
Abstract
Purpose of Review
Serum phosphorus is maintained in a narrow range by balancing dietary phosphate absorption, influx and efflux of phosphorus from bone and intracellular stores, and renal reabsorption of filtered phosphate. Acute hypophosphatemia, typically caused by transient increases in cellular uptake, can lead to severe complications such as cardiopulmonary dysfunction and rhabdomyolysis that can warrant parenteral phosphate repletion. Chronic hypophosphatemia, however, generally represents true phosphate deficiency and may result in long-term metabolic and skeletal complications, particularly in children due to the critical importance of phosphorus to skeletal mineralization and longitudinal growth.
Recent Findings
In addition to the well-characterized roles of vitamin D and parathyroid hormone (PTH), a new bone-kidney axis has been discovered that regulates phosphate homeostasis through the bone-derived hormone fibroblast growth factor 23 (FGF23) and its phosphaturic actions that are mediated by activation of fibroblast growth factor receptors (FGFRs) complexed with α-Klotho in renal tubules. Chronic hypophosphatemia can now be classified as FGF23 dependent or independent.
Summary
In cases of FGF23-dependent hypophosphatemia, traditional non-specific treatments with elemental phosphorus and 1,25(OH)2 vitamin D (calcitriol) can now be replaced with a targeted approach by using an FGF-23 blocking antibody (Burosumab).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phosphorus is a critical element that is necessary for physiological functions. The regulation of phosphorus homeostasis is multifaceted, relying on a complex interplay between (hormones from) multiple organ systems, most importantly bone, kidney, and intestine. Alteration in one and/or multiple facets of phosphorus regulation can result in both acute and chronic clinical consequences that require therapeutic intervention.
Phosphate Homeostasis
Phosphorus (P) is a highly reactive chemical element that binds to oxygen to form phosphate (PO43−). Phosphate plays an essential biological role in cellular and mineral metabolism. At the cellular level, phosphates are components of phospholipids in cell membranes, nucleic acids (DNA and RNA), energy production via adenosine triphosphate (ATP), oxygen delivery through 2,3-diphoshoglycerate (2,3-DGP) hemoglobin binding, and cell signaling through ubiquitous phosphorylation reactions. Systemically, phosphate binds to calcium to form hydroxyapatite to mineralize collagen in bone and cartilage. Phosphate also functions as a buffer to maintain acid-base balance (pH) [1, 2].
The serum phosphate concentration is maintained in the physiological range of 2.5 to 4.5 mg/dL in adults (0.81 to 1.45 mmol/L). In children, the serum phosphate levels are higher [2]. Hypophosphatemia is defined as a serum phosphate concentration below 2.5 mg/dL (0.81 mmol/L). Clinically relevant hyperphosphatemia is defined as a serum phosphate concentration above 6.5 mg/dL (2.10 mmol/L), a threshold associated with adverse outcomes and increased mortality in patients with chronic kidney disease (CKD) [3]. A higher serum phosphate is observed in children. Since the normal circulating calcium times phosphate (Ca × P) product is above the threshold for spontaneous calcification, soft-tissue calcifications are prevented by the presence of circulating inhibitors of mineralization, such as Matrix γ-carboxyglutamic acid Gla protein [4].
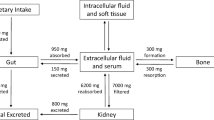
The serum phosphorus concentration is determined by the balance between intestinal absorption of phosphate from the diet (16 mg/kg/day), storage of phosphate in the skeleton (3 mg/kg/day), fluxes in and out of cells in response to metabolic demands, and excretion of phosphate through the urine (13 mg/kg/day) (Fig. 1) [2]. One of the key knowledge gaps in understanding phosphate homeostasis, however, is that a molecular mechanism whereby extracellular and/or intracellular phosphates are sensed has not been identified [5, 6].

Regulation of systemic phosphate homeostasis
Intestinal Absorption of Phosphate
The recommended dietary allowance of phosphate is 700 mg/day, and the maximal tolerated intake is 4000 mg/day in individuals with normal renal function. In the general population in the USA, phosphorus intakes are above the recommended amounts, with average daily intakes of 1602 mg in men and 1128 mg in women [7]. Nearly 70% of dietary phosphate is absorbed, resulting in transient postprandial increases in serum phosphate concentrations. The bioavailability of phosphorus in foods is variable, with phytate phosphorus found in plants being poorly absorbed because we lack phytases that release soluble phosphate (Fig. 1).
Dietary phosphate is actively transported transcellularly in intestinal cells through a sodium-dependent transporter SLC34a2 (Npt2b) [8] and passively absorbed through paracellular pathways. Npt2b-mediated active transport is important in phosphate-deficient states. Npt2b expression is increased by 1,25(OH)2D and low dietary phosphate and decreased by nicotinamide. The non-essential role of Npt2b in regulating phosphate homeostasis is revealed by the absence of hypophosphatemia in individuals with inactivating Npt2b mutations, which have pulmonary alveolar microlithiasis [9]. Malnutrition, malabsorption states, and phosphate binding medications may lead to hypophosphatemia through reduction in intestinal phosphate absorption. Niacin, which inhibits Npt2b, had small effects to reduce serum phosphate (i.e., 0.08 mg/dL (0.03 mmol/L) reduction per year of treatment) in patients with CKD [10].
Fluxes Been Phosphate Storage Pools
Once absorbed, extracellular phosphate levels are in a dynamic equilibrium between cellular and bone stores. Insulin, catecholamines, and alkalemia cause phosphate movement into cells and can lead to hypophosphatemia without phosphate deficiency (Fig. 1). Refeeding syndrome is characterized by cardiac arrhythmias, leading to multisystem organ dysfunction and death that is mediated in large part from effects of a nutritional load to induce hypophosphatemia. Phosphate flux in and out of bone, which is the major reservoir for phosphate, can also contribute to serum phosphate concentrations. In this regard, hungry bone syndrome that follows parathyroidectomy leads to hypocalcemia and hypophosphatemia due to uptake of calcium and phosphate into bone, whereas severe hyperparathyroidism in CKD can contribute to hyperphosphatemia due to efflux of phosphate from bone.
Renal Handling of Phosphate
Renal reabsorption and excretion of phosphate is the principal mechanism regulating phosphate homeostasis. Phosphate is filtered by the glomerulus and reabsorbed in the proximal tubule (PT) (Fig. 1). NPT2a (SLC34a1) and NPT2c (SLC34a3) located in the luminal brush border membrane of the PT reabsorb 80 and 20% of the total urinary phosphorus, respectively. The sodium/hydrogen exchanger regulatory factor 1 NHERF-1 binds to NPT2a and is an important regulator of membrane expression [11]. PTH and FGF23 lead to phosphorylation of NPT2a causing translocation to lysosomes and inhibition of phosphate reabsorption. CLCN5 gene encodes an endosomal H+/2Cl antiporter that regulates endosomal acidification and internalization of NPT2a. α-Klotho, which is predominantly expressed in the distal convoluted tubule, is released into the circulation as a soluble Kl1 + Kl2 biologically active fragment (sKl) by ADAM10, and ADAM17 sheddases may also be filtered by the glomerulus and regulate NPT2 membrane localization in the PT [12].
Primary Defects in Proximal Tubule Absorption of Phosphate
Several hypophosphatemic disorders are caused by inactivating mutation in the transporters NPT2a, and NPT2c as well as factors, such as NHERF-1, CLCN5, and OCRL, which regulate the endocytosis of these transporters, both resulting in direct defects in renal phosphate transport [13, 14]. Chronic and acute regulation of these renal transporters is modulated by changes in dietary and serum phosphate levels and by three major hormones: parathyroid hormone (PTH), 1,25-dihydroxy vitamin D3 (1,25(OH)2D3), and fibroblast growth factor 23 (FGF23). Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) is caused by loss of function of the solute carrier family 34, member 3 (SLC34A3) gene encoding a protein of the renal NPT2c located in the proximal straight tubule that results in hypophosphatemia, phosphaturia, and nephrocalcinosis and nephrolithiasis. Nephrolithiasis/osteoporosis and hypophosphatemic 1 and 2 (NPHLOP1 and NPHLOP2) are respectively caused by inactivating mutations in SLC34A1 encoding NPT2a and SLC9A3R1 encoding NHERF1 located in the proximal convoluted tubule [15]. Both cause an autosomal dominant hypophosphatemic disorder characterized by the coexistence of nephrolithiasis, osteoporosis, and hypophosphatemic rickets. X-linked recessive hypophosphatemic rickets, XLRHR, or Dent disease 1 is caused by inactivation mutation of the renal chloride channel CLCN5 gene that encodes the endosomal H+/2Cl− antiporter protein CIC-5 [16]. Mutations in CLCN5 lead to a renal proximal tubulopathy (Fanconi syndrome) characterized by defective reabsorption of phosphate as well as other solutes including amino acids, glucose, uric acid, potassium, and bicarbonate, and by low-molecular-weight proteinuria (LMWP) associated with hypercalciuria and/or its complications (nephrocalcinosis or nephrolithiasis) and progressive renal failure. Oculocerebrorenal syndrome of Lowe (OCRL), characterized by defects in the nervous system, eye, and kidney, is caused by mutations in the OCRL gene that encodes the inositol polyphosphate 5-phosphatase OCRL-1 that regulates membrane trafficking of transporters [17]. Fanconi-Bickel syndrome (FBS) is proximal renal tubular acidosis caused by mutations in the glucose transporter, Glut2, which results in severe hypophosphatemic rickets and failure to thrive due to proximal renal tubular dysfunction leading to glucosuria, phosphaturia, generalized aminoaciduria, bicarbonate wasting, and hypophosphatemia [18]. Primary defects in renal PT phosphate transport leads to secondary increments in 1,25(OH)2D levels, which is an important consideration in the selection of treatment options.
Hypophosphatemia Caused by Vitamin D Deficiency/PTH Excess
Hepatic 25-hydroxylase (CYP2R1) generates 25(OH)D. 1,25(OH)2D is produced in the renal proximal tubule from 25(OH) D by 1⎕α-hydroxylase (CYP27B1) and activates vitamin D receptors (VDR) in target tissues, including the intestines to regulate NPT2b-mediated phosphate absorption, the parathyroid gland to regulate PTH secretion, bone to stimulate bone resorption and inhibit bone mineralization, and in the kidney to regulate α-Klotho, to name a few of the effects of this hormone. Both 25(OH) D and 1,25(OH)2D are converted to 24,25(OH)2D by 24-hydroxylase (CYP24), leading to degradation. Genetic forms of vitamin D-dependent rickets (VDDRs) due to mutations impairing activation of vitamin D or decreasing vitamin D receptor responsiveness are associated with hypophosphatemia. Vitamin D-dependent rickets type 2A (VDDR2A) is caused by loss of function mutations in the vitamin D receptor (VDR) gene. Vitamin D-dependent rickets type 1 (VDDR1) is caused by inactivating mutations of CYP27B1. These disorders display decreased tissue responsiveness to 1,25(OH)2D. Very rare mutations of CYP2R1 have been associated with vitamin D-dependent rickets type 1B (VDDR1B) that is responsive to 25 (OH) D[19]. An activating mutation in CYP3A4 that oxidizes 1,25-dihydroxyvitamin D with a 2-fold greater activity than CYP24A1 leads to vitamin D deficiency [20]. The concomitant hypocalcemia and elevations in PTH and low levels of FGF23 distinguish these hypophosphatemic disorders to those caused by FGF23 excess.
FGF23 inhibits CYP27B1 and stimulates CYP24 leading to reduced levels of 1,25(OH)2D, consistent with its role as a vitamin D counter-regulatory hormone, whereas PTH has the opposite effect leading to increased 1,25(OH)2D, consistent with its role as a calcemic hormone [21, 22]. PTH stimulates phosphate absorption in the intestines and stimulates bone resorption to increase serum phosphate. PTH secreted by the parathyroid glands and FGF23 released from bone coordinately reduce NPT2-mediated phosphate reabsorption in the PT to promote phosphaturia. Clinical disorders leading to excess circulating PTH (primary and secondary hyperparathyroidism) and activating mutations of the PTH receptor in Jansen metaphyseal chondrodysplasia (JMC) and downstream mutant forms of GNAS1 in McCune-Albright Syndrome (MAS) associated with increased cAMP in osteocytes that cause elevated FGF23 all lead to hypophosphatemia. PTH excess and MAS also have increased bone turnover and osteolytic bone lesions.
Hypophosphatemia Caused by FGF23 Excess
FGF23 is a ~ 32 kDa protein with an N-terminal FGF-homology domain and a novel 71 amino acid C-terminus [23]. Both osteoblasts and osteocytes secrete FGF23 [23, 24]. FGF23 acts as a circulating hormone [25] that binds to α-Klotho/FGFR complexes in target tissues [26, 27]. α-Klotho is a type I membrane, β-glycosidase-like protein, which forms a trimeric complex with FGFRs and the C-terminus of FGF23 [28,29,30,31] leading to FGFR activation and tissue selectivity. FGF23 inhibits renal phosphate reabsorption by decreasing Npt2 co-transporters and suppresses 1,25(OH)2D levels by inhibiting Cyp27b1 and stimulating Cyp24 [26, 27, 32,33,34,35,36]. Excess FGF23 causes hypophosphatemia, suppression of 1,25(OH)2D, and rickets/osteomalacia [26, 33, 37,38,39]. Loss of Fgf23 function results in hyperphosphatemia, excess 1,25(OH)2D, and soft-tissue calcifications [32, 40,41,42,43,44,45].
Gene mutations resulting in a common hypophosphatemic rickets phenotype have been exploited to understand the molecular pathogenesis of elevated FGF23 [21] (Table 1). Autosomal dominant hypophosphatemic rickets (ADHR) is caused by mutations in an RXXR site of FGF23 that prevents its cleavage. X-linked hypophosphatemia [21], and its murine homolog condition, the Hyp mouse model, are caused by mutations in the PHEX endopeptidase [40, 46]. Phex is highly expressed in differentiated osteoblasts and osteocytes, and conditional deletion of Phex in the mature osteoblast lineage in vivo by Oc-Cre is sufficient to reproduce the Hyp mouse phenotype [47]. Although an initial study suggested that Phex processes FGF23 [48], subsequent studies failed to establish Phex-dependent cleavage of FGF23 [24, 49, 50]. Rather, Phex mutations lead to elevations of FGF23 due to increased FGF23 gene transcription in osteoblasts and osteocytes [51]. The increase in FGF23 is “intrinsic,” since increased FGF23 gene transcription is present in isolated osteoblasts, and bone mesenchymal stem cells (BMSCs) derived from Hyp mice and in Hyp bone explanted into wild-type mice continue to express high levels of FGF23 [46].
Autosomal recessive hypophosphatemic rickets (ARHR2) is caused by inactivating mutations in DMP1 [52]. The murine homolog Dmp1−/− mice [52, 53] shows an intrinsic stimulation of FGF23 expression in osteoblasts and osteocytes. DMP1 is a the small integrin-binding ligand N-linked glycoprotein (SIBLING) extracellular matrix protein that acts as a nucleator of mineralization [52]. Dmp1 is cleaved into 37- and 57-kDa fragments by BMP1 or cathepsin B. The NH2-terminal fragment is a proteoglycan with a chondroitin sulfate chain attached through Ser74 that binds to proMMP-9 and may sequester growth factors [54], whereas the C-terminal fragment has RGD and ASARM motifs. The ability of Dmp1 to bind to integrins through the RGD motif and Phex through ASARM provides a molecular basis for the interdependent functions of Phex and Dmp1 that we found in compound mutant mice. Compound mutant Hyp/Dmp1−/− mice exhibit non-additive effects on FGF23 expression that is mediated by a common pathway involving FGFR1 [55] and intrinsic abnormalities of mineralization and alterations of the extracellular matrix milieu [56,57,58,59]. Dmp1 and Phex mutations, which block the mineralization of extracellular matrix, lead to increased FGF23 production by osteoblasts and osteocytes [60, 61]. Coupling between mineralization and FGF23 occurs through the regulation of FGF23 expression in osteoblasts/osteocytes by “intrinsic factors” that leads to the activation of FGFR1 [62, 63].
Two additional genetic abnormalities in the ANK/ENPP1 also support a coupling of mineralization with FGF23 expression. Inactivating mutations in Enpp1, which causes hereditary generalized arterial calcification of infancy, also causes a variant of autosomal recessive hypophosphatemic rickets (ARHR2), characterized by FGF23-mediated hypophosphatemia [64, 65]. Enpp1 generates inorganic pyrophosphate (PPi), an inhibitor of calcification and substrate for alkaline phosphatase (TNAP), which converts PPi to Pi. The inactivation of this Enpp1 reduces the ratio of PPi to Pi in soft tissues leading to ectopic calcification, but local Pi depletion in bone that leads to osteomalacia. In addition, inactivation of Ank, a PPi transporter located in osteoblasts, results in impaired mineralization of extracellular matrix and nearly a 10-fold increase in Fgf23 expression in bone [66].
FAM20C is a dentin matrix protein that encodes a casein kinase that localizes to the Golgi apparatus and endosomes that phosphorylates SIBLING proteins. FAM20C also phosphorylates FGF23 and promotes its cleavage [67]. Inactivating mutations in FAM20C causes Raine syndrome, an autosomal recessive disorder with generalized osteosclerosis with periosteal bone formation and distinctive facial features. Fam20c−/− mice, however, develop hypophosphatemic rickets caused by elevated FGF23, but not osteosclerosis. Finally, a translocation causing increased α-Klotho expression leads to elevations of FGF23 and hypophosphatemic rickets [68]. The mechanism whereby overexpression of α-Kl increases FGF23 is not clear.
Diagnostic Approach to a Hypophosphatemic Patient
Although hypophosphatemia is typically defined as a serum phosphorus level below 2.5 mg/dl (0.81 mmol/L), it is reasonably common (occurs in up to 5% of hospitalized patients [69]), and there is quite often a delay in diagnosis. Specifically, the absence of serum phosphorus on routine chemistry panels and a complete metabolic profile (CMP or SMC20) reduces the likelihood of identification of these patients, as it must be separately ordered for each individual or as part of a renal function panel.
Etiology
The causes of hypophosphatemia in patients are quite heterogeneous but can be approached from a combination of acuteness and mechanistically based perspectives. Acute hypophosphatemia, which is loosely defined as developing over hours to days, generally occurs in hospitalized patients. In addition, it primarily is due to intracellular shifts in phosphorus, which is facilitated by insulin, catecholamines, and alkalosis due to hyperventilation as detailed above. Refeeding-induced hypophosphatemia is not uncommon and occurs more commonly with enteral versus parenteral nutrition [70]. Low phosphorus commonly occurs in burn patients as well, though it is not specifically associated with increased mortality [71]. Hypophosphatemia is also a marker of nutritional risk in older hospitalized patients [72].
In contradiction, chronic hypophosphatemia with very few exceptions is generally seen in outpatients and develops over months to years. Mechanistically, it can be categorized on a hereditarily versus acquired basis. In addition, it can be further sub-categorized based on the underlying mechanism of disease, those being FGF23-dependent and independent. This is particularly pertinent, as plasma FGF23 (sum of intact and carboxyterminal fragments) can be measured by several commercially available assays. As detailed in Table 1, enhanced FGF23 signaling due to gene mutations that result in increased production, decreased catabolism, augmented receptor activation, or increased co-ligand activity (α-Klotho) all cause renal phosphate wasting and impaired intestinal phosphorus absorption with resultant hypophosphatemia. In contrast, genetic causes of FGF23 independent hypophosphatemia are due to mutations in genes that either affect renal reabsorption or vitamin D-mediated intestinal absorption of phosphorus (Table 1). It is particularly important to discern the role of FGF23 in a patient with inherited renal phosphate wasting, as the therapeutic approach is necessarily altered to minimize the risk of complications. A diagnostic approach based on biochemical alterations can facilitate correctly identifying the disorder (Table 2).
Patients with acquired chronic hypophosphatemia may also be sub-categorized based on the presence or absence of underlying FGF23 pathology. Tumor-induced osteomalacia (TIO) is a classic but relatively rare disorder that is due to dramatic overproduction of FGF23 in an unregulated fashion by generally benign mesenchymal tumors that usually develop in connective tissue or bone. More recently recognized, reasonably common and, in contrast to other chronic hypophosphatemic disorders, parenteral iron-induced hypophosphatemia develops over days to weeks. Low serum phosphorus appears to be due to alterations in FGF23 catabolism but is generally restricted to iron carboxymaltose formulations based on available evidence [73•]. The most common cause of FGF23-independent chronic hypophosphatemia is primary hyperparathyroidism, which occurs in approximately one third of patients with the condition and is due to enhanced renal phosphate wasting. Drugs such as tenofovir can induce a generalized proximal tubulopathy resembling Fanconi’s syndrome that results in renal phosphate wasting [74]. Impairment of intestinal phosphorus absorption due to nutritional deficiencies (vitamin D, generalized malnutrition) or malabsorptive conditions (celiac disease, Roux-en-Y gastric bypass) can also cause hypophosphatemia. Finally, being non-renal and non-intestinal related, excessive deposition of phosphorus into bone that occurs in hungry bone syndrome after resolution of severe hyperparathyroidism can also result in hypophosphatemia. A more complete list of potential causes of acquired hypophosphatemia is available elsewhere [75].
Clinical Consequences of Hypophosphatemia
The signs and symptoms of hypophosphatemia can be quite variable, depending on the degree of phosphate deficit and the acuity of onset. Patients with inherited disorders, as well as those with acquired disease having modest hypophosphatemia (serum phosphorus ~ 1.5 to 2.5 mg/dL; 0.48 to 0.81 mmol/L), may complain of fatigue, bone pain, and proximal muscle weakness, although many patients are asymptomatic. Patients with similar levels of serum phosphorus may also have moderate symptoms, including stress or frank fractures. In addition, inherited hypophosphatemic disorders often confer different skeletal consequences based on the age of the patient. Children and adolescents often experience impaired longitudinal growth with or without lower extremity bowing, the former of which is not applicable to adults. Nonetheless, genetically affected adult patients often develop other musculoskeletal disease (i.e., enthesopathy), which can impair their quality of life [76••]. Patients with more severe hypophosphatemia (serum phosphorus < 1.0 to 1.2 mg/dl; 0.32 to 0.39 mmol/L), especially when acquired more acutely, may develop more severe organ dysfunction. These changes include marked decreases in cardiac and respiratory muscle function, intestinal ileus, and mental status changes. Patients with severe hypophosphatemia are also at risk for rhabdomyolysis and resultant myoglobin-induced acute kidney injury.
Treatment Considerations
Phosphorus/Vitamin D
Identification of the underlying cause of hypophosphatemia, in combination with the acuteness of onset and severity of the phosphorus deficit, will best guide the appropriate choice for clinical management of affected patients. Patients with severe hypophosphatemia (less than 1.0 mg/dl;0.32 mml/L), which generally occurs acutely in hospitalized patients as detailed above, may require intravenous phosphorus therapy that varies based on the degree of hypophosphatemia and associated clinical symptoms (for serum phosphorus above 1.25 mg/dL (0.39 mmol/L), 0.08 to 0.24 mmol/kg over 6 h up to a maximum total dose of 30 mmol; serum phosphorus below 1.25 mg/dL (0.40 mmol/L), 0.25 to 0.50 mmol/kg over 8 to 12 h up to a maximum total dose of 80 mmol). Once the serum phosphorus is above 1.5 mg/dL (0.48 mmol/L), patients are generally switched over to oral phosphorus (1 mmol/kg of elementary phosphorus (40–80 mmol/day) given in three to four doses over a 24-h period).
Hypophosphatemia in hospitalized patients is commonly due to redistribution of phosphorus as a result of respiratory alkalosis. Treatment with phosphate is generally not indicated. In diabetic ketoacidosis (DKA), insulin therapy results in transcellular shifts of transient hypophosphatemia, and phosphate administration has not been shown to affect the outcome and can result in hypocalcemia [77]. Whole-body phosphate deficiency of approximately 1 mmol/kg, however, can occur in DKA. Phosphate replacement is warranted if hypophosphatemia is severe (i.e., serum phosphate concentration less than 1.0 mg/dL; 0.32 mmol/L) or when hypophosphatemia is believed to contribute to cardiac dysfunction, anemia complications, rhabdomyolysis, or respiratory depression [78]. Identification and concomitant treatment of 25(OH) D insufficiency (i.e., 25(OH) D < 20 ng/mL) if present is also advised.
Patients with mild or moderate hypophosphatemia who are symptomatic, whether it is inherited or acquired, require a less aggressive treatment approach, which traditionally has been centered on the use of oral phosphate supplements and activated vitamin D analogues. In children with XLH, treatment with elemental phosphorus (generally available as 250 mg tablets or powder) at a dose of 20–40 mg/kg/day in three to five divided doses, is initially recommended, although higher does may be required [79]. Calcitriol is concomitantly given at a dose of 20–30 ng/kg/day in order to enhance intestinal phosphorus absorption and to limit the stimulus of phosphorus treatment to stimulate PTH release, although higher doses up to 70 ng/kg/day may need to be given initially to children with more severe osteomalacia based on clinical findings such as pain, alkaline phosphatase elevation, and degree of lower extremity bowing. Phosphorus and calcitriol doses may require adjustment, based on treatment response, as well as serum phosphorus, calcium, and parathyroid hormone levels. The most useful biomarker for skeletal response is serum alkaline phosphatase, which decreases in response to healing of osteomalacia. Pediatric patients with XLH should be monitored with serum and urine biochemistries every 3 months, including spot or 24-h urine calcium to assess for development of hypercalciuria, as nephrocalcinosis may occur independent of hypercalcemia. Renal ultrasonography is also recommended at 2- to 5-year intervals to assess for renal calcification. Radiographs of the distal femoral and proximal tibial sites every 2 years are also recommended to ensure epiphyseal correction in treated children. Although not specifically studied to date, this treatment approach is likely applicable to other inherited and acquired FGF23-dependent hypophosphatemic disorders as well.
The evidence supporting the treatment of adults with FGF23-dependent hypophosphatemia is less well established but generally follows the principles of pediatric treatment per above. Recommended indications for treatment of XLH in adults includes spontaneous insufficiency or stress fractures, pending orthopedic procedures, and skeletal pain with biochemical evidence of osteomalacia (i.e., elevated alkaline phosphatase) [79]. Calcitriol 0.50 to 0.75 mcg/day in two divided doses is initially given for 1 week, followed by 250 mg of elemental phosphorus twice daily, the latter of which is titrated up to 1000 mg/day in 2–4 divided doses over 1–2 weeks. For orthopedic procedures such as weight-bearing joint replacement, therapy is recommended 3–4 months before and 6–9 months after the operation. Laboratory and renal imaging safety monitoring are like those advised for children. It is important to note that phosphorus and calcitriol treatment will not improve enthesopathy in adults. In addition, FGF23 levels are increased in calcitriol-treated patients having XLH [80], which could theoretically exacerbate phosphate wasting or other adverse effects of excess FGF23 [81, 82]. Based on these observations, careful consideration of the risks and benefits of phosphorus and vitamin D therapy is merited in adult XLH patients considering treatment.
Regarding non-FGF23-mediated hypophosphatemic disorders, treatment is generally guided by the degree of serum phosphorus deficiency, as well as skeletal and systemic symptoms. Hereditary hypophosphatemic rickets with hypercalciuria, HHRH, is treated with elemental phosphorus at a dose of 20–50 mg/kg/day in three to five divided doses daily. Importantly, activated vitamin D analogues are not used, as serum calcitriol and urine calcium is elevated in these patients and would be worsened by such agents. Patients with vitamin D hydroxylation-deficient ricket types 1A and 1B have mild hypophosphatemia that does not require replacement. These patients are treated with calcitriol at daily divided doses of 10–20 mcg/kg/day, although patients with VDDR1A may require elemental calcium doses of 50–75 mcg/kg/day initially. Patients with vitamin D-dependent rickets, type 2A (VDDR2A) require much higher doses of calcitriol (1–6 mcg/kg/day) and 1 to 3 g of calcium per day, due to severe vitamin D receptor resistance [83]. Surveillance of pertinent biochemistries and renal ultrasonography, akin to management protocols in FGF23-dependent hypophosphatemic patients as detailed above, is also recommended for these patients. There is no available therapy for patients with Jansen metaphyseal chondrodysplasia.
Burosumab
Although phosphorus and calcitriol do improve biochemical and skeletal outcomes in patients with XLH, they do not target the underlying mechanism of disease, that being the disordered catabolism of FGF23. Additionally, as noted above, calcitriol treatment increases FGF23 levels in XLH patients and could undermine some of the benefits of treatment. Therefore, an XLH therapy that more directly targets the underlying pathophysiology and is easier to adhere to is preferred. Burosomab, a recombinant, fully human IgG1 monoclonal antibody that inhibits FGF23 activity and was approved for the treatment of children and adults with XLH, lowers FGF23 levels and establishes normal phosphorus and vitamin D homeostasis. At a dose of 0.8 mg/kg subcutaneously every 2 weeks, Burosumab safely improved linear growth and radiographic rickets severity score in children with XLH, while also concurrently improving physical function and reducing pain [84••]. In adult patients with XLH, 1.0 mg/kg Burosumab given subcutaneously every 4 weeks resolved hypophosphatemia and reduced stiffness. In addition, patients with insufficiency fractures before treatment were nearly 17-fold more likely to heal compared with placebo-treated patients [85]. Nonetheless, as is the case with conventional therapy with phosphorous and activated vitamin D, the benefits of therapy with Burosumab are less certain in adults compared with children. Burosumab is also currently under investigation as a therapy for the acquired FGF23-dependent disorder tumor-induced osteomalacia, although it has not been studied in other FGF23-dependent hypophosphatemic disorders to date. Recently, small-molecule inhibitors of FGF23 have been identified that increases serum phosphate in mouse models of FGF23 excess [86].
Conclusion
Acute severe phosphorus deficiency can impart significant clinical consequences, including severe organ dysfunction, whereas inherited and acquired chronic hypophosphatemia confer long-term metabolic and skeletal effects, including chronic pain and fractures, which can significantly impair longitudinal growth in children and physical function/quality of life in adults. Diagnostic approaches should determine the role that FGF23 plays in chronic disorders. Appropriate classification as an FGF23-independent or dependent disorder (e.g., XLH) facilitates a more tailored and individualized approach to the clinical management of inherited hypophosphatemic osteomalacia. Elevated FGF23 can greatly assist in correctly identifying the cause of acquired hypophosphatemic disorders in adults.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104.
Berndt T, Kumar R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda). 2009;24:17–25.
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31:607–17.
Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81.
Camalier CE, Yi M, Yu LR, Hood BL, Conrads KA, Lee YJ, et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J Cell Physiol. 2013;228:1536–50.
Michigami T, Kawai M, Yamazaki M, Ozono K. Phosphate as a signaling molecule and its sensing mechanism. Physiol Rev. 2018;98:2317–48.
Uribarri J, Calvo MS. Dietary phosphorus intake and health1-3. Am J Clin Nutr. 2014;99:247–8.
Sabbagh Y, O'Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–58.
Corut A, Senyigit A, Ugur SA, Altin S, Ozcelik U, Calisir H, et al. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet. 2006;79:650–6.
Malhotra R, Katz R, Hoofnagle A, Bostom A, Rifkin DE, McBride R, et al. The effect of extended release niacin on markers of mineral metabolism in CKD. Clin J Am Soc Nephrol. 2018;13:36–44.
Weinman EJ, Cunningham R, Wade JB, Shenolikar S. The role of NHERF-1 in the regulation of renal proximal tubule sodium-hydrogen exchanger 3 and sodium-dependent phosphate cotransporter 2a. J Physiol. 2005;567:27–32.
Hu MC, Shi M, Moe OW. Role of alphaKlotho and FGF23 in regulation of type II Na-dependent phosphate co-transporters. Pflugers Arch. 2019;471(1):99–108.
Michalus I, Rusinska A. Rare, genetically conditioned forms of rickets: differential diagnosis and advances in diagnostics and treatment. Clin Genet. 2018;94:103–14.
Forster IC, Hernando N, Biber J, Murer H. Proximal tubular handling of phosphate: a molecular perspective. Kidney Int. 2006;70:1548–59.
Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018;93:204–13.
Mansour-Hendili L, Blanchard A, Le Pottier N, Roncelin I, Lourdel S, Treard C, et al. Mutation update of the CLCN5 gene responsible for dent disease 1. Hum Mutat. 2015;36:743–52.
Inoue K, Balkin DM, Liu L, Nandez R, Wu Y, Tian X, et al. Kidney tubular ablation of Ocrl/Inpp5b phenocopies Lowe syndrome tubulopathy. J Am Soc Nephrol. 2017;28:1399–407.
Santer R, Schneppenheim R, Dombrowski A, Gotze H, Steinmann B, Schaub J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat Genet. 1997;17:324–6.
Molin A, Wiedemann A, Demers N, Kaufmann M, Do Cao J, Mainard L, et al. Vitamin D-dependent rickets type 1B (25-hydroxylase deficiency): a rare condition or a misdiagnosed condition? J Bone Miner Res. 2017;32:1893–9.
Roizen JD, Li D, O'Lear L, Javaid MK, Shaw NJ, Ebeling PR, et al. CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest. 2018;128:1913–8.
Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–8.
Quarles LD. The bone and beyond: ‘Dem bones’ are made for more than walking. Nat Med. 2011;17:428–30.
Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–8.
Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–26.
Suzuki M, Uehara Y, Motomura-Matsuzaka K, Oki J, Koyama Y, Kimura M, et al. {beta} klotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol Endocrinol. 2008;22:1006–14.
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–5.
White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–86.
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4.
Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, et al. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology. 2005;146:4647–56.
Li SA, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct. 2004;29:91–9.
Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H, et al. Anti-FGF23 neutralizing antibodies demonstrate the physiological role and structural features of FGF23. J Bone Miner Res. 2008;23:1509–18.
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–8.
Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289:F1088–95.
Tomiyama K, Maeda R, Urakawa I, Yamazaki Y, Tanaka T, Ito S, et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc Natl Acad Sci U S A. 2010;107:1666–71.
Beckman MJ, Tadikonda P, Werner E, Prahl J, Yamada S, DeLuca HF. Human 25-hydroxyvitamin D3-24-hydroxylase, a multicatalytic enzyme. Biochemistry. 1996;35:8465–72.
Hoenderop JG, Chon H, Gkika D, Bluyssen HA, Holstege FC, St-Arnaud R, et al. Regulation of gene expression by dietary Ca2+ in kidneys of 25-hydroxyvitamin D3-1 alpha-hydroxylase knockout mice. Kidney Int. 2004;65:531–9.
Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145:5269–79.
Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–94.
Fukumoto S, Yamashita T. Fibroblast growth factor-23 is the phosphaturic factor in tumor-induced osteomalacia and may be phosphatonin. Curr Opin Nephrol Hypertens. 2002;11:385–9.
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–49.
Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–90.
Larsson T, Davis SI, Garringer HJ, Mooney SD, Draman MS, Cullen MJ, et al. Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differentially processed. Endocrinology. 2005;146:3883–91.
Sitara D, Razzaque MS, St-Arnaud R, Huang W, Taguchi T, Erben RG, et al. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol. 2006;169:2161–70.
Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–32.
Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281:18370–7.
Liu S, Tang W, Zhou J, Vierthaler L, Quarles LD. Distinct roles for intrinsic osteocyte abnormalities and systemic factors in regulation of FGF23 and bone mineralization in Hyp mice. Am J Physiol Endocrinol Metab. 2007;293:E1636–44.
Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, et al. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. 2008;118:722–34.
Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R, et al. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun. 2001;284:977–81.
Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35:455–62.
Guo R, Liu S, Spurney RF, Quarles LD. Analysis of recombinant Phex: an endopeptidase in search of a substrate. Am J Physiol Endocrinol Metab. 2001;281:E837–47.
Quarles LD, Drezner MK. Pathophysiology of X-linked hypophosphatemia, tumor-induced osteomalacia, and autosomal dominant hypophosphatemia: a perPHEXing problem. J Clin Endocrinol Metab. 2001;86:494–6.
Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab. 2008;295:E254–61.
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–5.
Ogbureke KU, Fisher LW. Expression of SIBLINGs and their partner MMPs in salivary glands. J Dent Res. 2004;83:664–70.
Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. 2011;25:2551–62.
Quarles LD. Evidence for a bone-kidney axis regulating phosphate homeostasis. J Clin Invest. 2003;112:642–6.
Bai X, Miao D, Panda D, Grady S, McKee MD, Goltzman D, et al. Partial rescue of the Hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol Endocrinol. 2002;16:2913–25.
Erben RG, Mayer D, Weber K, Jonsson K, Juppner H, Lanske B. Overexpression of human PHEX under the human beta-actin promoter does not fully rescue the Hyp mouse phenotype. J Bone Miner Res. 2005;20:1149–60.
Liu S, Guo R, Tu Q, Quarles LD. Overexpression of Phex in osteoblasts fails to rescue the Hyp mouse phenotype. J Biol Chem. 2002;277:3686–97.
Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, et al. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol. 2009;23:1505–18.
Jean G, Bresson E, Terrat JC, Vanel T, Hurot JM, Lorriaux C, et al. Peripheral vascular calcification in long-haemodialysis patients: associated factors and survival consequences. Nephrol Dial Transplant. 2009;24:948–55.
Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One. 2014;9:e104154.
Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem. 2015;290:10447–59.
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86:267–72.
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86:273–8.
Chen IP, Wang L, Jiang X, Aguila HL, Reichenberger EJ. A Phe377del mutation in ANK leads to impaired osteoblastogenesis and osteoclastogenesis in a mouse model for craniometaphyseal dysplasia (CMD). Hum Mol Genet. 2011;20:948–61.
Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014;111:5520–5.
Brownstein CA, Adler F, Nelson-Williams C, Iijima J, Li P, Imura A, et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A. 2008;105:3455–60.
Halevy J, Bulvik S. Severe hypophosphatemia in hospitalized patients. Arch Intern Med. 1988;148:153–5.
Zeki S, Culkin A, Gabe SM, Nightingale JM. Refeeding hypophosphataemia is more common in enteral than parenteral feeding in adult in patients. Clin Nutr. 2011;30:365–8.
Yang HT, Yim H, Cho YS, Kim D, Hur J, Kim JH, et al. Change of serum phosphate level and clinical outcome of hypophosphatemia in massive burn patient. J Trauma Acute Care Surg. 2012;73:1298–302.
Pourhassan M, Muller MJ, Volkert D, Wirth R. Hypophosphatemia as a sign of malnutrition in older hospitalized patients. Eur J Clin Nutr. 2018. https://doi.org/10.1038/s41430-018-0251-6.
• Wolf M, Chertow GM, Macdougall IC, Kaper R, Krop J, Strauss W. Randomized trial of intravenous iron-induced hypophosphatemia. JCI Insight. 2018;3(23). https://doi.org/10.1172/jci.insight.124486. This randomized, double blinded controlled trial demonstrated an extremely high incidence (> 50% of patients with serum phosphorus less than 2.0 mg/dl) and persistence (29.1% affected through the end if five weeks) of hypophosphatemia with a single dose of ferric carboxymaltose, but not ferumoxytol, in adults with irone deficiency anemia.
Mateo L, Holgado S, Marinoso ML, Perez-Andres R, Bonjoch A, Romeu J, et al. Hypophosphatemic osteomalacia induced by tenofovir in HIV-infected patients. Clin Rheumatol. 2016;35:1271–9.
Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–101.
•• Che H, Roux C, Etcheto A, Rothenbuhler A, Kamenicky P, Linglart A, et al. Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms. Eur J Endocrinol. 2016;174:325–33 This prospective cohort study demonstrated a worse quality of life in adult patients with X-linked hypophopshatemic rickets compared with age and gender matched patients with axial spondyloarthritis. In addition, treatment with phosphorus and vitamin D was associated with improved mental health scores.
Wilson HK, Keuer SP, Lea AS, Boyd AE 3rd, Eknoyan G. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med. 1982;142:517–20.
Gosmanov AR, Gosmanova EO, Dillard-Cannon E. Management of adult diabetic ketoacidosis. Diabetes Metab Syndr Obes. 2014;7:255–64.
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26:1381–8.
Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ. Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2010;95:1846–50.
Liang G, Katz LD, Insogna KL, Carpenter TO, Macica CM. Survey of the enthesopathy of X-linked hypophosphatemia and its characterization in Hyp mice. Calcif Tissue Int. 2009;85:235–46.
Han X, Quarles LD. Multiple faces of fibroblast growth factor-23. Curr Opin Nephrol Hypertens. 2016;25:333–42.
Acar S, Demir K, Shi Y. Genetic causes of rickets. J Clin Res Pediatr Endocrinol. 2017;9:88–105.
•• Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378:1987–98 Children with XLH treated with the ant-FGF23 antibody burosumab achieved greater growth velocity, healing of radiographic rickets and improved phsyical performance, with greater effects occurring in the currently FDA approved every 2 week subcutanous dosing regimen than every 4 weeks.
Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, et al. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of Burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. J Bone Miner Res. 2018;33:1383–93.
Xiao Z, Riccardi D, Velazquez HA, Chin AL, Yates CR, Carrick JD, et al. A computationally identified compound antagonizes excess FGF-23 signaling in renal tubules and a mouse model of hypophosphatemia. Sci Signal. 2016;9:ra113.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Thomas J. Weber has received research funding, consultant fees and travel support from Ultragenyx.
L. Darryl Quarles has received research funding from Inozyme.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Molecular Control of Phosphorus Homeostasis
Rights and permissions
About this article

Cite this article
Weber, T.J., Quarles, L.D. Molecular Control of Phosphorus Homeostasis and Precision Treatment of Hypophosphatemic Disorders. Curr Mol Bio Rep 5, 75–85 (2019). https://doi.org/10.1007/s40610-019-0118-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-019-0118-1