
Abstract
Diastolic dysfunction (DD) refers to abnormalities in the mechanical function of the left ventricle (LV) during diastole. Severe LVDD can cause symptoms and the signs of heart failure (HF) in the setting of normal or near normal LV systolic function and is referred to as diastolic HF or HF with preserved ejection fraction (HFpEF). Pediatric cardiologists have long speculated HFpEF in children with congenital heart disease and cardiomyopathy. However, understanding the risk factors, clinical course, and validated biomarkers predictive of the outcome of HFpEF in children is challenging due to heterogeneous etiologies and overlapping pathophysiological mechanisms. The natural history of HFpEF varies depending upon the patient’s age, sex, race, geographic location, nutritional status, biochemical risk factors, underlying heart disease, and genetic-environmental interaction, among other factors. Pediatric onset HFpEF is often not the same disease as in adults. Advances in the noninvasive evaluation of the LV diastolic function by strain, and strain rate analysis with speckle-tracking echocardiography, tissue Doppler imaging, and cardiac magnetic resonance imaging have increased our understanding of the HFpEF in children. This review addresses HFpEF in children and identifies knowledge gaps in the underlying etiologies, pathogenesis, diagnosis, and management, especially compared to adults with HFpEF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A working definition of heart failure (HF) in children is “a progressive clinical and pathophysiological syndrome caused by cardiovascular and non-cardiovascular abnormalities that results in characteristic signs and symptoms including edema, respiratory distress, growth failure, and exercise intolerance and accompanied by circulatory, neurohormonal, and molecular derangements [1].” Heart failure can occur with preserved or a reduced left ventricular ejection fraction (LVEF). When a predominant or isolated abnormality in diastolic function accompanies HF symptoms, this clinical syndrome is called diastolic HF or HF with preserved EF (HFpEF). Diastolic dysfunction (DD) is defined as impaired ventricular relaxation and increased ventricle chamber stiffness, which increases cardiac filling pressures, but no overt symptoms of HF.
Diastolic dysfunction includes ventricular twist and deformation (diastolic strain), delay in untwisting, and reduced ventricular suction leading to impairment of early diastolic filling. The ventricular stiffness and impaired diastolic filling raise LV end-diastolic pressure (LVEDP) and left atrial (LA) pressure. Unlike systolic function, assessment of diastolic function in children is challenging because of considerable variability and inconsistency in echocardiographic criteria of DD. Conventionally, Doppler parameters such as mitral valve inflow, pulmonary venous flow, LV systolic-to-diastolic duration ratio, myocardial performance indices, and LA size are used to assess LVDD. However, these conventional noninvasive measures of DD may not be accurate, and the results are poorly correlated with invasive hemodynamics [2]. Recently, novel echocardiographic measures of LV stiffness, such as strain, and strain rate analysis by speckle-tracking echocardiography (STE) and tissue Doppler imaging (TDI), are better correlated with gold standard estimates of DD derived from pressure–volume loop analysis in children [3].
It is estimated that more than half of all adults with HF have HFpEF. Pediatric onset HFpEF is often not the same disease as in adults. Pediatric HFpEF has been described in patients with cardiomyopathy, patients with congenital heart disease (CHD), and patients who received cancer therapies, among other conditions [4,5,6,7,8,9,10]. The etiologies, risk factors, clinical course, biomarkers, and therapies in children with HFpEF may not only be different from adults but even within children. This review paper aims to describe HFpEF in children and identify knowledge gaps in the underlying etiology, pathogenesis, diagnosis, and management compared to adults with HFpEF.
Epidemiology
In 1983, one of the first papers was published that recognized that many adult patients hospitalized for HF had normal LVEF [11]. In another paper, in 1984, Dougherty et al. reported that in 188 adult patients with HF, the EF was ≥ 45% and coined the term HF with normal systolic function [12]. Then, the Acute Decompensated Heart Failure National Registry (ADHERE) reported that 46% of patients with HF had no impairment or mild systolic dysfunction [13]. It is currently estimated that about 6.2 million Americans over the age of 20 years carry an HF diagnosis, with half a million new diagnoses every year [14]. Estimates suggest that by 2030 the prevalence of HF will increase by 46%. Total medical costs in the US alone may exceed $53 billion, with HFpEF patients expected to outnumber HF with reduced EF (HFrEF) [15].
Epidemiological evidence suggests a latent phase (6 years) in which DD is present and progresses in severity before the symptoms of HF arise in adults [16]. Elevated LV stiffness is associated with diastolic filling abnormalities and exercise intolerance. LA pressures increase further when the LVDD progresses, resulting in pulmonary edema and dyspnea, classical features of left-sided HF. In one population-based study, asymptomatic LVDD was found in 21%, and moderate or severe DD was present in 7% of the general population without any signs of HF [17]. Also, the same study showed that both moderate and severe DD were associated with an increased risk of progression to end-stage HF in adults.
There is limited literature on the epidemiology of HFpEF in children beyond the classic phenotypes of hypertrophic cardiomyopathies (HCM) and restrictive cardiomyopathies (RCM). The difficulties in estimating the prevalence of HFpEF in children are due to heterogeneous etiologies, distinct clinical courses, and different genetic and biomarker profiles. Nevertheless, the recent epidemiological study of Acute Decompensated Heart Failure (ADHF) in children admitted to intensive care units (ICU) across 23 centers in the US showed 6% of 26,294 consecutive ICU admissions had either HFrEF or HFpHF requiring continuous vasoactive or diuretic infusion, respiratory support, or mechanical support [18]. The Pediatric Critical Care Consortium data are a resource to evaluate the prevalence of severe pediatric HFpEF requiring ICU admissions.
Maturation of Diastolic Function
Neonatal LVDD is common but often difficult to determine whether it is due to “normal physiology” or “abnormal pathology.” Therefore, it is essential to understand the maturation of diastolic LV function from fetal life to adolescence before appreciating DD in children. The neonatal myocardium is immature with impaired function of the sarcoplasmic reticular and T tubular function. Due to the limited ability to increase stroke volume due to reduced contractility, cardiac output depends predominantly on heart rate. Doppler echocardiography demonstrated sequential changes in the human fetal ventricular filling pattern at different gestational ages [19]. Doppler studies in 238 healthy neonates suggested that impaired LV relaxation and early diastolic filling were prevalent at birth, as noted by the reversal of the E to A wave’s velocity ratio [20]. The effect of heart rate on early LV filling appears to be negligible and does not account for the observed differences in diastolic function in the first two months of infancy [21]. In preterm infants, the maturation of diastolic function is prolonged and limits tolerance to any preload stressors [22]. Therefore, LVDD on echocardiography is relatively common in preterm infants, primarily due to volume overload from a patent ductus arteriosus in an already immature myocardium, while HFpEF is less frequent.
In a study of 121 echocardiograms in 31 infants, profound changes in diastolic function are reported between one week and one-month post-birth [23]. A study of cardiac growth in healthy children using TDI showed a correlation between changes in diastolic function and age [24, 25]. There are distinct age-dependent changes in myocardial responsiveness, structural changes such as hypertrophy and fibrosis, and systemic inflammation due to different comorbidities that are recognized as pivotal for the differences in severity of HFpEF [26].
Etiology of HFpEF in Children
In general, HFpEF in children is a unique phenotype caused by heterogeneous etiologies such as HCM or RCM, CHD, cancer therapy including radiotherapy and chemotherapy, HIV infection, renal failure, and obesity, among many etiologies. In many situations, there is a thin line between LVDD and HFpEF. For example, childhood cancer survivors who had LVDD with restrictive physiology and limited exercise tolerance due to radiation therapy and/or anthracycline therapy years earlier [27]. Patients are often asymptomatic at rest but develop symptomatic restrictive disease with exercise. Steiner et al. [5] pointed out that these children may have asymptomatic HFpEF early, which clinically manifests in later years. The etiologies of HFpEF in children are summarized in Table 1.
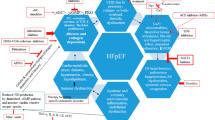
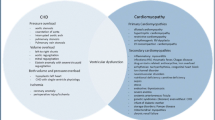
Pediatric HFpEF has heterogeneous and multifactorial elements that come into play (Fig. 1). Factors that can lead to or aggravate HFpEF in adults [28], such as prior CHD surgery, especially after Fontan procedure, valvular diseases, obesity, hypertension, diabetes mellitus, coronary artery disease, metabolic syndrome, severe anemia, chronic kidney disease, radiation or chemotherapy exposures, antiretroviral therapy (ART), and obstructive sleep apnea, are also some of the effectors of DD in children. However, multiple studies have demonstrated substantial differences in patient profiles and risk factors for HFpEF in CHD compared with those with cardiomyopathies and between pediatric and adult patients. Generally, most children with ADHF, either due to cardiomyopathy or CHD, have overlapping systolic dysfunction and DD [29, 30].

Venn diagram illustrating the heterogeneity and multifactorial elements in pediatric heart failure with a preserved ejection fraction
Congenital Heart Disease
The top three cardiovascular causes of HFpEF in children are complex CHD (53.4%), simple CHD (15.7%), and cardiomyopathies (7.4%) [31]. The pathophysiological causes of HFpEF in CHD are multifactorial. They involve ventricular fibrosis, intracardiac scarring, valvular abnormalities, arrhythmias, the presence of cyanosis, or pulmonary hypertension. There are five main classes of CHD, which results in diastolic dysfunction: (1) pressure–overload lesions such as aortic stenosis, (2) volume overload such as mitral regurgitation, (3) mixed pressure and volume overloads, such as aortic stenosis and regurgitation, (4) transposition of the great arteries leading to a unique situation in which the right ventricle (RV) is faced with increased afterload and the LV with a much lower pressure than normal, both of which may cause decreased compliance, and (5) single ventricle CHD, especially after Fontan operation.
Pressure Overload Caused by Left-Sided Lesions
Global and regional deformation of LV occur in response to obstruction of the LV outflow tract (LVOT) [32,33,34,35]. The LV remodels and develops hypertrophy. The LV hypertrophy results in delayed and incomplete relaxation of the LV wall, with decreased early diastolic filling with an increased dependence on late diastolic filling. With myocardial tagging cardiac magnetic resonance (CMR), Nagel et al. [32] showed that LV pressure overload was associated with a reduction in basal rotation and an increase in apical rotation of the heart, indicating increased torsion of the LV in severe aortic valve stenosis. It was also shown that diastolic untwisting was delayed and prolonged in patients with aortic valve stenosis. These findings explained the occurrence of DD in patients with left-sided obstructive lesions. Furthermore, the pressure overload by aortic stenosis causes increased interstitial fibrosis and LV stiffness and is related to a worse prognosis [35].
Volume Overload
Children with LV volume overload due to aortic or mitral regurgitation have shown dysfunction of myocardial contractility [36]. LV compliance usually decreases in isolated mitral regurgitation as the LV dilates to accommodate an increased volume. According to Laplace’s law, as the ventricular radius increases, myocardial wall tension also increases [37]. Over time, LV myocardial fibrosis occurs, resulting in relaxation impairment and DD.
Mixed Pressure and Volume Overload
Mixed pressure and volume overload can affect compliance, such as mixed aortic valve disease with aortic stenosis and aortic regurgitation. Similar to the effect of mixed aortic valve disease on the LV, RVDD can occur in repaired TOF with residual pulmonary valve stenosis and regurgitation. Right ventricular DD can also occur after surgical repair of tetralogy of Fallot (TOF) at intermediate follow-up [38]. More importantly, clinically asymptomatic patients with repaired TOF showed LV systolic dysfunction and DD and a lower than normal LV mass/volume ratio consistent with a pattern of eccentric LV remodeling [39].
Transposition of Great Arteries
Transposition of the great arteries (TGA) is one of the most common cyanotic CHD in neonates. The pulmonary artery (PA) is connected to the LV, and the aorta is connected to the RV. An arterial switch operation (ASO) is performed in TGA to convert the LV into the systemic ventricle, replacing the RV which was the systemic ventricle during the prenatal period. Therefore, the adaptation of LV and RV after ASO is an interesting issue. A study found impairment of the DD of both LV and RV by TDI and conventional Doppler parameters of DD during the short- to mid-term follow-up of pediatric patients who underwent ASO [40]. LV myocardial stiffening also was observed in adolescents and young adults with TGA after ASO and is associated with impairment of LV deformation and increased myocardial fibrosis leading to DD [41]. Reduced proximal aortic elasticity and aortic root dilatation are present in TGA patients post-ASO and are likely to contribute to LVDD. Also, impaired aortic elasticity is associated with age, suggesting it is useful to follow-up TGA patients for early onset LVDD.
Single Ventricle CHD
In the single ventricle CHD patients, myocardial hypertrophy, fibrosis, and DD often result from multiple, cumulative insults, which may include volume loading, pressure loading, chronic hypoxemia, coronary ischemia, chronic upregulation of the renin–angiotensin–aldosterone system, and chronic underfilling [29, 30, 42,43,44].
The Fontan patients with DD and normal EF had clinical evidence of decreased exercise tolerance suggestive of early HFpEF [45]. Diastolic dysfunction appears to be a progressive phenomenon in patients with single ventricles, with elevated filling pressures, sometimes already being present in childhood and deteriorating over time. Michel et al. [46] showed that children who have undergone Fontan surgery for hypoplastic left heart syndrome demonstrated progressive impairment of diastolic function at follow-up. Several factors may contribute to this impairment, including chronic deprivation of preload to the single ventricle with insufficient ventricular filling and resultant progressive myocardial stiffness. Alternatively, myocardial fibrosis may be a progressive phenomenon contributing to diastolic dysfunction. Amino-terminal procollagen type III, a surrogate marker of fibrosis, is more prevalent with impaired diastolic function in patients with dilated cardiomyopathy [47,48,49]. Hypoxemia and volume overload before the Fontan procedure are associated with elevated amino-terminal procollagen type III levels, and pre-Fontan amino-terminal procollagen type III levels correlate significantly with post-Fontan end-diastolic pressures [49].
Cardiomyopathies
Among all types of cardiomyopathies, HCM and RCM are two common causes of DD in children. Left ventricular hypertrophy induces LVDD [50]. Varying degree of DD is common in children with HCM with or without LVOT obstruction. The etiology of DD in HCM is multifactorial. At the molecular level, changes in Ca2+ affinity between the various contractile proteins, and at the tissue level, myocyte hypertrophy, myocyte disarray, and endocardial and interstitial fibrosis contribute to DD in HCM patients [51]. On the other hand, DD with normal to near normal systolic performance and little or no increase in end-diastolic or end-systolic dimensions of either RV or LV is the primary abnormality in RCM. The DD in RCM can result from myocardial or endomyocardial disease, the etiologies of which may be known or idiopathic. Diastolic dysfunction that is essentially myocardial can be idiopathic, infiltrative (myocardial interstitium), or within myocardial cells (storage diseases). Diastolic dysfunction that results from endomyocardial disease is typified by endomyocardial fibrosis or the hypereosinophilic syndrome, although carcinoid, metastatic malignancies, radiation, and anthracycline toxicity may be accompanied by endomyocardial restriction [52]. The DD in RCM leads to impaired ventricular filling affecting either or both ventricles and may result in HFpEF.
Heart Transplantation
Diastolic dysfunction is a well-recognized complication early after heart transplantation. After the first few days to months after heart transplantation, the right atrial (RA) pressure and LA pressure are often elevated, reflecting decreased compliance of the transplanted heart. This early diastolic impairment usually improves due to a general reduction of inflammation within the allograft. The incidence of DD gradually decreases within the first year after heart transplantation. Diastolic dysfunction often progresses to HFpEF after a few years of heart transplantation due to coronary microvascular dysfunction, interstitial fibrosis, and increased myocardial stiffness [53,54,55]. Children with mild cardiac allograft vasculopathy detected by angiography and HFpEF are at increased risk of graft loss or death [55]. Pulmonary capillary wedge pressure (PCWP) measured at rest and after exercise revealed a high prevalence of exercise-induced HFpEF after heart transplantation [56]. Early treatment with sirolimus is associated with improved diastolic function and filling pressures than calcineurin-inhibitor therapy [57]. The exact mechanism of progression from DD to HFpEF in post-transplanted hearts is unknown. Allograft vasculopathy, endothelial dysfunction, low-grade inflammation, epicardial and endocardial fibrosis, chronotropic incompetence, and immunological mechanisms are suggested as possible mechanisms [58].
Sepsis
Myocardial dysfunction is seen in patients with sepsis and carries significant mortality compared with those who do not have myocardial dysfunction [59]. Recent studies have demonstrated isolated DD and HFpEF in children with sepsis [60,61,62]. In children with sepsis-induced DD, this is reported to be an independent risk factor for poor outcomes [63].
Human Immunodeficiency Virus Infection
Cardiomyopathy and subclinical cardiac abnormalities have been reported in children and adolescents infected with the human immunodeficiency virus (HIV) [64, 65]. HIV infection is a primary cause of acquired heart diseases, notably accelerated atherosclerosis, symptomatic HF, and pulmonary arterial hypertension (PH). Cardiac complications often occur in late-stage HIV infections as the prolonged viral infection becomes more relevant with improved longevity. Thus, multi-agent HIV therapies that help sustain life may also increase the risk of cardiovascular events. In recent years, prospective studies in HIV-positive patients on combined antiretroviral therapy (ART) have reported a higher prevalence of DD, elevated LV mass index, accelerated atherosclerosis, and dysautonomia [66, 67]. Children exposed to ART in utero have subclinical yet significant differences in specific LV diastolic indices. Follow-up with serial echocardiograms is recommended in this population to assess further the potential cardiac toxicity of perinatal exposure to ART [68].
The cardiometabolic abnormalities in HIV-infected children include high rates of unfavorable lipid profiles, insulin resistance, cardiovascular inflammation, vascular stiffness, and the phenotypic features of truncal adiposity and facial/extremity wasting [69]. Children who received ART prenatally may have reduced LV mass, LV dimension, and septal wall thickness z-scores and increased LV fractional shortening and contractility up to age two years [70]. Cardiac structure and function were relatively preserved in perinatally HIV-infected children exposed to highly active antiretroviral therapy (HAART) but declined over time when compared to those of similar children from the pre-HAART era [71]. The perinatal exposure to HAART therapies blocks normal cardiac growth resulting in hearts being too small for body surface area as they grow older.
Cancer Therapy
A continuum of changes starting with cardiac biomarker increase, myocardial structural deformation, and asymptomatic LVDD occurs during chemotherapy leading to HF in children and adults [9, 72]. Children treated with doxorubicin had decreased wall thickness relative to somatic growth compared to normal healthy children at a median follow-up of 11.8 years [7]. Cardiac abnormalities are persistent and progressive after doxorubicin therapy. Inadequate ventricular mass with chronic afterload excess was associated with the progressive contractile deficit and possibly reduced cardiac output due to restrictive physiology [73]. Typically, adult number of myocytes is present before the first year of life, and subsequent myocardial growth occurs by increasing the size of the cells. Therefore, damage to or loss of these cardiomyocytes might impair the heart's ability to generate an average adult myocardial mass. During long-term follow-up after completing doxorubicin treatment, a particular pattern of cardiac dysfunction has been noted in children. Early on, doxorubicin-treated children appeared to have dilated-like cardiomyopathy, which seemed to resolve but instead progressed to restrictive-type cardiomyopathy in later years [74]. The progressive restrictive-like cardiomyopathy was persistent because of a progressive fall in LV mass and cavity size that became inadequate for body size. Cardiomyopathy, marked by shrinking myocardial mass and cavity size for body size (“Grinch Syndrome”), appears to be a long-term risk in this population [75].
Myocardial injury induced by chemotherapies in children with malignancy starts with subclinical LVDD and increases cardiac biomarkers before progression to more advanced HFpEF or HFrEF, similar to some adult cancer patients, even though the subsequent course of progressive late cardiotoxicity in survivors may differ [76, 77]. Serum biomarkers of myocardial injury and increased myocardial stress during anthracycline therapy for childhood cancer are associated with abnormal LV structure and function years later as long-term survivors [78]. In pediatric Hodgkin lymphoma patients treated with mediastinal radiation therapy, DD during follow-up resulted in the loss of physical functioning as monitored by the Short-Form 36 quality-of-life instrument [9]. Alehan et al. [79] screened survivors of childhood Hodgkin lymphoma with Doppler echocardiography and confirmed that DD is common after anthracycline therapy, even without radiotherapy. Their findings are even more remarkable when considering that the median dose of doxorubicin was only 150 mg/m2 and that 69% of patients received < 300 mg/m2, a cut-off used to designate those at the highest risk for systolic dysfunction. Dexrazoxane, a cardioprotective iron chelator that reduces mitochondrial iron levels in children receiving anthracycline without reducing chemotherapeutic efficacy, is approved for this indication with increasing use based on multiple pediatric clinical studies demonstrating long-term cardioprotection [80, 81]. Pediatric cardio-oncology issues related to cardiac structure and function have been comprehensively reviewed and available in the literature for those interested in details [81,82,83].
Chronic Renal Failure
Elevated cardiac troponin T levels identify subclinical cardiac damage in patients with chronic renal failure without coronary artery diseases and are associated with depressed LV load-independent contractility [84]. Recently, TDI in 128 patients with chronic kidney disease showed significant LVDD compared to healthy children [85]. Early identification of LVDD in patients with uremia may prevent further progression to HFpEF and reduce cardiovascular morbidity and mortality in this high-risk population [86]. The underlying pathophysiological mechanisms of LVDD are LV hypertrophy with myocardial interstitial fibrosis due to chronic volume overload and uncontrolled hypertension. Several other factors, including secondary hyperparathyroidism, over-activation of the renin-angiotensin-aldosterone system, and inflammation, may influence LVH [87, 88]. LVDD causes an increase in LV filling pressure, which may lead to pulmonary congestion.
Obesity
Diastolic dysfunction is common in children with obesity or overweight [89, 90]. Obesity adversely affects morbidity and mortality in children with HF [91]. Many overweight children had three or more metabolic syndrome risk factors, indicating that being overweight in early adolescence may put children at risk for adult-onset cardiovascular disease and type 2 diabetes before they become teenagers [92]. Early bariatric surgical intervention not only improves body mass index but, more importantly, also improves cardiovascular and metabolic comorbidities of severe obesity, including HFpEF in adolescents [93]. Studies have also demonstrated LVDD in obstructive sleep apnea in obese and non-obese children and young adults [94, 95]. Obese children and adolescents often have prediabetes, a risk factor for LVDD leading to HFpEF [96,97,98].
Hemolytic Anemia
LV hypertrophy and LVDD in children with hereditary hemolytic anemia, sickle cell anemia, and thalassemia have been well documented [99, 100]. About one-third of children with sickle cell anemia had TDI evidence of LVDD, which correlated with hemoglobin levels [101]. Adding serial assessments of LV function with TDI may detect early DD, especially in children with severe anemia. In a study by Nanjegowda et al., a higher serum ferritin level (greater than 2076 ng/mL) is associated with an increased incidence of LVDD [102]. Diastolic dysfunction has been reported in children with Kawasaki disease [103], Marfan syndrome [104], juvenile rheumatoid arthritis [105], systemic lupus erythematosus [106], and other rare non-cardiovascular etiologies [107].
Pathophysiology and Mechanism of HFpEF in Children
Pediatric HFpEF is a complex heterogeneous clinical finding for which our pathophysiological understanding is limited. Recently, experimental models of HFpEF have demonstrated an interaction between metabolic stress and chronic inflammation, resulting in alterations in systemic and cardiac immune responses that contribute to HFpEF pathophysiology [108]. There is also evidence of elevated circulating inflammatory biomarkers such as interleukin-1, C-reactive protein, tumor necrosis factor-α, and soluble suppression of tumorigenesis-2 in HFpEF. Inflammatory cells express transforming growth factor β, interferon-γ, Galectin-3, connective tissue growth factor, and ACE, promoting the conversion of fibroblasts to myofibroblasts and collagen deposition [109].
The myocardial response to biochemical stress is different between pediatric and adult HF patients [110]. Differences in pediatric and adult HFrEF are also supported by a lack of adequate response to adult HF therapies [111,112,113,114]. Titin is a sarcomere protein and has been recognized as a determinant of myocardial relaxation. Titin is expressed in two isoforms: a smaller, stiffer N2B, and a larger, more compliant N2A. Nagueh et al. found an increase in the N2B:N2A titin isoform ratio compared with healthy controls in children with DCM [115]. This shift to a smaller (N2B) titin isoform can cause myocardial stiffness and DD. Moreover, titin is modulated by phosphorylation. The cGMP-protein kinase (PK) G-dependent pathway has been suggested to play an important role in regulating diastolic relaxation through titin phosphorylation and troponin I phosphorylation [116]. In pre-clinical models, many cardiac myosin-binding protein C mutations increase DD by increasing myofilament Ca2+ sensitivity and cross-bridge turnover between actin and myosin filaments [117, 118]. The pathophysiology of pediatric HFpEF depends upon age-related myocardial responsiveness, energy metabolism at the cellular level, the systemic inflammatory response, and the interaction of genes with environmental elements [16].
In children with CHD, remodeling processes in the LV depend upon hemodynamic stressors: pressure overload, volume overload, and pressure and volume overload [119, 120]. There are overlapping changes in the LV in CHD and cardiomyopathies due to hemodynamic overload, including collagen deposition, increased extracellular matrix deposition, myocardial and perivascular fibrosis, activation of the local tissue renin-angiotensin, secretion of pro-inflammatory chemokines, altered neurohormonal response, and increased matrix metalloproteinase activity (including collagenase) [121,122,123]. In addition, the impairment of sarcoplasmic Ca2 + -ATPase activity is a significant determinant of myocardial stiffness, leading to LVDD and ultimately to HFpEF [124]. The extent of fibrosis differs depending on the physiological age. Understanding these developmental differences in cardiomyocytes’ response to injury has important therapeutic implications [125].
In an autopsy study, Perens et al. found that in children with end-stage HF who underwent heart transplantation, the explanted hearts showed interstitial and perivascular fibrosis in most cases [126]. The authors also examined their failed pediatric allografts and found a different pattern of severe epicardial fibrosis and relatively sparing of the myocardium, which could explain the HFpEF with relatively preserved systolic function after heart transplantation.
Masutani et al. reported that children with HFpEF had abnormal systolic and diastolic function at rest, arterial stiffening, and dissociation of ventricular-vascular coupling at baseline long before the development of HF [127]. In addition, they had limited diastolic reserve capacity in response to enhanced preload and inotropic, lusitropic, and ventricular–arterial coupling responses to beta-adrenergic stimulation. Furthermore, children with HFpEF also had an abnormal coronary supply/demand balance, associated with ventricular diastolic stiffening and poor beta-adrenergic responsiveness. These findings in children support shared overlapping pathways in the pathogenesis of HFpEF in both children and adults [128, 129]. Pathophysiologic mechanisms that contribute to the development of HFpEF in both children and adults include systemic inflammation, endothelial dysfunction, decreased nitric oxide production, increased reactive oxygen species, increased sensitivity of the myocardium to Ca2+, cardiomyocyte hypertrophy, and ultimately deposition of interstitial and perivascular collagen deposition leading to fibrosis [42, 121]. These shared pathophysiological processes speculated to develop HFpEF in children are summarized in Fig. 2.

Pathophysiology of HFpEF in children (TNF-α = tumor necrosis factor-α; IL-6 = interleukin-6; IL-1β = interleukin-1β; sST2 = soluble ST2 (receptor for IL-1 and 33); NO = nitric oxide; ROS = reactive oxygen species; cGMP = cyclic guanosine monophosphate; PKG = protein kinase G; SNS = sympathetic nervous system; RAAS = renin–angiotensin–aldosterone system)
In HFpEF, concentric remodeling and increased stiffness of the LV lead to an increase in LA pressure to maintain the gradient of transmitral pressure that allows LV filling. The elevated LA pressure, in turn, causes passive PH. In addition, increased LA pressure leads to alveolar capillary stress failure and vascular remodeling, further increasing pulmonary vascular resistance and a mixed type of pre- and post-capillary PH. Compared to patients with pure passive PH (i.e., caused by isolated LA hypertension or post-capillary PH). HFpEF patients are at increased risk of death due to structural remodeling in the lung vasculature that may not be reversible after the management of LVDD alone. Patients with HFpEF and pulmonary vascular disease display unique pathophysiology. The inability to enhance RV ejection increases right heart enlargement and LV underfilling, even though LV filling pressure is elevated. Even among patients with HFpEF who display normal pulmonary vascular resistance at rest, there is inadequate pulmonary vasodilation during exercise in HFpEF. This is associated with adverse clinical outcomes and impairment in RV functional reserve. The development of RV dysfunction identifies patients with HFpEF with a markedly increased risk of death [130]. The RV is dilated in response to chronic pressure overload, functional tricuspid incompetence, and further RV dilation progression, ultimately resulting in RV failure.
Diagnosis of HFpEF in Children
Making a diagnosis of HFpEF in children remains a challenge. The diagnosis of HFpEF requires history, clinical symptoms, signs of HF, and evidence of DD with normal or mildly reduced LVEF. It is also important to exclude other medical conditions like lung diseases and PH, which may mimic HFpEF. Elevated natriuretic peptides support, but normal levels do not exclude a diagnosis of HFpEF. Pediatric and adult dilated cardiomyopathy patients display distinct biomarker profiles, and pediatric-specific biomarkers may be more useful in future [131].
Cardiac Catheterization
Cardiac catheterization measurements of elevated PCWP, which reflects LV filling pressure, can diagnose HFpEF at rest but may be more apparent after an intravenous fluid challenge. In the early stages of LVDD, mildly elevated LVEDP is the only abnormality noted. With tachycardia and/or increased LV afterload with intravenous Dobutamine, mean PCWP and LA pressure increase becomes more prominent, providing the basis for the diastolic stress test. Changes in PCWP, an indirect estimate of LV diastolic pressure, during exercise can be assessed by right heart catheterization through the right internal jugular vein. Right heart catheterization is also useful to differentiate between pre-, post-, and mixed-type PH. Hemodynamics including mean PA pressure, diastolic PA pressure, pulmonary vascular resistance, and vasodilator testing are crucial for the management of PH associated with HFpEF.
Left heart catheterization measures the time constant, tau (τ), representing the LV relaxation delay. When the log of the diastolic pressure is plotted as a time function, τ is equal to the inverse of the slope of this linear relationship. When τ > 48 ms, it reflects significant LVDD. Thus, τ is considered the best index for evaluating LV relaxation [132]. However, performing a cardiac catheterization is an invasive procedure, and serial cardiac catheterization is not suitable for clinical surveillance for HFpEF in children.
Echocardiography
Echocardiography is the primary testing modality for diagnosing LVDD in children. Based on the conventional pulsed Doppler study of the mitral inflow, there are four phases of diastole: Phase1: isovolumic relaxation, Phase 2: Rapid passive filling (E wave), Phase 3: Slow filling and mild stasis, and Phase 4: Active filling (A wave) due to atrial contraction. According to the 2016 American Society of Echocardiography (ASE) guidelines for adults, four variables are obtained to assess LVDD: E′ velocity, E/E′ ratio, LA volume indexed to body surface area, and tricuspid regurgitation peak velocity [133]. The 2016 ASE criteria have simplified the echocardiographic assessment of diastolic function and reduced inconclusive diagnoses [134]. However, the adult echocardiographic guidelines are yet to be validated in children with cardiomyopathy and CHD [135, 136].
Tissue Doppler Imaging
The LV diastolic function is better determined by TDI evaluating the longitudinal movement at the mitral, tricuspid, and septal annulus levels, calculating early (E') and late diastolic (A') velocities, respectively. Normal TDI parameters for children are available for comparison [25]. TDI-Based myocardial deformation imaging evaluate LV stiffness and LVDD in children with CHD. Lateral E:E'/end-diastolic volume (EDV) appeared to correlate well with LVEDP obtained by simultaneous cardiac catheterization in pediatric patients after Fontan Surgery [137]. TDI has also been proven to be helpful in children with dilated cardiomyopathy to predict cardiac events [138, 139], characterize LV diastolic function in LV non-compaction cardiomyopathy in children [140, 141], estimate filling pressures in HCM and RCM in both children and adults [142,143,144,145], and to distinguish RCM from constrictive pericarditis in children and adults [146, 147].
Speckle Tracking Echocardiography
In adult patients, two-dimensional STE has been applied to assess LA function, and LA strain (LAS) has recently been proposed as a novel technique to be used in clinical practice [147,148,149]. Strain imaging aims to provide a high-resolution, real-time measure of LV contractility and relaxation in children with normal loading conditions [150,151,152]. Strain imaging can be used to distinguish between restrictive cardiomyopathy and constrictive pericarditis [153], as well as helping to distinguish the athletic heart from HCM [154]. The three-dimensional STE has proven to be a relatively accurate and efficient method for assessing early abnormalities in LV systolic deformation (peak LV global longitudinal, circumferential, and radial strain); abnormalities in strain analysis are known to predict adverse outcomes in HFpEF [155, 156]. Diastolic strain and strain rates measured by STE are reduced in adult patients with HFpEF [157]. The STE imaging has also been shown to correlate with invasively measured pressure–volume loops, which might achieve an accurate diagnosis [157, 158]. Some data are available on LAS in children to be helpful for the assessment of LV filling pressures and LVDD [159, 160]. All components of the LAS decline modestly with age. Ghelani et al. have reported normative data for 3-dimensional echocardiography-derived LAS, which may be very useful in comparing LAS obtained in children with HFpEF [161].
Cardiac Magnetic Resonance (CMR) Imaging
CMR imaging is the modality of choice in diagnosing and following up cardiomyopathy patients. Myocardial tissue characterization enables the evaluation of the presence and extent of myocardial fibrosis [162, 163]. After administration of intravenous gadolinium-based contrast, late gadolinium enhancement (LGE) is the gold standard noninvasive tool for detecting focal areas of fibrosis and myocardial disarray. The LGE may not detect diffuse myocardial fibrosis. Parametric tissue mapping with myocardial T1 relaxation time and extracellular volume evaluation helps detect diffuse fibrosis even in the absence of focal LGE [162].
The abnormalities in LGE, T1 relaxation times, and extracellular volume are vital findings in differentiating HCM from athlete’s heart or hypertensive heart disease [163]. In asymptomatic or mildly symptomatic HCM patients, the presence of LGE was associated with ventricular arrhythmia detected on Holter monitoring [164]. With better visualization, CMR can assess additional phenotypic features such as septal hypertrophy, apical hypertrophy, sigmoidal and reverse curve HCM, involvement of specific LV segments, involvement of papillary muscles, presence of myocardial crypts, aneurysm, presence of dynamic LVOT obstruction and LA dilation. CMR is also the gold standard in assessing LA and LV volumes, biventricular function, and LV myocardial mass [165]. Specifically, maximum wall thickness, focal or diffuse fibrosis, LVOT obstruction, and LV mass are markers for risk stratification in HCM. Quantification of LGE (% of myocardial mass involved) provides additional prognostic values. Every 10% increase in LGE was associated with a 36% relative risk for sudden cardiac death. CMR evidence of myocardial fibrosis with T1 imaging in Fontan patients relates to DD, increased liver stiffness, and elevated circulating fibrosis biomarkers [42]. The CMR with T2* imaging is helpful as a noninvasive diagnostic tool for quantifying myocardial iron overload in children with hemochromatosis [166]. Hence, multimodality imaging such as echocardiography and CMR provides important information to assess the etiology of HFpEF.
Chronic hypoxemia, altered loading conditions, and genetic predisposition have been linked to diffuse myocardial fibrosis in CHD [167]. The application of CMR is crucial in evaluating DD in children with CHD, such as TOF [168], single ventricle physiology with systemic RV [169], after Fontan [170], and congenital aortic valve stenosis [171]. While the pediatric CMR community is moving toward implementing parametric mapping, challenges remain, the most important being the lack of standardization in pediatric parametric mapping. However, the Society of CMR has published a consensus recommendation in its 2020 update on reporting results of parametric mapping in clinical practice [172].
Treatment of HFpEF in Children
The treatment for HFpEF in children is extrapolated from adult clinical trials and is predominantly used for symptomatic relief. Diuretics are commonly used to relieve congestion in HFpEF. Large clinical trials testing neurohormonal inhibition failed to reach positive outcomes in children [107,108,109]. Theoretically, an ideal drug should facilitate myocardial relaxation and improve ventricular compliance. The drug should relieve congestion, increase cardiac output, improve quality of life, and decrease hospitalization and cardiac-related events, including death or heart transplantation. However, currently, there is no such ideal pharmacological agent available. Lifestyle factors such as poor diet, obesity, lack of physical activity, smoking, heavy alcohol consumption, and increased emotional stress likely contributes for development of the HFpEF in the younger age group [173]. Physical inactivity and poor cardiopulmonary fitness are modifiable risk factors. Exercise training and rehabilitation have been proven helpful in both HFpEF and HFrEF. Treatment of HFpEF must include lifestyle changes and management of comorbidites such as hypertension, diabetes, obesity, chronic renal failure, anemia, and chronic obstructive pulmonary diseases. The noncardiac comorbidities cause a chronic systemic inflammatory response and increase oxidative stress, are major factors that promote the development of HFpEF pathologies.
In children who receive cancer therapy, it is crucial to identify patients at high risk of cardiotoxicity. Early detection of systolic dysfunction or DD after initiation of cancer therapy with the use of newer echocardiographic modalities and use of cardioprotectives such as dexrazoxane in high-risk cancer patients will help minimize cardiac dysfunction. Oncology patients should be referred to cardiology for evluation and treatment for cardiac dysfunction before, during and after chemotherapy.
While most HFpEF treatment predominantly blocks neuroendocrine activation, newer therapies target inflammation and oxidative stress pathways and may be more effective in the future.
Pharmacologic Therapy
Diuretics are commonly used to relieve congestion in HFpEF. Betablockers are helpful by decreasing heart rate, and increasing diastolic duration and reducing myocardial oxygen consumption in patients with HFpEF [174]. Milrinone is a phosphodiesterase-3 inhibitor, that increaes cAMP, enhances myocardial contractility, promotes myocardial relaxation and increases cardiac output. Prophylactic intravenous use of high-dose milrinone after cardiac surgery in children significantly reduced the prevalence of low cardiac output syndrome [175]. Angiotensin-converting enzyme inhibitors [176], angiotensin receptor blockers [177], and calcium channel blockers [177,178,179,180] are only of limited use for symptomatic relief in pediatric HFpEF. However, spironolactone may be effective in HFpEF treatment [180,181,182,183,184]. The beneficial effect of spironolactone may be due to the afterload reducing impact, changes in serum electrolytes (potassium-sparing effect), reduction in LV mass, and the anti-fibrotic action on the myocardium. New therapies have been developed to interfere with the mediator pathways of HFpEF at the cellular and molecular level, including soluble guanylate cyclase modulators and angiotensin receptor-neprilysin inhibitors (ARNI) [185, 186]. ARNI is highly beneficial in treating adult HFrEF patients, but the PARAGON-HF trial revealed that it does not significantly lower the rate of total hospitalizations for HF and death from cardiovascular diseases in HFpEF patients [187]. A new class of sodium glucose transporter protein 2 (SGLT2) inhibitor drugs is beneficial in various trials in adults across the HF spectrum, including HFpEF [188].
Surgical Therapy
In pediatric patients with CHD, acquired, and congenital myopathies presenting with HF, the treatment should be directed at the underlying etiology and precipitant causes and individualize the therapeutic approach. For those with valvular diseases causing volume overload, such as in TOF, early intervention, including pulmonary valve replacement, may be beneficial. A similar strategy should be used for chronic aortic or mitral regurgitation associated with LV dysfunction. Pediatric patients with LVOT obstructive lesions causing LV hypertrophy and dysfunction would benefit from surgical or interventional catheterization relief of the mechanical obstruction. Restrictive cardiomyopathy is usually progressive and may need earlier consideration for cardiac transplantation. Patients with HCM may benefit from the ease of relief of outflow obstruction through pharmacological or surgical interventions and automated internal cardioverter placement to prevent sudden death due to cardiac arrhythmias. Constrictive pericarditis is a form of HFpEF as a fibrotic, thickened, and adherent pericardium restricts the diastolic filling of the heart. The symmetrical constricting effect of the pericardium results in elevation and equilibrium of diastolic pressures in all four cardiac chambers. Surgical resection of the pericardium results in hemodynamic and symptomatic improvement in patients with constrictive pericarditis. The role of left ventricular assist devices (LVAD) is limited in pediatric patients with HFpEF [189, 190]. Prior studies in children reported that when LVAD was used in HF patients due to HCM or RCM, their mortality rate was comparable to those with dilated or ischemic cardiomyopathy [191]. However, complications including right heart failure, prolonged inotropic use, and central venous catheter infections are more common in patients with RCM and HCM treated with LVAD [191, 192].
While LVAD implantation is technically feasible in patients with severe restrictive physiology and HFpEF, it requires additional thoughts for patient selection, cannulation strategy, and monitoring for PH and RV failure. Patients may also have a small LV cavity in HCM and RCM, raising concern for inflow obstruction and suction events with typical LV apex cannulation [193]. Also, in the HCM and RCM patients, apical cannulation may not be ideal due to contraction around the inflow cannula compromising flow. Alternatively, atrial cannulation may be considered. This strategy also avoids ventriculotomy and cardiopulmonary bypass [194].
Conclusions
The increase in LVEDP represents the final common pathway to HF in children, either due to HFpEF or HFrEF. However, importantly, there may be a rapid progression of PH and RV failure with HFpEF. For this reason, the evaluation of LV diastolic function in children is an important first stage in diagnosis, monitoring of therapy, and prognosis in HFpEF. Echocardiography is the primary tool to assess LVDD. However, heterogeneity of pediatric patient diagnoses, age at presentation, and anatomy of CHD require pediatric-specific DD criteria for early diagnosis of HFpEF in children. To better define pediatric HFpEF, pediatric cardiologists should use CMR imaging modalities and novel echocardiographic parameters, including STE, TDI, and LAS, that may reliably predict prognosis and avoid those twin traps of either overtreatment or therapeutic nihilism. There is a need for research to organize multicenter studies to standardize the newer echocardiographic modalities and CMR imaging criteria to diagnose pediatric HFpEF. In conclusion, a significant understanding of the basic mechanisms of pediatric LVDD and therapies for adults with HFpEF exists. Clinical trials evaluating adult HFpEF therapies in children are needed to establish their safety and efficacy. Until then, diuretics remain as mainstay of therapy for symptomatic treatment reducing symptoms of congestion in children with HFpEF.
References
Hsu DT, Pearson GD (2009) Heart failure in children: part I: history, etiology, and pathophysiology. Circ Heart Fail 2:63–70
Dragulescu A, Mertens L, Friedberg MK (2013) Interpretation of left ventricular diastolic dysfunction in children with cardiomyopathy by echocardiography: problems and limitations. Circ Cardiovasc Imaging 6:254–261
Chowdhury SM, Butts RJ, Hlavacek AM et al (2018) Echocardiographic detection of increased ventricular diastolic stiffness in pediatric heart transplant recipients: a pilot study. J Am Soc Echocardiogr 31:342-348.e1
Masutani S, Saiki H, Kurishima C, Ishido H, Tamura M, Senzaki H (2013) Heart failure with preserved ejection fraction in children: hormonal imbalance between aldosterone and brain natriuretic peptide. Circ J 77:2375–2382
Steiner R (2013) Increasing exercise in long-term survivors of pediatric cancer and their siblings: should treatment be a family affair? Pediatr Blood Cancer 60:529–530
Lipshultz SE, Colan SD, Gelber RD et al (1991) Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med 324:808–815
Lipshultz SE, Lipsitz SR, Sallan SE et al (2005) Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol 23:2629–2636
Chang HY, Lee CH, Su PL et al (2021) Subtle cardiac dysfunction in lymphoma patients receiving low to moderate dose chemotherapy. Sci Rep 11:7100. https://doi.org/10.1038/s41598-021-86652-x
Adams MJ, Lipsitz SR, Colan SD et al (2004) Cardiovascular status in long-term survivors of Hodgkin’s disease treated with chest radiotherapy. J Clin Oncol 22:3139–3148
Adams MJ, Ng AK, Mauch P, Lipsitz SR, Winters P, Lipshultz SE (2015) Peak oxygen consumption in Hodgkin’s lymphoma survivors treated with mediastinal radiotherapy as a predictor of quality of life 5 years later. Prog Pediatr Cardiol 39:93–98
Echeverria HH, Bilsker MS, Myerburg RJ, Kessler KM (1983) Congestive heart failure: echocardiographic insights. Am J Med 75:750–755
Dougherty AH, Naccarelli GV, Gray EL, Hicks CH, Goldstein RA (1984) Congestive heart failure with normal systolic function. Am J Cardiol 54:778–782
Adams KF, Fonarow GC, Emerman CL et al (2005) Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 149:209–216
Virani SS, Alonso A, Aparicio HJ et al (2021) Heart disease and stroke statistics-2021 update: a report from the American Heart Association. Circulation 143:e254–e743
Ziaeian B, Fonarow GC (2016) Epidemiology and etiology of heart failure. Nat Rev Cardiol 13:368–378
Kane GC, Karon BL, Mahony DW et al (2011) Progression of left ventricular diastolic dysfunction and the risk of heart failure. JAMA 306:856–863
Redfield MM, Jacobsen SJ, Burnett JC Jr, Mahoney DW, Bailey KR, Rodeheffer RJ (2003) Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA 289:194–202
Lasa JJ, Gaies M, Bush L et al (2020) Epidemiology and outcomes of acute decompensated heart failure in children. Circ Heart Fail 13:e006101
Miyake T (2001) Doppler echocardiographic studies of diastolic cardiac function in the human fetal heart. Kurume Med J 48:59–64
Schmitz L, Xanthopoulos A, Koch H, Lange PE (2004) Doppler flow parameters of left ventricular filling in infants: how long does it take for the maturation of the diastolic function in a normal left ventricle to occur? Pediatr Cardiol 25:482–491
Mori K, Nakagawa R, Nii M et al (2004) Pulsed wave Doppler tissue echocardiography assessment of the long axis function of the right and left ventricles during the early neonatal period. Heart (British Cardiac Society) 90:175–180
Schmitz L, Stiller B, Koehne P, Koch H, Lange PE (2004) Doppler echocardiographic investigation of left ventricular diastolic function in preterm infants with and without a patent ductus arteriosus. Klin Paediatr 216:36–40
de Waal K, Costley N, Phad N, Crendal E (2019) Left ventricular diastolic dysfunction and diastolic heart failure in preterm infants. Pediatr Cardiol 40:1709–1715
Erickson CT, Meyers B, Li L et al (2021) Progression of left ventricular diastolic function in the neonate and early childhood from transmitral color M-mode filling analysis. Pediatr Res 89:987–995
Eidem BW, McMahon CJ, Cohen RR et al (2004) Impact of cardiac growth on Doppler tissue imaging velocities: a study in healthy children. J Am Soc Echocardiogr 17:212–221
Tini G, Cannata A, Canepa M et al (2021) Is heart failure with preserved ejection fraction a “dementia’ of the heart? Heart Fail Rev. https://doi.org/10.1007/s10741-021-10114-9
Miller AM, Lopez-Mitnik G, Somarriba G et al (2012) Exercise capacity in long-term survivors of pediatric cancer: an analysis from the Cardiac Risk Factors in Childhood Cancer Survivor Study. Pediatr Blood Cancer 60:663–668
Sagel PS, Tawil JN, Boettcher BT et al (2021) Heart failure with preserved ejection fraction: a comprehensive review and update of diagnosis, pathophysiology, treatment, and perioperative implications. J Cardiovasc Anaesth 35:1839–1859
Hinton RB, Ware SM (2017) Heart failure in pediatric patients with congenital heart disease. Circ Res 120:978–994
Fahed AC, Roberts AE, Mital S, Lakdawala NK (2014) Heart failure in congenital heart disease: a confluence of acquired and congenital. Heart Fail Clin 10:219–227
Pan B, Hu D, Sun H, Lv T, Xu W, Tian J (2022) Pediatric diastolic heart failure: clinical features description of 421 cases. Front Pediatr 2022(10):846408
Nagel E, Stuber M, Burkhard B et al (2000) Cardiac rotation and relaxation in patients with aortic valve stenosis. Eur Heart J 21:582–589
Friedman KG, McElhinney DB, Rhodes J et al (2013) Left ventricular diastolic function in children and young adults with congenital aortic valve disease. Am J Cardiol 111(2):243–249
Milano AD, Faggian G, Dodonov M et al (2012) Prognostic value of myocardial fibrosis in patients with severe aortic valve stenosis. J Thorac Cardiovasc Surg 144(4):830–837
Moskowitz WB, Schieken RM, Mosteller M, Bossano R (1990) Altered systolic and diastolic function in children after “successful” repair of coarctation of the aorta. Am Heart J 120:103–109
Gentiles TL, Colan SD, Wilson NJ, Niosa R, Neutze JM (2001) Left ventricular mechanics during and after acute rheumatic fever: contractile dysfunction is closely related to valve regurgitation. J Am Coll Cardiol 37:201–207
Stillwell GK (1973) The law of laplace. Some clinical applications. Mayo Clin Proc 48:863–869
Yasuoka K, Harada K, Toyono M et al (2004) Tei index determined by tissue Doppler imaging in patients with pulmonary regurgitation after repair of tetralogy of Fallot. Pediatr Cardiol 25:131–136
Andrade AC, Jerosch-Herold M, Wegner P et al (2019) Determinants of left ventricular dysfunction and remodeling in patients with corrected Tetralogy of Fallot. J Am Heart Assoc 8:e009618
Öner T, Özdemir R, Güven B et al (2016) Evaluation of myocardial function in pediatric patients with transposition of great arteries after arterial switch operation. Anatol J Cardiol 16:55–61
Wang C, Li VW-Y, So EK-F, Cheung Y-F (2020) Left ventricular stiffness in adolescents and young adults after arterial switch operation for complete transposition of the great arteries. Pediatr Cardiol 41:747–754
Budts W, Ravekes WJ, Danford DA, Kutty S (2020) Diastolic heart failure with Fontan circulation: a review. JAMA Cardiol 5:590–597
Alsaied T, Moore RA, Lang SM et al (2020) Myocardial fibrosis, diastolic dysfunction and elevated liver stiffness in the Fontan circulation. Open Heart 7:e001434
Hui W, Abd E, Rahman MY et al (2013) Diastolic asynchrony and myocardial dysfunction in patients with univentricular heart after Fontan operation. J Echocardio 11:130–137
Chowdhury SM, Graham EM, Taylor CL et al (2022) Diastolic dysfunction with preserved ejection fraction after the Fontan procedure. JAHA 10:e024095
Michel M, Logoteta J, Entenmann A et al (2016) Decline of systolic and diastolic 2D strain rate during follow-up of HLHS patients after Fontan palliation. Pediatr Cardiol 37:1250–1257
Rossi A, Cicoira M, Golia G et al (2004) Amino-terminal propeptide of type III procollagen is associated with restrictive mitral filling pattern in patients with dilated cardiomyopathy: a possible link between diastolic dysfunction and prognosis. Heart 90:650–654
Kaufman BD, Videon N, Zhang X, Harris MA, Shaddy RE, Goldmuntz E (2015) Procollagen type III amino-terminal propeptide: a serum biomarker of left ventricular remodeling in pediatric dilated cardiomyopathy. Cardiol Young 25:228–236
Sugimoto M, Masutani S, Seki M, Kajino H, Fujieda K, Senzaki H (2009) High serum levels of procollagen type III N-terminal amino-peptide in patients with congenital heart disease. Heart 95:2023–2028
Kattel S, Memon S, Saito K, Narula J, Saito Y (2016) An effect of left ventricular hypertrophy on mild-to-moderate left ventricular diastolic dysfunction. Hellenic J Cardiol 57:92–98
Menon SC, Eidem BW, Dearani J, Ommen SR, Ackerman MJ, Miller D (2009) Diastolic dysfunction and its histopathological correlation in obstructive hypertrophic cardiomyopathy in children and adolescents. J Am Soc Echocardigr 22:1327–1334
Lipshultz SE, Cochran TR, Briston DA, Brown SR, Sambatakos PJ, Miller TL et al (2013) Pediatric cardiomyopathies: causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiol 9:817–848
Kao AC, Trigt PV, Shaeffer-McCall GS et al (1995) Allograft diastolic dysfunction and chronotropic incompetence limit cardiac output response to exercise two to six years after heart transplantation. J Heart Lung Transplant 14:11–22
Korang-Asante A, Fickey M, Boucek MM et al (2004) Diastolic performance assessed by tissue Doppler after pediatric heart transplantation. J Heart Lung Transpl 23:865–872
Kindel SJ, Law YM, Chin C et al (2015) Improved detection of cardiac allograft vasculopathy: a multi-institutional analysis of functional parameters in pediatric heart transplant recipients. J Am Coll Cardiol 66:547057
Meluzin J, Hude P, Leinveber P et al (2014) High prevalence of exercise-induced heart failure with normal ejection fraction in post-heart transplant patients. Biomed pap Med Fac Univ Palacky Olomouc Czech Repub 158:295–302
Alnsasra H, Asleh R, Oh JK et al (2021) Impact of sirolimus as a primary immunosuppressant on myocardial fibrosis and diastolic function following heart transplantation. J Am Heart Assoc 10:e018186
Tallaj JA, Kirklin JK, Brown RN et al (2007) Post-heart transplant diastolic dysfunction is a risk factor for mortality. J Am Coll Cardiol 50:1064–1069
Blanco J, Muriel-Bombin A, Sagredo V et al (2008) Incidence, organ dysfunction and mortality in severe sepsis: a Spanish multicenter study. Crit Care (London, England) 12:R158
Raj S, Killinger JS, Gonzalez JA, Lopez L (2014) Myocardial dysfunction in pediatric septic shock. J Pediatr 164:72–77
Jain A, Sankar J, Anubhuti A, Yadav DK, Sankar MJ (2018) Myocardial dysfunction in children with “Sepsis” “With” and “Without Shock”- a prospective observational study. J Trop Pediatr 64:501–509
Vallabhajosyula S, Pruthi S, Shah S, Wiley BM, Mankad SV, Jentxer JC (2018) Basic and advanced echocardiographic evaluation of myocardial dysfunction in sepsis and septic shock. Anaesth Intensive Care 46:13–24
Patel MD, Mariano K, Dunbar T, Cornell TT, Punn R, Haileselassie B (2020) Cardiac dysfunction identified by strain echocardiography is associated with illness severity in pediatric sepsis. Pediatr Crit care Med 21:e192–e199
Lipshultz SE, Miller TL, Wilkinson JD et al (2013) Cardiac effects in perinatally HIV-infected and HIV-exposed but uninfected children and adolescents: a view from the United States of America. J Int AIDS Soc 16:18597
Perez-Atayde AR, Kearney DI, Bricker JT, P2C2 HIV Study Group et al (2004) Cardiac, aortic, and pulmonary arteriopathy in HIV-infected children: the Prospective P2C2 HIV Multicenter Study. Pediatr Dev Pathol 7(1):61–70
Antony I, Kannichamy V, Banerjee A, Gandhi AB, Valaiyaduppu SS, Hamid P (2020) An outlook on the impact of HIV infection and highly active antiretroviral therapy on the cardiovascular system -a review. Cureus 12:e11539
Casaretti L, Paolillo S, Formisano R et al (2011) Metabolic and cardiovascular effects of combined antiretroviral therapy in patients with HIV infection. Systematic review of literature. Monaldi Arch Chest Dis 76:175–182
Lipshultz SE, Sasaki N, Thompson B et al (2020) Left ventricular diastolic dysfunction in HIV-uninfected infants exposed in utero to antiretroviral therapy. AIDS 34:529–537
Miller TL, Grant YT, Almeida DN, Sharma T, Lipshultz SE (2008) Cardiometabolic disease in human immunodeficiency virus-infected children. J Cardiometab Syndr 3:98–105
Lipshultz SE, Shearer WT, Thompson B et al (2011) Cardiac effects of antiretroviral therapy in HIV-negative infants born to HIV-positive mothers: NHLBI CHAART-1 (National Heart, Lung, and Blood Institute Cardiovascular Status of HAART Therapy in HIV-Exposed Infants and Children cohort study). J Am Coll Cardiol 57:76–85
Lipshultz SE, Wilkinson JD, Thompson B et al (2017) Cardiac effects of highly active antiretroviral therapy in perinatally HIV-infected children: the CHAART-2 study. J Am Coll Cardiol 70:2240–2247
Nicol M, Baudet M, Cohen-Solal A (2019) Subclinical left ventricular dysfunction during chemotherapy. Card Fail Rev 5:31–36
Lipshultz SE, Franco VI, Miller T et al (2015) Cardiovascular disease in adult survivors of childhood cancer. Annu Rev Med 66:161–176
Simbre VC, Adams MJ, Deshpande SS, Duffy SA, Miller TL, Lipshultz SE (2001) Cardiomyopathy caused by antineoplastic therapies. Curr Treat Options in Cardiovasc Med 3:493–505
Lipshultz SE, Scully RE, Stevenson KE et al (2014) Hearts too small for body size after doxorubicin for childhood ALL: grinch syndrome. J Clin Oncol 32:10021 (Abstract)
Yoldaş T, Yeşil Ş, Karademir S et al (2019) Evaluation of long-term cardiac side effects of anthracycline chemotherapy by conventional and non-conventional echocardiographic methods in childhood cancer survivors. Cardiol Young 29:904–909
Shigemitsu S, Takahashi K, Yazaki K et al (2019) New insight into the intraventricular pressure gradient as a sensitive indicator of diastolic cardiac dysfunction in patients with childhood cancer after anthracycline therapy. Heart Vessels 34:992–1001
Lipshultz SE, Miller TL, Scully RE et al (2012) Changes in cardiac biomarkers during doxorubicin treatment of pediatric patients with high-risk acute lymphoblastic leukemia: associations with long-term echocardiographic outcomes. J Clin Oncol 30:1042–1049
Alehan D, Sahin M, Varan A, Yildirim I, Küpeli S, Büyükpamukçu M (2012) Tissue Doppler evaluation of systolic and diastolic cardiac functions in long-term survivors of Hodgkin lymphoma. Pediatr Blood Cancer 58:250–255
Chow EJ, Aplenc R, Vrooman LM et al (2022) Late health outcomes after dexrazoxane treatment: a report from the Children’s Oncology Group. Cancer 28:788–796
Lipshultz SE, Adams MJ, Colan SD, American Heart Association Congenital Heart Defects Committee of the Council on Cardiovascular Disease in the Young, Council on Basic Cardiovascular Sciences, Council on Cardiovascular and Stroke Nursing, Council on Cardiovascular Radiology et al (2013) Long-term cardiovascular toxicity in children, adolescents and young adults who receive cancer therapy: pathophysiology, course, monitoring, management, prevention, and research directions: a scientific statement from the American Heart Association. Circulation 128:1927–95
Babiker HM, McBride A, Newton M et al (2018) Cardiotoxic effects of chemotherapy: a review of both cytotoxic and molecular targeted oncology therapies and their effect on the cardiovascular system. Critical Rev Oncol/Hematol 126:186–200
Bansal N, Blanco JG, Sharma UC, Pokharel S, Shisler S, Lipshultz SE (2020) Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev 39:55–68
Lipshultz SE, Somers MJ, Lipsitz SR et al (2003) Serum cardiac troponin and subclinical cardiac status in pediatric chronic renal failure. Pediatrics 112:79–86
Doyon A, Haas P, Erdem S et al (2019) Impaired systolic and diastolic left ventricular function in children with chronic kidney disease—results from the 4C study. Sci Rep 9:11462
Mitsnefes MM, Kimball TR, Border WL et al (2004) Impaired left ventricular diastolic function in children with chronic renal failure. Kidney Int 65:1461–1546
Mitsnefes MM (2008) Cardiovascular complications of pediatric chronic kidney disease. Pediatr Nephrol (Berlin, Germany) 23:27–39
Unger ED, Dubin RF, Deo R et al (2016) Association of chronic kidney disease with abnormal cardiac mechanics and adverse outcomes in patients with heart failure and preserved ejection fraction. Eur J Heart Fail 18:103–112
Qureshi MY, Wilkinson JD, Lipshultz SE (2012) The relationship of childhood obesity with cardiomyopathy and heart failure in pediatric metabolic syndrome. Springer Science & Business Media, Berlin, pp 192–215
Lipshultz SE, Messiah SE, Miller TL (2012) Pediatric metabolic syndrome: comprehensive clinical review and related health issues. Springer-Verlag London Ltd., London, pp 1–378
Mehta SK, Holliday C, Hayduk L et al (2004) Comparison of myocardial function in children with body mass indexes 25 versus those < 25 kg/m2. Am J Cardiol 93:1567–1569
Messiah SE, Arheart KL, Luke B, Lipshultz SE, Miller TL (2008) Relationship between body mass index and metabolic syndrome risk factors among US 8- to 14-year-olds, 1999 to 2002. J Pediatr 153:215–221
Sarno LA, Lipshultz SE, Harmon C et al (2020) Short- and long-term safety and efficacy of bariatric surgery for severely obese adolescents: a narrative review. Pediatr Res 87:202–209
Amin RS, Kimball TR, Kalra M et al (2005) Left ventricular function in children with sleep-disordered breathing. Am J Cardiol 95:801–804
Hui W, Slorach C, Guerra V et al (2019) Effect of obstructive sleep apnea on cardiovascular function in obese youth. Am J Cardiol 123:341–347
Ekiloglu BS, Atabek ME, Akyurek N, Alp H (2016) Prediabetes and cardiovascular parameters in obese children and adolescents. J Clin Res Pediatr Endocrinol 8:80–85
Casagrande SS, Menke A, Linder B, Osganian SK, Cowie CC (2018) Cardiovascular risk factors in adolescents with prediabetes. Diab Med. https://doi.org/10.1111/dme.13661
Procar-Almela M, Codoner-Franch P, Tuzon M, Navarro-Solera M, Carrasco-Luna J, Ferrando J (2015) Left ventricular diastolic function and cardiometabolic factors in obese normotensive children. Nutr Metab Cardiovasc Dis 25:108–185
Seliem MA, Al-Saad HI, Bou-Holaaigah IH, Khan MN, Palileo MR (2002) Left ventricular diastolic dysfunction in congenital chronic anemias during childhood as determined by comprehensive echocardiographic imaging including acoustic quantification. Eur J Echocardiogr 3:103–110
Allen KY, Jones S, Jackson T et al (2019) Echocardiographic screening of cardiovascular status in pediatric sickle cell disease. Pediatric Cardiol 40:1670–1678
Eddine AC, Alvarez O, Lipshultz SE et al (2012) Ventricular structure and function in children with sickle cell disease using conventional and tissue Doppler echocardiography. Am J Cardiol 109:1358–1364
Nanjegowda CK, Kamath SP, Kamath P et al (2019) Comparison of diastolic function in children with transfusion-dependent beta-thalassemia major by tissue and conventional Doppler imaging indices and its correlation with serum ferritin levels. Turk J Pediatr 61:250–256
Tierney ESS, Newburger JN, Graham D et al (2011) Diastolic function in children withKawasaki disease. Int J Cardiol 148:309–312
Das BB, Taylor AL, Yetman AT (2006) Left ventricular diastolic dysfunction in children and young adults with Marfan syndrome. Pediatr Cardiol 27:256–258
Oguz D, Ocal B, Ertan U, Narin H, Karademir S, Senocak F (2000) Left ventricular diastolic functions in juvenile rheumatoid arthritis. Pediatr Cardiol 21:374–377
Chang JC, White BR, Wlias MD et al (2019) Echocardiographic assessment of diastolic dysfunction in children with incident systemic lupus erythematosus. Pediatr Cardiol 40:1017–1025
Shaddy RE, George AT, Jaecklin T et al (2018) Systematic literature review on the incidence and prevalence of heart failure in children and adolescents. Pediatr Cardiol 39:415–436
Schiattarella GG, Alcaide P, Condorelli G et al (2022) Immunometabolic mechanisms of heart failure with preserved ejection fraction. Nat Cardiovasc Res 1:211–222
Aoki T (2019) Failure with preserved ejection fraction (HFpEF) patients: HFpEF as a manifestation of systemic disease. Circ J 83:277–278
Nakano SJ, Sucharov J, van Dusen R et al (2017) Cardiac adenylyl cyclase and phosphodiesterase expression profiles vary by age, disease, and chronic phosphodiesterase inhibitor treatment. J Card Fail 23:72–80
Shaddy RE, Boucek MM, Hsu DT, Pediatric Carvedilol Study Group et al (2007) Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA 298:1171–79
Lipshultz SE, Lipsitz SR, Sallan SE et al (2002) Long-term enalapril therapy for left ventricular dysfunction in doxorubicin-treated survivors of childhood cancer. J Clin Oncol 20:4517–4522
Grenier MA, Fioravanti J, Truesdell SC, Mendelsohn AM, Vermilion RP, Lipshultz SE (2000) Angiotensin-converting enzyme inhibitor therapy for ventricular dysfunction in infants, children and adolescents: a review. Prog Ped Cardiol 12:91–111
Miyamoto SD, Sucharov C, Woulfe KC (2018) Differential response to heart failure medications in children. Prog Pediatr Cardiol 49:27–30
Nagueh SF, Shah G, Wu Y et al (2004) Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110:155–162
Fukuda N, Wu Y, Nair P, Granzier HL (2005) Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J Gen Physiol 125:257–271
Fraysse B, Weinberger F, Bardswell SC, a.l, (2012) Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol 52:1299–1307
Barefield D, Sadayappan S (2010) Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol 48:866–875
Ohuchi H, Hasegawa S, Yasuda K, Yamada O, Ono Y, Echigo S (2001) Severely impaired cardiac autonomic nervous activity after the Fontan operation. Circulation 104:1513–1518
Senzaki H, Masutani S, Kobayashi J et al (2002) Ventricular afterload and ventricular work in Fontan circulation: comparison with normal two-ventricle circulation and single-ventricle circulation with Blalock-Taussig shunts. Circulation 105:2885–2892
Rathod RH, Powell AJ, Geva T (2016) Myocardial fibrosis in congenital heart disease. Circ J 80:1300–1307
Tian J, An X, Niu L (2017) Myocardial fibrosis in congenital and pediatric heart disease. Exp Ther Med 13:1660–1664
Ghonim S, Voges I, Hatehouse PD et al (2017) Myocardial architecture, mechanics, and fibrosis in congenital heart disease. Front Cardiovasc Med 4:30. https://doi.org/10.3389/fcvm.2017.00030
Jaffe R, Flugelman MY, Halon DA, Lewis BS (1997) Ventricular remodeling: from bedside to molecule. Adv Exp Med Biol 430:257–266
Spinale FG, Zile MR (2013) Integrating the myocardial matrix into heart failure recognition and management. Circ Res 113:725–738
Perens G, Li F, Meier S, Kaur R, Alejos JC, Fishbein M (2008) Clinic-pathological findings in end-stage pediatric transplant grafts. Pediatr Transplant 13:887–891
Masutani S, Kuwata S, Kirishima C et al (2016) Ventricular-vascular dynamics in pediatric patients with heart failure and preserved ejection fraction. Int J Cardiology 225:306–312
Simmonds SJ, Cuijpers I, Heymans S, Jones EAV (2020) Cellular and molecular differences between HFpEF and HFrEF: a step ahead in an improved pathological understanding. Cells 9:242. https://doi.org/10.3390/cells9010242
Gevaert AB, Kataria R, Zannad F et al (2022) Heart failure with preserved ejection fraction: recent concepts in diagnosis, mechanisms and management. Heart 12:319605. https://doi.org/10.1136/heartjnl-2021-319605
Berglund F, Pina P, Herrera CJ (2020) Right ventricle in heart failure with preserved ejection fraction. Heart 106:1798–1804
Gropler MRF, Lipshultz SE, Wilkinson JD et al (2021) Pediatric and adult dilated cardiomyopathy are distinguished by distinct biomarker profiles. Pediatr Res. https://doi.org/10.1038/s41390-021-01698-x
Zile MR, Brutsaert DL (2002) New concepts in diastolic dysfunction and diastolic heart failure: Part I: diagnosis, prognosis, and measurements of diastolic function. Circulation 105:1387–1393
Nagueh SF, Smiseth OA, Appleton CP et al (2016) Recommendations for the evaluation of left ventricular diastolic function by echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 29:277–314
Sorrentino R, Esposito R, Santoro C et al (2020) Practical impact of new diastolic recommendations on noninvasive estimation of left ventricular diastolic function and filling pressures. J Am Soc Echocardiogr 33:171–181
Ezon DS, Maskatia SA, Sexson-Tejtel K, Dreyer WJ, Jeewa A, Denfield SW (2015) Tissue Doppler imaging measures correlate poorly with left ventricular filling pressures in pediatric cardiomyopathy. Congenit Heart Dis 10:203–209
Cantinotti M, Lopez L (2013) Nomogram for blood flow and tissue Doppler velocities to evaluate diastolic dysfunction in children: a critical review. J Am Soc Echocardiogr 26:126–141
Menon SC, Gray R, Tani LY (2011) Evaluation of ventricular filling pressures and ventricular function by Doppler echocardiography in patients with functional single ventricle: correlation with simultaneous cardiac catheterization. J Am Soc Echocardiogr 24:1220–1225
Rihal CS, Nishimura RA, Hatle LK et al (1994) Systolic and diastolic dysfunction in patients with a clinical diagnosis of dilated cardiomyopathy: relation to symptoms and prognosis. Circulation 90:2772–2779
Dujardin KS, Tei C, Yeo T et al (1998) Prognostic value of a Doppler index combining systolic and diastolic performance in idiopathic dilated cardiomyopathy. Am J Cardiol 82:1071–1076
McMohan CJ, Pignatelli RH, Naguesh SF et al (2007) Left ventricular non-compaction cardiomyopathy in children: characterization of clinical status using tissue Doppler-derived indices of left ventricular diastolic relaxation. Heart 93:676–681
Niemann M, Liu D, Hu k, et al (2012) Echocardiographic quantification of regional deformation helps to distinguish isolated left ventricular non-compaction from dilated cardiomyopathy. Eu J Heart Failure 14:155–161
Nagueh SF, Lakkis NM, Middleton KJ et al (1999) Doppler estimation of left ventricular filling pressures in patients with hypertrophic cardiomyopathy. Circulation 99:254–261
Geske JB, Sorajja P, Nishimura RA et al (2007) Evaluation of left ventricular filling pressures by Doppler echocardiography in patients with hypertrophic cardiomyopathy: correlation with direct left atrial pressure measurement at cardiac catheterization. Circulation 116:2702–2708
Haland TF, Edvardsen T (2020) The role of echocardiography in management of hypertrophic cardiomyopathy. J Echocardiogr 18:77–85
Ryan TD, Madueme PC, Jefferies JL et al (2017) Utility of echocardiography in the assessment of left ventricular diastolic function and restrictive physiology in children and young adults with restrictive cardiomyopathy: a comparative echocardiography-catheterization study. Pediatr Cardiol 38:381–389
Butz T, Piper C, Langer C et al (2010) Diagnostic superiority of a combined assessment of the systolic and early diastolic mitral annular velocities by tissue Doppler imaging for the differentiation of restrictive cardiomyopathy from constrictive pericarditis. Clin Res Cardiol 99:207–215
Garcia MJ, Rodriguez L, Ares M, Griffin BP, Thomas JD, Klein AL (1996) Differentiation of constrictive pericarditis from restrictive cardiomyopathy: assessment of left ventricular diastolic velocities in longitudinal axis by Doppler tissue imaging. J Am Coll Cardiol 27:108–114
Badano LP, Kolias TJ, Muraru D et al (2018) Standardization of left atrial, right ventricular, and right atrial deformation imaging using two-dimensional speckle-tracking echocardiography: a consensus document of the EACVI/ASE/Industry Task Force to standardize deformation imaging. Eur Heart J Cardiovasc Imaging 19:591–600
Inoue K, Khan FH, Remme EW et al (2021) Determinants of left atrial reservoir and pump strain and use of atrial strain for evaluation of left ventricular filling pressure. Eur Heart J Cardiovasc Imaging 23:61–70
Yuda S (2021) Current clinical applications of speckle tracking echocardiography for assessment of left atrial function. J Echocardiogr 193:129–140
Chowdhury SM, Butts RJ, Taylor CL et al (2018) Longitudinal measures of deformation are associated with a composite measure of contractility derived from pressure-volume loop analysis in children. Eur Heart J Cardiovasc Imaging 19:562–568
Støylen A, Slørdahl S, Skjelvan GK, Heimdal A, Skjaerpe T (2001) Strain rate imaging in normal and reduced diastolic function: comparison with pulsed Doppler tissue imaging of the mitral annulus. J Am Soc Echocardiogr 14:264–274
Palka P, Lange A, Donnelly JE, Nihoyannopoulos P (2000) Differentiation between restrictive cardiomyopathy and constrictive pericarditis by early diastolic Doppler myocardial velocity gradient at the posterior wall. Circulation 102:655–662
Afonso L, Kondur A, Simegn M et al (2012) Two-dimensional strain profiles in patients with physiological and pathological hypertrophy and preserved left ventricular systolic function: a comparative analyses. BMJ Open 2:e001390
Sengelov M, Jorgensen PG, Jensen JS et al (2015) Global longitudinal strain is a superior predictor of all-cause mortality in heart failure with reduced ejection fraction. JACC Cardiovasc Imaging 12:1351–59
Adamo L, Perry A, Novak E, Maken M, Lindman BR, Mann DL (2017) Abnormal global longitudinal strain predicts future deterioration of left ventricular function in heart failure patients with a recovered left ventricular ejection fraction. Circ Heart Fail 10:e003788
Wang J, Khoury DS, Trohan V, Torre-Amone G, Nagueh SF (2007) Global diastolic strain rate for the assessment of left ventricular relaxation and filling pressure. Circulation 115:1376–1383
Steflik D, Butts RJ, Baker GH et al (2017) A preliminary comparison of two-dimensional speckle tracking echocardiography and pressure-volume loop analysis in patients with Fontan physiology: the role of ventricular morphology. Echocardiography 34:1353–1359
Margossian R, Schwartz ML, Prakash A et al (2009) Comparison of echocardiographic and cardiac magnetic resonance imaging measurements of functional single ventricular volumes, mass, and ejection fraction (from the Pediatric Heart Network Fontan Cross-Sectional Study). Am J Cardiol 104:419–428
Matsubara D, Chang J, Kauffman HL et al (2022) Longitudinal assessment of cardiac outcomes of multisystem inflammatory syndrome in children associated with COVID-19 infections. J Am Heart Assoc 19:e023251
Ghelani SJ, Brown DW, Kuebler JD, Perrin D, Shakti D, Williams DN (2018) Left atrial volumes and strain in healthy children measured by three-dimensional echocardiography: normal values and maturational Changes. J Am Soc Echocardiogr 31:187–193
Messroghli DR, Moon JC, Ferreira VM, Grosse-Wortmann L, He T, Kellman P (2017) Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson 19(1):75
Rao S, Tseng SY, Pednekar A et al (2022) Myocardial parametric mapping by cardiac magnetic resonance imaging in pediatric cardiology and congenital heart disease. Circ Cardiovasc Imaging 15:e012242
Adabag AS, Maron BJ, Appelbaum E et al (2008) Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol 51:1369–1374
Brenes JC, Doltra A, Prat S (2018) Cardiac magnetic resonance imaging in the evaluation of patients with hypertrophic cardiomyopathy. Glob Cardiol Sci Pract 18:22. https://doi.org/10.21542/gcsp.2018.22
Wood JC, Origa R, Agus A, Matta G, Coates TD, Galanello R (2008) Onset of cardiac iron loading in pediatric patients with thalassemia major. Haematologica 93:917–920
Riesenkampff E, Messroghli DR, Redington AN, Grosse-Wortmann L (2015) Myocardial T1 mapping in pediatric and congenital heart disease. Circ Cardiovasc Imaging 8:e002504
Yim D, Riesenkampff E, Caro-Dominguez P, Yoo SJ, Seed M, Grosse-Wortmann L (2017) Assessment of diffuse ventricular myocardial fibrosis using native T1 in children with repaired tetralogy of Fallot. Circ Cardiovasc Imaging 10:e005695
Payment CM, Sado DM, Taylor AM et al (2013) Diffuse myocardial fibrosis in the systemic right ventricle of patients late after Mustard or Senning surgery: an equilibrium contrast cardiovascular magnetic resonance study. Eur Heart J Cardiovasc Imaging 14:963–968
Kato A, Riesenkampff E, Yim D, Yoo SJ, Seed M, Grosse-Wortmann L (2017) Pediatric Fontan patients are at risk for myocardial fibrotic remodeling and dysfunction. Int J Cardiol 240:172–177
Dusenbery SM, Lunze FI, Jerosch-Herold M, Geva T, Newburger JW, Colan SD, Powell AJ (2015) Left ventricular strain and myocardial fibrosis in congenital aortic stenosis. Am J Cardiol 116:1257–1262
Schulz-Menger J, Bluemke DA, Bremerich J et al (2020) Standardized image interpretation and post-processing in cardiovascular magnetic resonance-2020 update: society for cardiovascular magnetic resonance (SCMR): board of trustees task force on standardized post-processing. J Cardiovasc Magn Reson 22:19
Aggarwal M, Bozkurt B, Panjrath G, Aggarwal B, Ostfeld RJ, Barnard ND et al (2018) Lifestyle modifications for preventing and treating heart failure. J Am Coll Cardiol 72:2391–2405
Cho MJ, Lim RK, Kwak MJ et al (2015) Effects of beta-blockers for congestive heart failure in pediatric and congenital heart disease patients-a meta-analysis of published studies. Minerva Cardioangio 63:495–505
Hoffman TM, Wernovsky G, Atz AM et al (2003) Efficacy and safety of milrinone in preventing low cardiac output syndrome in infants and children after corrective surgery for congenital heart disease. Circulation 107:996–1002
Brilla CG, Rupp H, Funck R, Maisch B (1995) The renin-angiotensin-aldosterone system and myocardial collagen matrix remodeling in congestive heart failure. Eur Heart J. https://doi.org/10.1093/eurheartj/16.suppl_O.107
Vincent L, Cinca J, Vazquez-García R, Gonzalez-Juanatey JR, Rivera M, Segovia J (2019) Discharge treatment with angiotensin-converting enzyme inhibitor/angiotensin receptor blocker after a heart failure hospitalization is associated with a better prognosis irrespective of left ventricular ejection fraction. Intern Med J 49:1505–1513
Setaro JF, Zaret BL, Schulman DS, Black HR, Soufer R (1990) Usefulness of verapamil for congestive heart failure associated with abnormal left ventricular diastolic filling and normal left ventricular systolic performance. Am J Cardiol 66:981–986
Betocchi S, Chiariello M (1993) Effects of calcium antagonists on left ventricular structure and function. J Hypertens Suppl 11:S33-37
Iliceto S (1997) Left ventricular dysfunction: which role for calcium antagonists? Eur Heart J Suppl A. https://doi.org/10.1093/eurheartj/18.suppl_A.87
Degre S, Detry JM, Unger P, Cosyns J, Brochet C, Kormos N (1998) Effects of spironolactone-altizide on left ventricular hypertrophy. Acta Cardiol 53:261–267
Edelmann F, Wachter R, Schmidt AG et al (2013) Effects of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection function: the Aldo-DHF randomized controlled trial. JAMA 309:781–791
Pitt B, Pfeffer MA, Assmann SF et al (2014) Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 370:1383–1392
Pfeffer MA, Claggett B, Assmann SF et al (2015) Regional variation in patients and outcomes in the treatment of preserved cardiac function heart failure with aldosterone antagonist (TOPCAT) trial. Circulation 131:34–42
Armstrong PW, Lam CSP, Anstrom KJ, VITALITY-HFpEF Study Group et al (2020) Effect of Vericiguat vs. Placebo on quality of life in patients with heart failure and preserved ejection fraction: The VITALITY-HFpEF Randomized Clinical Trial. JAMA 324:1512–52
Cunningham JW, Claggett BL, O’Meara E et al (2020) Effect of Sacubitril/Valsartan on biomarkers of extracellular matrix regulation in patients with HFpEF. J Am Coll Cardiol 76:503–514
Solomon SD, McMurray JJV, Anand IS et al (2019) Angiotensin-Neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 381:1609–1620
Das SR, Everett BM, Birtcher KK et al (2020) 2020 expert consensus decision pathway on novel therapies for cardiovascular risk reduction in patients with type 2 diabetes: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol 76:1117–1145
Su JA, Manteer J (2017) Outcomes of Berlin Heart Excor pediatric ventricular assist device support in patients with restrictive and hypertrophic cardiomyopathy. Pedatr Transpl 21:28905470. https://doi.org/10.1111/petr.13048
Grupper A, Park S, Pereira N et al (2015) Role of ventricular assist therapy for patients with heart failure and restrictive physiology: Improving outcomes for a lethal disease. J Heart Lung Transplant 34:1042–1049
Topilsky Y, Pereira NL, Shah DK et al (2011) Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail 4:266–267
Sreenivasan J, Kaul R, Khan MS et al (2021) Left ventricular assist device implantation in hypertrophic and restrictive cardiomyopathy: a systematic review. ASAIO J 67:239–244
Patel SR, Saeed O, Naftel D et al (2017) Outcomes of restrictive and hypertrophic cardiomyopathies after LVAD: An INTERMACS Analysis. J Card Fail 23:859–867
Sundararajan S, Thiruchelvam T, Hsia TY, Karimova A (2014) New 15-mL ventricular assist device in children with restrictive physiology of the left ventricle. J Thorac Cardiovasc Surg 147:e79-80
Funding
“Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM121334." Dr. Lipshultz’s work is supported by grants from the National Heart, Lung, and Blood Institute (HL53392, HL111459, and HL109090), in part from the National Institutes of Health (HL139968, HL137558, HL 152090, HD104909, AI170252, HL140443, and CA211996), and the Children’s Cardiomyopathy Foundation (CCF), the Children’s Discovery Institute Foundation (CDIF), Sofia’s Hope, Inc. (SHI), and the Kyle John Rymiszewski Foundation (KJRF). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI, NIH, CCF. CDIF, SHI, or KJRF.
Author information
Authors and Affiliations
Contributions
B.D. drafted the manuscript and revised it; S.D., J. A.V., D. S., W. M., S.L.-collected references, contributed for revisions, and added to the paper; S.L contributed to supervision and critical review of multiple versions of this manuscript. All agreed to the final version submitted.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Das, B., Deshpande, S., Akam-Venkata, J. et al. Heart Failure with Preserved Ejection Fraction in Children. Pediatr Cardiol 44, 513–529 (2023). https://doi.org/10.1007/s00246-022-02960-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-022-02960-7