Nat Methods 7:335–336
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Edgar R, Haas B, Clemente J, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
R Core Team (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. Available from http://www.R-project.org/
Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson G, Solymos P, Stevens M, Wagner H (2009) Vegan: Community ecology package. R package version 1.15–2. URL:http://CRAN.R-project.org/package=vegan
Cleary DFR (2003) An examination of scale of assessment, logging and ENSO-induced fires on butterfly diversity in Borneo. Oecologia 135:313–321
Legendre P, Gallagher ED (2001) Ecologically meaningful transformations for ordination of species data. Oecologia 129:271–280
de Voogd NJ, Cleary DRF, Polónia ARM, Gomes NCM (2015) Bacterial community composition and predicted functional ecology of sponges, sediment and seawater from the thousand island reef complex, West-Java, Indonesia. FEMS Microbiol Ecol. 91(4):1:12. pii: fiv019. doi:10.1093/femsec/fiv019
Friedrich AB, Fischer I, Proksch P, Hacker J, Hentschel U (2001) Temporal variation of the microbial community associated with the Mediterranean sponge Aplysina aerophoba. FEMS Microbiol Ecol 38:105–113
Reveillaud J, Maignien L, Murat Eren A, Huber JA, Apprill A, Sogin ML, Vanreusel A (2014) Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J 8:1198–1209. doi:10.1038/ismej.2013.227
Hawlena H, Rynkiewicz E, Toh E, Alfred A, Durden LA, Hastriter MW, Nelson DE, Rong R, Munro D, Dong Q, Fuqua C, Clay K (2013) The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J 7:221–223. doi:10.1038/ismej.2012.71
Dinasquet J, Kragh T, Schrøter ML, Søndergaard M, Riemann L (2013) Functional and compositional succession of bacterioplankton in response to a gradient in bioavailable dissolved organic carbon. Environ Microbiol 15:2616–2628. doi:10.1111/1462-2920.12178
Ngugi DK, Antunes A, Brune A, Stingl U (2012) Biogeography of pelagic bacterioplankton across an antagonistic temperature-salinity gradient in the Red Sea. Mol Ecol 21:388–405. doi:10.1111/j.1365-294X.2011.05378.x
Schmitt S, Angermeier H, Schiller R, Lindquist N, Hentschel U (2008) Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. App Environ Microb 74:7694–7708
Giles EC, Kamke J, Moitinho-Silva L, Taylor MW, Hentschel U, Ravasi T, Schmitt S (2013) Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol Ecol 83:232–241
Webster NS, Luter HM, Soo RM, Botte ES, Simister RL, Abdo D, Whalan S (2012) Same, same but different: symbiotic bacterial associations in GBR sponges. Front Microbiol 3:444. doi:10.3389/fmicb.2012.00444
Han M, Liu F, Zhang F, Li Z, Lin H (2012) Bacterial and archaeal symbionts in the South China Sea sponge Phakellia fusca: community structure, relative abundance, and ammonia-oxidizing populations. Mar Biotechnol 14:701–713
Polónia ARM, Cleary DRF, Freitas R, de Voogd NJ, Gomes NCM (2015) The putative functional ecology and distribution of archaeal communities in sponges, sediment and seawater in a coral reef environment. Mol Ecol 24:409–423. doi:10.1111/mec.13024
Preston CM, Wu KY, Molinski TF, DeLong EF (1996) A psychrophilic crenarchaeon inhabits a marine sponge: Cenarchaeum symbiosum gen. nov., sp. nov. Proc Natl Acad Sci U S A 93:6241–6246
Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, Perez T, Rodrigo A, Schupp PJ, Vacelet J, Webster N, Hentschel U, Taylor MW (2011) Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J 6:564–576
Erpenbeck D, Sutcliffe P, Cook SC, Dietzel A, Maldonado M, van Soest RWM, Hooper JNA, Wörheide G (2012) Horny sponges and their affairs: on the phylogenetic relationships of keratose sponges. Mol Phylogenet Evol 63:809–816. doi:10.1016/j.ympev.2012.02.024
Easson CG, Thacker RW (2014) Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front Microbiol 5:532. doi:10.3389/fmicb.2014.00532
Desqueyroux-Faúndez R, Valentine C (2002) Family Petrosiidae Van Soest, 1980. In: Hooper JNA, Van Soest RWM (eds) Systema Porifera. A guide to the classification of sponges. 1 (Kluwer Academic/ Plenum Publishers, New York, pp 906–917
Van Soest RWM, Erpenbeck D, Alvarez B (2002) Family Dictyonellidae Van Soest, Diaz & Pomponi, 1990. In: Hooper JNA, Van Soest RWM, Willenz P (eds) Systema Porifera. Springer, US, pp 773–786
Kennedy J, Flemer B, Jackson SA, Morrissey JP, O’Gara F, Dobson ADW (2014) Evidence of a putative deep sea specific microbiome in marine sponges. PLoS One 9:e91092. doi:10.1371/journal.pone.0091092
Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, Ravasi T, Hentschel U (2014) Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol Ecol 23:1348–1363
Gloeckner V, Hentschel U, Ereskovsky AV, Schmitt S (2013) Unique and species-specific microbial communities in Oscarella lobularis and other Mediterranean Oscarella species (Porifera: Homoscleromorpha). Mar Biol 160:781–791. doi:10.1007/s00227-012-2133-0
Thacker RW, Freeman CJ (2012) Sponge-microbe symbioses: recent advances and new directions. Advances in Sponge Science: Physiology, Chemical and Microbial Diversity, Biotechnology. Becerro MA, Uriz MJ, Maldonado M, Turon X. San Diego, Elsevier Academic Press Inc. 62: 57–111
Weisz JB, Lindquist N, Martens CS (2008) Do associated microbial abundances impact marine demosponge pumping rates and tissue densities. Oecologia 155:367–376. doi:10.1007/s00442-007-0910-0
Ribes M, Jimenez E, Yahel G, Lopez-Sendino P, Diez B, Massana R, Sharp JH, Coma R (2012) Functional convergence of microbes associated with temperate marine sponges. Environ Microbiol 14:1224–1239
Levipan HA, Molina V (2014) Fernandez C (2014) Nitrospina-like bacteria are the main drivers of nitrite oxidation in the seasonal upwelling area of the Eastern South Pacific (Central Chile ~36°S). Environ Microbiol Rep. doi:10.1111/1758-2229.12158
Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, Ishihama A (2005) Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J Biol Chem 280:1448–1456
Laub MT, Goulian M (2007) Specificity in two-component signal transduction pathways. Annu Rev Genet 41:121–145
Cooper TF, Heinemann JA (2000) Postsegregational killing does not increase plasmid stability but acts to mediate the exclusion of competing plasmids. Proc Natl Acad Sci U S A 97:12643–12648. doi:10.1073/pnas.220077897
Schureck MA, Maehigashi T, Miles SJ, Marquez J, Cho SE, Erdman R, Dunham CM (2014) Structure of the Proteus vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J Biol Chem 289:1060–1070. doi:10.1074/jbc.M113.512095
Wen Y, Behiels E, Devreese B (2014) Toxin–Antitoxin systems: their role in persistence, biofilm formation, and pathogenicity. Pathog Dis 70:240–249. doi:10.1111/2049-632X.12145
Pandey DP, Gerdes K (2005) Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res 33:966–976
Li D, Xu Y, Shao CL, Yang RY, Zheng CJ, Chen YY, Fu XM, Qian PY, She ZG, de Voogd NJ, Wang CY (2012) Antibacterial bisabolane-type sesquiterpenoids from the sponge-derived fungus Aspergillus sp. Mar Drugs 10:234–241. doi:10.3390/md10010234
Thakur NL, Hentschel U, Krasko A, Pabel CT, Anil AC, Müller WEG (2003) Antibacterial activity of the sponge Suberites domuncula and its primmorphs: potential basis for epibacterial chemical defense. Aquat Microb Ecol 31:77–83
Bell JJ, Smith D, Hannan D, Haris A, Jompa J, Thomas L (2014) Resilience to disturbance despite limited dispersal and self-recruitment in tropical barrel sponges: implications for conservation and management. PLoS One 9:e91635. doi:10.1371/journal.pone.0091635
de Voogd NJ, Cleary DFR (2009) Variation in sponge composition among Singapore reefs. Raffles B Zool Supp 22:59–67
Swierts T, Peijnenburg KTCA, Cleary DFR, Hörnlein C, Setiawan E, Wörheide G, Erpenbeck D, de Voogd NJ (2013) Lock, stock and two different barrels: morphological and genetic variation of the Indo-Pacific sponge Xestospongia testudinaria around Lembeh Island, Indonesia. PLoS One 8:e74396. doi:10.1371/journal.pone.0074396
Carstens J, Heinrich MR, Steglich W (2013) Studies on the synthesis and biosynthesis of the fungal alkaloid necatorone. Tetrahedron Lett 54:5445–5447
Kibet JK, Khachatryan L, Dellinger B (2013) Molecular products from the pyrolysis and oxidative pyrolysis of tyrosine. Chemosphere 91:1026–1034
Hill RA (2007) Marine natural products. Annu Rep Prog Chem Sect B: Org Chem 102:123–137. doi:10.1039/B515100G
Nguyen XC, Longeon A, Pham VC, Urvois F, Bressy C, Trinh TT, Nguyen HN, Phan VK, Chau VM, Briand JF, Bourguet-Kondracki ML (2013) Antifouling 26, 27-cyclosterols from the Vietnamese marine sponge Xestospongia testudinaria. J Nat Prod 76:1313–1318. doi:10.1021/np400288j
Youssef DTA (2005) Hyrtioerectines A − C, Cytotoxic Alkaloids from the Red Sea Sponge Hyrtios erectus. J Nat Prod 68:1416–1419. doi:10.1021/np050142c
Youssef DTA, Shaala LA, Asfour HZ (2013) Bioactive compounds from the Red Sea Marine sponge Hyrtios species. Mar Drugs 11:1061–1070. doi:10.3390/md11041061
Zhou X, Lu Y, Lin X, Yang B, Yang X, Liu Y (2011) Brominated aliphatic hydrocarbons and sterols from the sponge Xestospongia testudinaria with their bioactivities. Chem Phys Lipids 164:703–706. doi:10.1016/j.chemphyslip.2011.08.002
Patel K, Laville R, Martin MT, Tilvi S, Moriou C, Gallard JF, Ermolenko L, Debitus C, Al-Mourabit A (2010) Unprecedented stylissazoles A-C from Stylissa carteri: another dimension for marine pyrrole-2-aminoimidazole metabolite diversity. Angew Chem Int Edit 49:4775–4779. doi:10.1002/anie.201000444
Rohde S, Gochfeld D, Ankisetty S, Avula B, Schupp P, Slattery M (2012) Spatial variability in secondary metabolites of the indo-pacific sponge Stylissa massa. J Chem Ecol 38:463–475. doi:10.1007/s10886-012-0124-8
Wang X, Morinaka BI, Molinski TF (2014) Structures and solution conformational dynamics of stylissamides G and H from the Bahamian sponge Stylissa caribica. J Nat Prod 77:625–630
Yamaguchi M, Miyazaki M, Kodrasov MP, Rotinsulu H, Losung F, Mangindaan REP, de Voogd NJ, Yokosawa H, Nicholson B, Tsukamoto S (2013) Spongiacidin C, a pyrrole alkaloid from the marine sponge Stylissa massa, functions as a USP7 inhibitor. Bioorg Med Chem Lett 23:3884–3886. doi:10.1016/j.bmcl.2013.04.066
Acknowledgments
This research was supported by the Portuguese Foundation for Science and Technology (FCT) under grant PTDC/AAC-AMB/115304/2009 (LESS CORAL) and a PhD Fellowship SFRH/BD/33391/2008. Samples were collected under a research permit issued by the Indonesian State Ministry for Research and Technology (Kementerian Riset Dan Teknologi Republik Indonesia (RISTEK)). We thank the Indonesian Institute of Sciences (PPO-LIPI) for their support and especially Yos Tuti.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(PDF 113 kb)
Online Resource 2
Stacked barplot showing the relative abundances of the nine most abundant phyla sampled from the six biotopes, a) S. carteri, b) S. massa, c) X. testudinaria, d) H. erectus, e) Sediment and f) Seawater. The site codes (x axis) are Lae: Lae Lae, Sam: Samalona, Kud: Kudingkareng Keke, Bak: Bone Baku and Lan: Langkai (PDF 7.10 kb)
Online Resource 3
List of most abundant OTUs (>300 sequences) including OTU-numbers; total sequences (Sum) and the subtotals for samples from seawater (Wt); sediment (Sd); S. massa (Sm); S. carteri (Sc); X. testudinaria (Xt); H. erectus (He); taxonomic affiliation of OTU, GenBank GenInfo sequence identifiers (GI) of closely related organisms identified using BLAST; sequence identity (Sq ident) of these organisms with our representative OTU sequences; isolation source (Source) of closely related organisms identified using BLAST; location where the isolation source was sampled (Location). (XLS 18 kb)
Online Resource 4
Maximum Likelihood phylogenetic tree (16S rRNA gene sequences) of the most dominant OTUs(Table 1) assigned to the phylum Proteobacteria and their cultured closest relatives (gi = GeneBank sequence identification number). Symbols represent samples from seawater (Wt), sediment (Sd), S. massa (Sm), S. carteri (Sc), X. testudinaria (Xt) and H. erectus (He). Bootstrap values generated from 500 replicates. Bootstrap values lower than 50 % were omitted. For the analysis, selected 16S rRNA gene sequences of the most dominant OTUs (≥300 sequences) and their cultured closest relatives in GenBank [http://www.ncbi.nlm.nih.gov/] were aligned using ClustalW and a phylogenetic analysis conducted using MEGA 6 software (http://www.megasoftware.net/) (Tamura et al., 2011). Phylogenetic trees were constructed according to the maximum-likelihood statistical method using the general time reversible (GTR) model with a discrete Gamma distribution (5 categories (+G, parameter = 0.4305). In the results, we present a bootstrap consensus tree based on 500 replicates. The bootstrap value is shown next to each branch when this exceeds 49 %. This value represents the percentage of replicate trees in which the associated taxa clustered together. For tree inference we used the nearest neighbor interchange (NNI) heuristic method and automatic initial tree selection. (PDF 18 kb)
Online Resource 5
Maximum Likelihood phylogenetic tree (16S rRNA gene sequences) of the most dominant OTUs(Table 1) assigned to the non-proteobacterial phyla and their cultured closest relatives (gi = GeneBank sequence identification number). Symbols represent samples from seawater (Wt), sediment (Sd), S. massa (Sm), S. carteri (Sc), X. testudinaria (Xt) and H. erectus (He). Bootstrap values generated from 500 replicates. Bootstrap values lower than 50 % were omitted. (PDF 10 kb)
Online Resource 6
Stacked bar plots showing the estimated gene count (Y axis) of a set of KOs and the contribution of selected orders to the gene count; unclassified OTUs at the order level and OTUs belonging to all other orders are pooled and indicated by ‘Other’. a) K00087 (xanthine dehydrogenase molybdenum-binding subunit), b) K03409 (chemotaxis protein CheX), c) K04561 (nitric oxide reductase) and d) K10535 (hydroxylamine oxidase) for all samples (X axis). (PDF 7 kb)
Online Resource 7
Stacked bar plots showing the estimated gene count (Y axis) of a set of KOs and the contribution of selected orders to the gene count; unclassified OTUs at the order level and OTUs belonging to all other orders are pooled and indicated by ‘Other’. a) K05522 (endonuclease VIII), b) K07239 (heavy metal exporter), c) K07334 (proteic killer suppression protein) and d) K07665 (copper resistance phosphate regulon response regulator CusR) for all samples (X axis). (PDF 9 kb)
Rights and permissions
About this article

Cite this article
Cleary, D.F.R., de Voogd, N.J., Polónia, A.R.M. et al. Composition and Predictive Functional Analysis of Bacterial Communities in Seawater, Sediment and Sponges in the Spermonde Archipelago, Indonesia. Microb Ecol 70, 889–903 (2015). https://doi.org/10.1007/s00248-015-0632-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0632-5