
Abstract
Digital PCR methods were recently introduced in food analysis. To run these methods within the scope of ISO17025, they have to be validated. Although several guidelines are available, each laboratory has to implement these guidelines in an adapted validation scheme. We present here one possible implementation. We chose 13 GMO traits which were predominantly detected in the past. The results show that in the range of 1% GMO content, the digital PCR has a little superior performance compared to real time PCR. In the range of the detection limit, measurement uncertainty remains comparable to real time PCR. During validation, a conversion factor was determined for each trait suggesting that the calculation from % copies/copies to % weight/weight may be possible. This shows that GMO contents can be measured without the use of reference material by the validation described here and determination of a conversion factor, which is a great improvement in terms of expense and storage capacity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Establishment of digital PCR can be done in a straight forward process and within reasonable time [1,2,3]. Duplex [4] and even 12-plex assays were recently successfully realized [5]. The optimization of primers and probes as well as the other conditions were investigated extensively [6]. In consequence the quantification of GMO contents without the use of reference material may be possible. The previously gained results seem to exhibit a lower measurement uncertainty than when applying real time PCR [7, 8]. The main advantage seems to be a better precision. To convert measured copy number and their percentage to percentage by weight to weight [9] a so called conversion factor (earlier called zygosity factor) has to be applied. This factor can be determined using a certified reference material. For some new traits, this was already done by the European Joint Research Center (JRC), Ispra, Italy (http://gmo-crl.jrc.ec.europa.eu/StatusOfDossiers.aspx). These factors are available in the appropriate validation report of the transgene traits, published after successful validation by the JRC. Unfortunately, these factors are not available for the older GMO traits we used in this study.
To assess specificity, e.g., false positive signals, in silico investigations can give hints but remain incomplete. Extensive testing of DNA from different GMO traits and other plant and animal species is required. The specificity of digital PCR can be higher as mostly only one target DNA copy is enclosed per reaction chamber (droplet). This reduces the chance of amplification of not targeted primer sites or of unexpected DNA-hybrids.
For routine application in the scope of ISO 17025 full validation has to be done to show the suitability of such methods. Several guidelines were released for the validation of PCR systems already [10,11,12]. Although they are an important help to resolve the different aspects during each validation step, they remain unclear in the concrete steps and details and do not address digital PCR specifically. This gives space for interpretation. In this work we focussed on a validation scheme to comply with the guidelines with reasonable input. The final goal was to assess the lower limit of quantification and the measurement uncertainty. In addition, conversion factors were also determined and were compared with previously determined ones.
Results from real time PCR suggested, that below 0.1% the quantification has a high measurement uncertainty (often in the range of ±30%) and becomes rather arbitrary. We expected that the measurement uncertainty to be smaller with digital PCR.
The selection of GMO traits and duplex droplet digital PCR (ddPCR™) systems were made by identified positive samples from the last 10 years of GMO) analysis at our laboratory.
Materials and methods
Reference material
Genetically modified reference material was purchased from the following source: Sigma-Aldrich, St. Louis, USA: RoundupReady (RR, ERM 410dk), Bt176 (ERM BF 411f), Bt11 (ERM BF 412f), Mon810 (ERM BF 413ck), GA21 (ERM BF 414f), NK603 (ERM BF 415f), Mon863 (ERM BF 416d), 59122 (ERM BF424d), Mir604 (ERM BF423d), TC1507 (ERM BF 418d). AgrEvo GmbH Berlin, Germany: T25 (Reference C/F/95/12-07). AOCS, Urbana, USA: Mon89788 (Cat. No. 0906 B), 2704 (Cat. No. 0707 B6).
Plant and animal material
Plant and animal material was purchased from local retailer as whole recognizable material. Animal tissue was sequenced to ensure correct species identification. From this material DNA was extracted and this DNA was used as non-target template to assess the specificity of the here validated assays.
Equipment
The QX200 Droplet Digital PCR (ddPCR™) system of Bio-Rad Laboratories Inc. USA was used to perform the droplet digital PCR in this work. For pipetting we used manual Gilson Pipetman of 20, 100 and 200 μl volume. To transfer the Droplets from the droplet generating cartridge to the PCR-tubes we used ART 100 μl filter tips (Cat No. 713116) or Biosphere plus tips 200 μl (Sarstedt, Cat. No. 70.760.211) or Clear Line low binding tips 100 μl (Milian, Cat. No. 713116) filter tips in combination with a Gilson 8 channel manual Pipetman L P8 × 200L. All combinations lead to successful generation of 10,000 droplets or more.
DNA extraction
DNA extraction from all sample matrices was performed using the column based Wizard Plus Miniprep DNA purification system (Promega, Madison, USA). 200 mg of ground sample material were extracted and the DNA was eluted into 50 μl elution buffer according to the supplier’s manual. The concentration was determined spectrophotometrically (denatured single strand DNA, 320/260 nm) and adjusted by dilution to 20 ng/μl using PCR-grade water.
Primers and probes
Primers and hydrolysis-probes were published earlier or published by the European Union Reference Laboratory for Genetically Modified Food & Feed (EURL-GMFF, http://gmo-crl.jrc.ec.europa.eu/StatusOfDossiers.aspx, see Table 1). Primer concentrations were optimised by titration. All primers and hydrolysis-probes were synthesized by Microsynth AG, Balgach, Switzerland. The sequences of primers and probes as well as the labelling are listed in Table 1.
Other material
All plastic material was of PCR-grade quality. To avoid cross-contamination, all pipetting steps were performed using filter tips. Solutions were prepared with DNA/RNA free water. To diminish contaminations, a three room concept was applied.
Droplet digital PCR procedure (ddPCR™)
5 μl DNA extracts were added to 17 μl of reaction mix containing 11 μl Supermix for Probes (Cat No. 186-3024) and the primers and probes. 20 μl of these final 22 μl were used to generate a water/oil emulsion. The final primer concentrations for the applied ddPCR systems are listed in Table 1, the concentration of the probes was always 250 nM. The setup was according to the manual of the QX200 ddPCR™ system. To generate water in oil emulsion microfluidic cartridges for the ddPCR™ were used. After pipetting the 40 µl emulsion to the 96 multiwell-plates, they were sealed using the heat sealer PX1 from Bio-Rad. The emulsion-PCR was performed on a Master-Cycler Nexus gradient from Eppendorf. The cycling was done according to the manual except that 50 cycles were performed (see below). The ramp rate was fixed to 2.5 °C/s, the annealing temperature was set at 55 °C for all systems. The whole cycling required approximately 2.5 h. The reading of the droplets was then performed using the droplet reader of the QX200-system. All steps were performed according to manual of the QX200 ddPCR™ system of Bio-Rad Laboratories Inc. USA.
Calculation of % GMO content
The percentage of GMO content was calculated as following:
cf: conversion factor (no unit); target (droplet/μl); housekeeping (droplet/μl)
Determination of the optimal primer concentration and annealing temperature
Cycling numbers must be enough to amplify a signal also in cases where the sample matrix is inhibiting the polymerase. To assure this, we decided to cycle 50 times in all cases. The optimization of the primer concentration was done according to a previous publication [4]. All ddPCR™ systems were run in duplex format using lectin (for soy traits) or mhmg (for maize traits) as housekeeping gene. For some of the PCR systems negative and positive droplet populations were difficult to distinguish due to “rain”. Using an annealing temperature of 55 °C for all systems leads to sufficient signal separation to set a clear threshold. This finding corresponds to observations during extensive optimizations previously described [6]. Optimised primer concentrations are listed in Table 1.
Results and discussion
Problems during validation
During validation the following issues had to be assayed: optimal template concentration, achievable lower limit of quantification (LOQ), no template control (ntc) and conversion factor to convert percent values of droplets to weight percent values. In a previous establishment we experienced rapid first results and were, therefore, confident to finish validation in a reasonable time. Optimal template concentration was at 100 ng total DNA per assay. For two (GA21 and Bt176) of the total 13 Systems we faced serious problems when assaying the ntc samples. A slight but resistant rain of false positive droplets were observed making distinction of positive to negative samples in the low range impossible. These two systems [15] and an unpublished in house system served in multiplex real time PCR faithfully since years. We therefore think that these false positive results are due to difference in cycling parameters, annealing temperature and the DNA-polymerase used in digital PCR. We decided to exchange these PCR systems by systems from the JRC.
Specificity
All digital PCR systems were tested for cross reactions to a wide selection of gm traits, plant and animal DNA. To reduce the reactions and costs we used pooled templates DNA’s of eight species (100 ng of total DNA per assay, 12.5 ng per species). DNA of the following organisms were tested (100 ng total DNA per assay): genetically modified traits: Soybean MON87769 100%, Soybean MON87708 100%, Soybean MON87705 100%, Soybean FG72 100%, Maize MON810 5%, Maize MON863 9.9%, Maize T25 100%,, Maize CBH351 1%, Maize NK603 5%, Maize Bt176 5%, Maize Ly038 E-7, Maize Bt11 5%, Maize GA21 4.3%, Maize 59122 10%, Maize MON98034 100%, Maize MON88017 100%, Maize ES3272 10%, Maize Mir604 10%, Maize TC1507 10%, Maize DP98140 10%, Maize DAS40278-9 1%, Maize Mir162 100%, Maize MON87460 100%, Maize Bt10 100%, Soybean RRS 5%, Soybean A2704 100%, Soybean MON89788 100%, Soybean A5547 100%, Soybean DP356 10%, Soybean DP305 10%, Soybean MON87701 100%, Soybean CV127 100%, Rice KMD1
The following cross reactions were observed: The GA21-system exhibit cross reaction when applying DNA from Starlink (2%) and Mon89034 (0.1%). These cross reactions are unlikely (Starlink, cultivation stopped) or may come from contaminated reference material (Mon 89034) and have to be considered when examining such samples.
Bt10 Maize can not be distinguished from Bt11 by the Bt11-system. This was expected as Bt10 and Bt11 are very close relatives. However, Bt10 maize is very unlikely to be detected as cultivation was stopped years ago.
Animals Cattle (Bos taurus), turkey (Meleagris gallopavo), horse (Equus caballus), porc (Sus scrofa), chicken (Gallus gallus), swordfish (Xiphias gladius).
No cross reactions were observed.
Plants Almond (Prunus dulcis), apricot (Prunus armeniaca), walnut (Juglans regia), hazelnut (Corylus avellana), peanut (Arachis hypogaea), cinnamon (Cinnamomum verum), beans (Phaseolus vulgaris), rice (Oryza spp.), wheat (Triticum spp.), sesame (Sesamum indicum), lentil (Lens culinaris), lupin (Lupinus), maize (Zea mays), cauliflower (Brassica oleracea), peas (pisum sativum), tomato (Solanum lycopersicum), rape (Brassica napus), mango (Mangifera indica), banana (Musa), broccoli (Brassica oleracea), rye (Secale cereale) durum wheat (Triticum durum), avocado (Persea americana), lupine (Lupinus albus), papaya (Carica papaya), cashew (Anacardium occidentale)
The following cross reactions were observed: The 2704-system detects 100% cinnamon with 0.2%. This has to be considered when assaying samples containing high level of cinnamon. Radish (0.3%) and rye (1.4%) lead to cross reactions in the Mon863-system. This has also to be considered. No other unspecific signal was observed. Cross-contamination for these findings could be an explanation, however, this was considered to be very unlikely. For all traits only one batch was tested.
Measurement range
For the housekeeping assays, the lambda factor was below 0.5 positive droplets/total droplets. For the transgene target systems, the lambda factor was below 0.05.
Most transgene target systems showed a consistent positive signals, at a level of 11 positive droplets (0.5 copies/μl) or more. We consider that the limit of detection (LOD) is close to this value. Expressed in %, 11 or more positive droplets were gained at a level of about 0.1% (prerequisite: 100 ng of total template DNA, spectrophotometrically determined) The system for the corresponding housekeeping gene exhibited about 1500 copies/μl positive droplets of housekeeping gene, e.g., mhmg, if the reference material contained 100% transgene DNA). In practice the upper limit was limited by the maximal amount of template DNA used and the quantification range of the QX200 system. Considering the average droplet number of 10 000, the upper quantification limit was experienced to be at about 5000 copies/μl. In our hands the lower limit of quantification was, therefore, not lower than when using real time PCR (also at 0.1%). In many cases, the available reference material does not consist of 100% GMO, in consequence, the tested quantification range became smaller, e.g., 0.1–1%. Values higher than 1% have to be determined by extrapolation or dilution. This was not investigated here as usually the most samples contain low amounts of GMO. The labelling threshold for authorized traits in Switzerland and EU is at 0.9% and is covered by this measurement range. And in cases of unauthorized gm traits the product has to be withdrawn from the market, when a reproducible positive signal is measured, a quantification is not required.
Accuracy, precision, measurement uncertainty (MU) and robustness
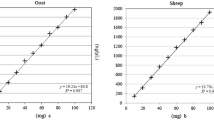
Limit of quantification (LOQ), precision and measurement uncertainty of the ddPCR™ was tested at three levels of DNA concentration: e.g., if the content was 100% GMO: 2 ng/μl corresponds to 10%, 0.2 ng/μl corresponds to 1% and 0.02 ng/μl corresponds to 0.1%. But the proportion of GMO relative to the housekeeping lectin (for soy) or mhmg (for maize) was not always 100%. In cases where the level was, e.g., only 5% (depending on the available reference material), this material could only be diluted 1: 50 to reach the lower LOQ of 0.1% for the GMO marker gene, which had to be reached. This low level of 0.1% was estimated to be close to the limit of quantification usually obtained for GMO quantification methods using real time PCR methods [12]. We therefore determined measurement uncertainty performing 12 duplex experiments for each transgene soy trait. These experiments were performed at different days by 3 persons to assess the robustness of the systems. Further robustness testing was not done. Recommendations of varying annealing temperature and primer concentration is not helpful for ddPCR™ as this is an endpoint method and such variations have only minor impact, which was shown during optimization experiments [6]. To assay false positive rate, e.g., by possible cross-contamination during the droplet procedure, no template controls (ntc) were run during four different assays. For the systems that were finally selected, no false positive droplets were observed. However, from a total of 13 systems, two systems showed consistent false positive signals during development and had to be replaced (GA21 and Bt176).
At the level of 0.1% GMO, all assays always showed positive results and we conclude, therefore, that 0.1% GMO content is covered by the quantification range.
The determined measurement uncertainty (MU), including the precision and accuracy, ranged from 9 to 43%. The guidance document for method performance requirements of the European network of GMO Laboratories sets a limit of maximal ±50% [12] for the MU. The determined MU is, therefore, acceptable at this low concentration level. For the higher concentration level of 1% GMO, data for precision and measurement uncertainty are also acceptable (measurement uncertainty ranging from 7.5 to 26%).
Comparing to internal validation data, in six cases the ddPCR™ exhibited a lower measurement uncertainty in the low range than real time PCR. But in other six cases the real time PCR exhibited lower measurement uncertainty. We estimate, therefore, that ddPCR™ and real time PCR have comparable measurement uncertainty in general in the low range. In the higher range of 1% GMO we observed for ddPCR™ in 8 cases superior MU and for real time PCR only 4 times. This suggests that ddPCR™ can exhibit lower measurement uncertainty comparing to real time PCR, only in cases where the template concentration is high. The high expectations for ddPCR were, therefore, not fulfilled in this case.
Zygosity/conversion factor
Reference material for GMO analysis is often a mixture of flour with and without GMO trait by weight to weight, leading to a certain percentage of GMO. Digital PCR determines copy numbers leading to percentage of GMO as well but not weight to weight. Surprisingly, for all tested soy traits only minor difference were observed [13, 14]. The zygosity or conversion factor for the tested soy traits was always close to 1. For maize traits this seems not to be the case. For GMO maize traits the factor is often higher than 1. Usually the PCR systems to determine the genetic modification use a single integration site. The second gene, often called housekeeping, determines the plant species, in this case the major high mobility group gene (mhmg) of maize. This gene may have multiple sites and the number of sites may be different from one trait to the other. Earlier publications report that the mhmg used here should be present in all maize traits at comparable amounts and is therefore widely used as a housekeeping gene [15]. In principle the difference between percentage measured by copy numbers and the reference material weight by weight may also come from use of not appropriate traits or different weights of the kernels or zygosity in the F1 generation. But these arguments also apply to GM soy. But for soy the conversion factor seems to be close to 1 for all investigated traits [8]), see Table 2. We suspect therefore the different conversion factors of GM maize traits to be mainly a result from difference in copy number of the mhmg housekeeping gene in maize traits. An additional explanation may be the fact that maize is not self-pollinating, in contrast to soy. In addition, maize is mostly sold as hybrid trait. As a consequence, first generation kernels are not 100% homozygous and the zygosity may be therefore affected.
We determined the conversion factors using the actually available reference material described above (section reference material). Once determined, the conversion factor can be used for the appropriate primer system without using reference material for each analysis. However, when changing the PCR system for the housekeeping gene or the reference material, we expect that this factor has to be determined again.
Results from proficiency studies
Earlier experiences showed that ddPCR™ may be superior to real time PCR results. To assess this, we analysed proficiency test samples in parallel using real time PCR and ddPCR™ (applying the determined conversion factors). The results are compiled in Table 3. In 25 cases ddPCR was superior to real time PCR, in 23 cases the real time PCR was superior. We consider this as not significant. Earlier results showed comparable MU at low concentrations, confirming this result. Looking at the performance of individual PCR systems, e.g., Mon863 and 59122, the general finding of equal MU and closeness to the true value seem to depend mainly on the PCR-system applied. This could be a hint for future ameliorations.
Conclusion
We validated 13 droplet digital PCR systems according to guidelines [10,11,12], fulfilling ISO17025 requirements using certified reference material. The lower range of quantification could be confirmed for all systems at a level of 0.1% GMO. At this level total positive droplets were in the range of 10–20 per assay which is statistically sufficient. At a level of 1%, measurement uncertainty was significantly lower than when using real time PCR in the majority of cases. The accuracy for maize samples had to be corrected by a conversion factor to get the true value of the GMO content by weight to weight. For soy samples this factor was close to one. Published values for soy traits are until now are also close to one, confirming our findings. This seems to be a principal difference between maize and soy traits. Earlier work attempted to use a sequence for the housekeeping gene, which should lead to comparable signals for all maize traits. This mhmg-gene, however, seem to vary significantly. This effect is not observed when analysing soy traits. All effects discussed concerning mixing and selection of the material may therefore not have an impact on the zygosity and conversion factor as they apply to both traits, soy and maize. We therefore consider the difference in mhmg as the major impact for the different conversion factors in maize traits. The presented conversion factors only apply to the PCR systems and reference material in this study. And they may need to be confirmed by a greater effort including more defined reference material and statistical calculations as described by the European Joint Research Center [23].
The results presented here were generated without enzymatic cutting of the template DNA suggesting that this step is not required. As real samples in general have degraded template DNA (therefore sensitive PCR systems usually amplify amplicons below 150 bp) and make such an enzymatic degrading step unnecessary. The guidelines to perform robustness testing should be adapted for digital PCR as the recommendations don’t apply for digital PCR as the amplification is intrinsically less dependent on annealing temperature and primer concentrations.
Finally, the validated ddPCR™-systems for the most prevalent GMO traits we present here are fit for routine analytic. The assessed MU and performance, analysing proficiency test samples revealed in general similar performance as real time PCR. In contrast to earlier findings ddPCR™ was not superior to real time PCR. A remaining advantage is still that results can be generated without the use of reference material once the conversion factors were determined.
References
Sanders R, Huggett JF, Bushell CA, Cowen S, Scott DJ, Foy CA (2011) Evaluation of digital PCR for absolute DNA quantification. Anal Chem 83(17):6474–6484
Pinheira LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, Emsli KR (2012) Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 84:1003–1011
Gürtler P, Gerdes L, (2014) Digitale polymerasekettenreaktion, ddPCR; BIOspektrum | 06.14 | 20. Jahrgang, p. 632–635
Köppel René, Bucher Thomas (2015) Rapid establishment of droplet digital PCR for quantitative GMO analysis. Eur Food Res Technol 241(3):427–439
Dobnik D, Spilsberg B, Bogožalec Košir A, Holst-Jensen A, Žel J (2015) Multiplex quantification of 12 European union authorized genetically modified maize lines with droplet digital polymerase chain reaction. Anal Chem 87(16):8218–8226
Gerdes L, Iwobi A, Busch U, Pecoraro S (2016) Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol Detect Quantif 7:9–20
Corbisier P, Bhat S, Partis L, Rui V, Xie D, Emslie KR (2010) Absolute quantification of genetically modified MON810 maize (Zea mays L.) by digital polymerase chain reaction. Anal Bioanal Chem 396(6):2143–2150
Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Rufa IK, Benjamin J, Hindson R, Vessella L, Tewari M (2013) Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods 10:1003–1005
Urquiza MP, Silva AA (2014) Copy number ratios determined by two digital polymerase chain reaction systems in genetically modified grains. Metrologia 51(1):61–66
European Union Reference Laboratory for GM Food and Feed (2008) Definition of minimum Performance requirements for analytical methods of GMO testing. http://gmo-crl.jrc.ec.europa.eu/doc/Min_Perf_Requirements_Analytical_methods.pdf Accessed 29 Jan 2013
S. Trapman, Burns M, Broll H, Macarthur R, Wood R, Zel J (2009) Guidance document on measurement uncertainty for GMO testing laboratories. In: European Commission—Joint Research Centre—Institute for Reference Materials and Measurements, vol 1111, pp 1–41 (ISBN: 978-92-79-11228-7; p18, Equation 10)
Broedersa S, Huberb I, Grohmannc L, Berbend G, Tavernierse I, Mazzaraf M, Roosensa N, Morisset D (2014) Guidelines for validation of qualitative real-time PCR methods. Trends Food Sci Technol 37:115e126
Scholdberg TA, Norden TD, Nelson D, Jenkins RG (2009) Evaluating precision and accuracy when quantifying different endogenous control, reference genes in maize using real-time PCR. J Agric Food Chem 57(7):2903
Zhang Nina Papazova David, Gruden Kristina, Vojvoda Jana, Yang Litao, Gašparič Meti Buh, Blejec Andrej, Fouilloux Stephane, De Loose Marc, Taverniers Isabel (2010) Evaluation of the reliability of maize reference assays for GMO quantification. Anal Bioanal Chem 396(6):2189–2201
Schaldberg TA, Norden TD, Nelson D, Jenkins GR (2009) Evaluating precision and accuracy when quantifying different endogenous control reference genes in maize using real time PCR. J Agric Food Chem 57:2903–2911
Köppel R, Dvorak V, Zimmerli F, Breitenmoser A, Eugster A, Waiblinger HU (2010) Two tetraplex real-time PCR for the detection and quantification of DNA from eight allergens in food. Eur Food Res Technol 230:367–374
Rönning SB (2003) Event specific real-time quantitative PCR for genetically modified Bt11 maize (Zea mays). Eur Food Res Technol 216:347–354
Nielsen Christer R, Berdal Knut G, Holst-Jensen Arne (2004) Charcterisation of the 5’ integration site and development of an event-specific real-time PCR assay for NK603 maize from a low starting copy number. Eur Food Res Technol 219:421–427
Lee S-H, Yi B-Y, Kim S-J (2009) Event-specific analytical methods for biotech maize MIR 604 and DAS-59122-7. J Sci Food Agric 89(15):2616–2624
Kim J-H, Kim H-Y (2009) Event-specific detection methods for genetically modified maize MIR604 using real-time PCR. Food Sci Biotechnol 18(5):1118–1123
La Paz J-L, Garca-Muniz N, Nadal A, Esteve T, Puigdomnech P, Pla M (2006) Interlaboratory transfer of a real-time polymerase chain reaction assay for quantitative detection of genetically modified maize event TC-1507. J AOAC Int 89(5):1347–1352
Köppel R, Bucher T, Frei A, Waiblinger H-U (2015) Droplet digital PCR versus multiplex real-time PCR Method for the detection and quantification of DNA from the four transgenic soy traits Mon87769, Mon87708, MON87705, FG72 and lectin. Eur Food Res Technol 241(4):521–527
Corbisier P, Barbante A, Berben G, Broothaerts W, De Loose M, Emons H, Georgieva TZ, Lievens A, Mazzara M, Papazova N, Perri E, Sowa S, Stebih D, Terzi V, Trapmann S (2017) Recommendation for the unit of measurement and the measuring system to report traceable and comparable results expressing GM content in accordance with EU legislation. Publ Office Eur Union. doi:10.2760/177516
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Compliance with ethics requirements
This article does not contain any studies with human or animal subjects.
Rights and permissions
About this article

Cite this article
Köppel, R., Bucher, T., Bär, D. et al. Validation of 13 duplex droplet digital PCR systems for quantitative GMO analysis of most prevalent GMO traits. Eur Food Res Technol 244, 313–321 (2018). https://doi.org/10.1007/s00217-017-2957-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-017-2957-4