
Abstract
The developed method for simultaneous detection of aflatoxin B1 (AFB1) and aflD genes can effectively monitor from the source and reduce the safety problems and economic losses caused by the production of aflatoxin, which can be of great significance for food safety regulations. In this paper, we constructed a sensitive and convenient fluorescent biosensor to detect AFB1 and aflD genes simultaneously based on fluorescence resonance energy transfer (FRET) between quantum dots (QDs) and a black hole quenching agent. A stable “Y” shaped aptasensor was employed as the detection platform and a double quantum dot labeled DNA fragment was utilized to be the sensing element in this work. When the targets of AFB1 and aflD genes were presented in the solution, the aptamer in the “Y” shaped probe is specifically recognized by the target. At this time, both Si-carbon quantum dots (Si-CDs) and CdTe QDs are far away from the BHQ1 and BHQ3 to recover the fluorescence. The linear range of the prepared fluorescence simultaneous detection method was as wide as 0.5–500 ng·mL−1 with detection lines of 0.64 ng·mL−1 for AFB1 and 0.5–500 nM with detection lines of 0.75 nM for aflD genes (3σ/k). This fabricated fluorescent biosensor was further validated in real rice flour and corn flour samples, which also achieved good results. The recoveries were calculated by comparing the known and found amounts of AFB1 which ranged from 88.4 to approximately 115.32% in the rice flour samples and 90.7 ~ 102.58% in the corn flour samples. The recoveries of aflD genes ranged from 84.32 to approximately 109.3% in the rice flour samples and 89.48 ~ 100.99% in the corn flour samples. Therefore, the proposed biosensor can significantly improve food safety and quality control through a simple, fast, and sensitive agricultural product monitoring and detection system.
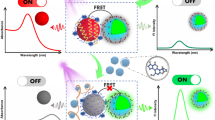
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mycotoxins are a group of secondary metabolites that are produced by fungi and can be detected at every stages of the food chain, contaminating approximately 25% of the world’s agricultural products, resulting in a reduction in their quality [1]. Nearly 100 species of toxin-producing fungi are in the record, which can produce about 400 types of metabolites [2]. Many mycotoxins show strong toxicity and are difficult to be degraded by cooking, baking, or frying due to their high heat stability [3]. These mycotoxins [4] were reported as the most common in our daily life including aflatoxin (AF), fumonisin (FB), ochratoxin A (OTA), T-2 toxin, and zearalenone (ZEN) [5, 6]. Especially, the AFB1 (aflatoxin B1) is regarded as the strongest carcinogenic mycotoxin and has been labeled as a group I carcinogen by the International Agency for Research on Cancer (IARC) [7]. AFB1 is characterized by high structural diversity and small molecular weight, which is harmful to humans and animals at low concentrations [8]. Therefore, it is very important to monitor AFB1 and its toxin-producing gene in foodstuff and its products. Currently, AFB1 detection methods commonly used in China include liquid chromatography-mass spectrometry [9], thin-layer chromatography [10], and high-performance liquid chromatography (HPLC) [11]. Nevertheless, these methods require complex pre-treatments, professional staff and laboratory environment, as well as expensive instruments. In addition to traditional detection methods, electrochemistry, fluorescence methods, and enzyme-linked immunosorbent assay (ELISA) [12] have been reported in recent years [13]. Förster resonance energy transfer (FRET) is a non-radiative energy transfer process between energy donors and energy acceptors that occurs between 10 nm, often referred to as a “FRET pair” [14]. Energy acceptors are usually chosen to absorb fluorescence from materials such as graphene oxide (GO) [15], black hole quenching agent (BHQ) [16], gold nanoparticles (AuNPs) [17, 18], and silver nanoparticles (AgNPs) [19]. Meanwhile, various FRET fluorescence energy donors have been successively reported, such as carboxyfluorescein [20], quantum dots (QDs) [21], upconversion nanoparticles (UCNPs) [22, 23], and metal nanoclusters [24, 25]. QDs are the energy donors chosen by many researchers due to their good biocompatibility and excellent photostability, which facilitate the recognition and detection of fluorescent biomarkers and various fungal toxins [15].
An aptamer is a single-stranded nucleic acid, systematically evolved by systematic evolution of ligands by exponential enrichment (SELEX) technique, capable of binding tightly and selectively as a natural antibody target [13]. Aptamers have outstanding characteristics compared to antibodies, including higher specificity and affinity [26,27,28], better thermochemical stability [29], high reproducibility, and ease of modification of multiple chemical groups [30]. Aptamers have several advantages over antibodies despite the fact that aptamers are similar in character to antibodies. Firstly, the production of aptamers is a chemical process, which avoids the problem of contamination during the production process and makes the production method simpler. Secondly, the aptamer is more specific and has a higher affinity for the target molecule. Ultimately, the aptamer is also more stable than the antibody, facilitating storage and making it a good alternative [31].
This paper presents the construction of a “Y” shaped fluorescent biosensor based on aptamer/ssDNA modified QDs and BHQ-labeled complementary DNA (cDNA). The use of FRET detection strategy to simultaneously monitor dual targets (AFB1 and aflD genes) in one step can be regulated at the source and has important implications for food safety. A stable “Y” shaped aptasensor was used as the detection platform and the FRET between the double QDs labeled aptamer and the quenching agents (BHQ1 and BHQ3) were used as the sensing element. When the targets are present in solution, they will bind specifically to the aptamer on the “Y” shaped probes. The QDs will be released away from the bursting agent to restore fluorescence. This novel fluorescent biosensor has high sensitivity and specificity, and it was further validated by detecting AFB1 and aflD genes in real samples. The developed strategy is promising to be a tool for the detection of food products contaminated with mycotoxins. By conducting dual target simultaneous detection, especially for toxin-producing genes, food crops can be prevented from being contaminated with aflatoxin in advance. Grains with potential for contamination can be picked out and processed to avoid further contamination.
Experimental
Reagents and apparatus
All reagents are of analytical grade: trisodium citrate (99%), 3-aminopropyltriethoxysilane (APTES), tellurium powder (99%), NaBH4 (99%), CdCl2 (99%), 3-mercaptopropionic acid (MPA, 99%), sodium hydroxide (NaOH, 96%), acetone (99%), 1-ethyl-3-(3-dimethyllaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS). The distilled water was used in the whole experiments.
The fluorescence spectrophotometer FS5 (UK) was used for fluorescence detection of samples. The UV-visible spectrophotometer UV-2700 (Japan) was used to record the UV-vis absorption spectrum. Sample morphology was examined by transmission electron microscopy (TEM, Tecnai G20, USA). Infrared spectra were obtained using a Fourier infrared spectrometer (Spectrum One, USA). Zeta potential was obtained with the zeta potential analyzer (Zetasizer Advance, UK).
All DNA sequences (Table S1) were purchased from Sangon Biotech Co., Ltd. (Shanghai, China).
Preparation of Si-CDs
On the basis of previously reported protocol with slight modifications, the Si-CDs were synthesized by the one-pot hydrothermal method [16, 32]. Briefly, 2.065 g trisodium citrate was dissolved in a beaker with 32 mL of deionized water and degassed with N2 for 15 min. 7.9 mL of APTES was injected into the beaker and kept stirring for 10 min. After stirring, the precursors obtained were sealed in a Teflon-lined stainless-steel autoclave of 50 mL followed by a hydrothermal treatment at 200 ℃ for 2 h. When the reaction was finished, the solution was naturally cooled to room temperature, opened the autoclave, and decanted the canary yellow solution into a 1-kDa dialysis bag to purify the unreacted substance with deionized water. After purified for 24 h, the solution was collected and stored in the refrigerator at 4 ℃.
Synthesis of CdTe QDs
The CdTe QDs were synthesized via an alternative scheme reported earlier, with slight modifications [33]. First, 0.05 g of NaBH4 and 0.0075 g of tellurium powder were added to 3 mL of deionized water, reconstituted under a nitrogen atmosphere for 15 min, and stored in a refrigerator for 4 h. During the reaction, NaBH4 was reduced and a purple NaHTe solution was generated. Meanwhile, 0.1140 g of CdCl2, 50 mL of distilled water, and 75 µL of MPA were added sequentially to a 100-mL three-neck flask. Stirring rapidly under nitrogen at 300 rpm, 1.0 M NaOH was dropped to regulate the pH to between 9 and 10. Then, 2 mL of purple NaHTe was quickly pipetted into the solution and continued to be protected with a nitrogen atmosphere. Eventually, the solution was refluxed in a constant temperature water bath at 100 ℃ for 4 h to obtain the MPA-modified red CdTe quantum dots and washed with acetone, the resulting deposit was re-dispersed in deionized water and vacuum freeze-dried to obtain a red powder, which was also kept at 4 ℃ in the refrigerator protected from light.
Conjugation of QDs with aptamers
EDC and NHS powders were dissolved in PBS buffer (10 mM, pH = 7.4) solution to obtain concentrations of 20 mg·mL−1 and 10 mg·mL−1, respectively. Three milliliters of Si-CDs/CdTe QDs, 150 µL of EDC, and 150 µL of NHS were mixed and incubated at 25 ℃ for 1 h to activate the surface carboxylic group, followed by adding 150 µL of aptamer/ssDNA to the solution and whisking gentler for further 12 h at 37 ℃. Subsequently, the solution was centrifuged at 10,000 rpm for 5 min and washed 3 times. Dispersion of the Si-CDs aptamer/CdTe-ssDNA probes was carried out in 3 mL of PBS buffer and stored at 4 ℃ [34].
The fabrication of the “Y” shaped biosensor
Before constructing the nanobiosensor, the DNA probes should be annealed at 95 °C and gradient cooled to room temperature which can avoid folding of the DAN probes [35]. For fabrication of the “Y” shaped aptasensor, 20 µL of the Si-CDs-aptamer and CdTe-ssDNA nanobiological mixture (1:1, v/v) was incubated with 5 µL of the BHQ1-cDNA-BHQ3 with PBS buffer for 1 h at 37 ℃. At this point, the BHQ1-cDNA-BHQ3 hybridized with the aptamer/ssDNA and some of the bases complementarily paired, causing a fluorescence burst at the QDs.
Detection of AFB1 and aflD genes in real samples
The practicality of the method was verified by quantifying AFB1 and aflD genes in the rice flour samples and corn flour samples with known concentrations of AFB1 and aflD genes [36]. Initially, 800 µL of the “Y” shaped probes was absorbed into centrifuge tubes. Then the samples were accurately weighed at 0.1 g ± 0.005 g and 100 µL of AFB1 and aflD genes was added. The mixture is subsequently mixed thoroughly using a shaker and centrifuged at 10,000 rpm for 10 min. The supernatants after centrifugation were filtered through 0.22-µm filters and diluted with PBS buffer for recovery experiments.
Results and discussion
Principle of the “Y” shaped biosensor
An overview of the “Y” shaped biosensor for AFB1 and aflD gene detection based on FRET strategy was schematically illustrated in Fig. 1. In this system, the aptamer was labeled with hydrothermal synthesis of Si-CDs and ssDNA was labeled with water bath reflux synthesis of CdTe QDs. When no target was available, the Si-CDs-aptamer and CdTe-ssDNA would hybridize to BHQ1-cDNA-BHQ3 via partial base complementary pairing to form the “Y” shaped aptasensor. The fluorescence of the Si-CDs and CdTe QDs was quenched by BHQ1 and BHQ3 due to FRET. When the targets were added, the AFB1 and aflD genes competed specifically for the aptamer and ssDNA and hybridized to form dsDNA-QDs owing to fluorescence recovery.

Schematic illustration of the aptasensor for the detection of AFB1 and aflD genes
Characterization of quantum dots and their DNA conjugations
The prepared Si-CDs, CdTe QDs, and quantum dot–conjugated aptamers were firstly studied using fluorescence spectra. Setting the fluorescence detector to a slit width of 2 and the excitation emission peak to 330 nm, the fluorescence emission peaks of the Si-CDs and CdTe QDs were obtained at 445 nm and 647 nm, respectively [21]. When the Si-CDs and CdTe QDs were red-shifted by 2 nm and 20 nm after coupling with the aptamer, the fluorescence emission peaks were 447 nm and 667 nm, respectively. As shown in Fig. 2A and C, the difference in wavelength between the emission peaks amounted to 220 nm, which indicates that they would be perfectly suited as fluorescent markers for the simultaneous dual-target detection. The prepared Si-CDs and CdTe QDs and the coupled aptamers/ssDNA were studied using UV-vis spectra. Figure 2B and D shows the UV-vis absorption spectra of the Si-CDs and CdTe QDs with characteristic peaks at 341 nm and 607 nm. The peaks at 270 nm for these two quantum dots were formed due to ssDNA binding, suggesting that these double QDs were successfully functionalized by aptamer/ssDNA through amide covalent bond formation. When excited with a 365-nm UV lamp, solutions of the Si-CDs and CdTe QDs emitted strong blue and red fluorescence, respectively (inset of Fig. 2A and C).

A The fluorescence emission spectra of Si-CDs and Si-CDs-Apt; B the UV-vis absorption spectra of Si-CDs and Si-CDs-Apt. Inset: photograph of Si-CDs solution under visible light and 365-nm UV light; C the fluorescence emission spectra of CdTe and CdTe-ssDNA; D the UV-vis absorption spectra of CdTe and CdTe-ssDNA. Inset: photograph of CdTe solution under visible light and 365-nm UV light
For the characterization of the shape and size of the Si-CDs and CdTe QDs, TEM imaging interpretation showed that the synthesized Si-CDs (Fig. 3A) and CdTe QDs (Fig. 3B) were uniformly distributed, well dispersed, and had a distinct lattice structure. The particle sizes of the quantum dots were calculated [37] from the relevant equations as 1.836 nm and 2.51 nm with ImageJ (Fig. 3C and D). The insets of Fig. 3A and B showed high-resolution transmission electron microscopy (HRTEM) of the Si-CDs and CdTe QDs which were nearly spherical with a lattice spacing of ca. 0.18 nm and 0.31 nm, respectively [38].

A TEM image of Si-CDs. Inset: HRTEM images of Si-CDs; B TEM image of CdTe QDs. Inset: HRTEM images of CdTe QDs; C statistical diagram of particle size distribution of Si-CDs; D statistical diagram of particle size distribution of CdTe
The FTIR spectra of the Si-CDs are shown in Fig. S1A. The formation of successful chemical functionalization of Si-CDs surfaces can be demonstrated by the characteristic peaks at 1027 cm−1 and 1140 cm−1 (v Si-O-CH) and 768 cm−1 (v Si-CH2). The N-H stretching vibration peaks appeared near 1409 cm−1, respectively, indicating the binding of amino groups on the surface of Si-CDs successfully. In addition, O-H stretching vibration peak appeared at 3411 cm−1, a -CH2- stretching vibration peak appeared at 2933 cm−1, and a C = O stretching vibration peak appeared near 1612 cm−1, which proved a large number of hydrophilic groups such as hydroxyl and carboxyl groups on the surface of Si-CDs. The surface chemistry of CdTe QDs was analyzed using FT-IR spectroscopy. The FT-IR spectrum of CdTe QDs showed the presence of amidogen was evidenced by the broad and peak at 3445 cm−1 (Fig. S1B). Moreover, C-H could be found at 2931 cm−1 and C = O could be found at 1565 cm−1 and 1565 cm−1, respectively. From the XRD pattern of Si-CDs (Fig. S1C), the broad diffraction bands of 19.7° and 31.5° can be attributed to disordered carbon atoms and (002) graphite lattice, respectively. Figure S1D shows the XRD pattern of CdTe QDs; the three diffraction peaks in the figure are the characteristic diffraction peaks of (111), (220), and (311) of CdTe QDs, respectively [39].
To further confirm the elemental composition of the Si-CDs and CdTe, XPS was employed to illustrate the study. As shown in Fig. S2A, the strong O 1 s and C 1 s peaks indicate that the Si-CDs are mainly composed of carbon and oxygen. The N 1 s and Si 2p peaks indicate that APTES is involved in the reaction of Si-CDs. In Fig. S2B, C 1 s can be decomposed into three peaks, with characteristic peaks appearing mainly at 284.8, 286.2, and 287.8 eV, corresponding to C-C, C-O-C, and O-C = O, respectively. N 1 s contains two peaks located at 399.8 and 400.38 eV, representing -NH, N-C, and C-NH2 (Fig. S2C). O 1 s contains two peaks located at 531.18 and 535.38 eV, respectively, representing O = C-O and C = O (Fig. S2D). Si 2p can be decomposed into two peaks at 101.8 and 103.78 eV, corresponding to Si3N4 and Si-(O)4, respectively (Fig. S2E). Figure S3A shows mainly four typical characteristic peaks including C 1 s (284.8 eV), Cd 3d (405.08 eV), O 1 s (531.08 eV), and Te 3d (618.08 eV). C 1 s contains three peaks located at 284.8, 286.08, and 288.5 eV, respectively, representing C-C, C-O-C, and O-C = O (Fig. S3B). O 1 s contains two peaks located at 531.28 and 535.68 eV, respectively, representing O-H and O = C-O (Fig. S3C). From Fig. S3D, it can be seen that the binding energies of Cd2+ are 404.98 and 411.68 eV, respectively. The binding energies of Te2− are 572.48 and 582.48 eV, respectively, with corresponding elemental valence states of Te 3d3/2 and Te 3d5/2 (Fig. S3E). In the meantime, high-resolution XPS of P 2p was used to verify the successful preparation of QDs labeled aptamer and ssDNA (Fig. S4A and B). The maximum signal peaks of Si-CDs-Apt and CdTe-ssDNA appeared at 133 eV, and it was attributed to the phosphate backbone in DNA.
By characterizing two quantum dots, the zeta potentials of Si-CDs and CdTe under neutral pH conditions were divided into − 9.359 and − 14.167 mV, indicating that both surfaces carry negative charges. At the same time, DNA is composed of a phosphate backbone, which carries negative charges. After coupling DNA with quantum dots, the size increases, resulting in a significant decrease in zeta potential emission, with potentials of − 14.523 and − 23.055 eV, respectively (Fig. S5).
Optimization of detection conditions
It is essential to adjust the relevant key parameters such as the type of buffer solution, incubation time [40], and pH value of the aptasensor to obtain the most optimum analytical performance. Four buffer solutions, which were PBS, Tris-HCl, Te, and deionized water, were selected to investigate the fluorescence intensity of the QDs which had been quenched. However, the substances in the TE buffer solution would chelate with the Cd2+ in the CdTe QDs, which would result in the complete quenching of fluorescence of the CdTe QDs. The PBS buffer solution had the lowest fluorescence intensity and was selected as the best buffer solution for the sensor construction process, consequently (Fig. S6A). Since the reaction time affects the detection efficiency and sensitivity of the “Y” shaped aptasensor, different reaction times were evaluated. As the reaction time was increased from 20 to 100 min, the fluorescence signal intensity decreased first and then increased and was lowest at 60 min, indicating that the “Y” shaped aptasensor had bound and stabilized, and the fluorescence intensity had turned off. Consequently, 60 min was selected as the incubation time for this experiment (Fig. S6B).
Furthermore, the concentration of the burster was fixed and the efficiency of fluorescence recovery after addition of the target was measured by varying the concentration of the aptamer/ssDNA. As shown in Fig. S6C, the fluorescence recovery efficiency increased significantly at 50 nM as the concentration of aptamer and ssDNA increased from 5 to 500 nM, indicating that a concentration of 50 nM was the best choice for constructing the “Y” shaped aptasensor to detect mycotoxins (F refers to the value of fluorescence recovery, and F0 refers to the value of fluorescence quenching) [2]. In order to make the detection performance of the sensor more stable, the influence of different pH on the target-induced fluorescence signal intensification was further investigated after identifying PBS as the buffer solution throughout the detection process. The highest fluorescence intensity was observed at pH = 7.4 after the addition of the target substance. Besides, pH = 7.4 could maintain the best value for biological activity. Therefore, pH = 7.4 was chosen as the optimum pH of the solution (Fig. S6D).
Finally, the hybridization time for fluorescence recovery upon addition of the target was optimized. The hybridization time from 40 to 100 min was investigated and is displayed in Fig. S6E. Apparently, the fluorescence intensity of solution decreased with the increasing hybridization time. When the hybridization time was 80 min, the fluorescence intensity of the solution changed most significantly and reached a plateau period. Therefore, 80 min was selected as the incubation time for the following analysis.
Simultaneous determination of AFB1 and aflD genes
Under optimum conditions, the proposed aptasensor was used to incubation with different concentrations of targets to simultaneous quantification of AFB1 and aflD genes. We added 5 µL of AFB1 and 5 µL of aflD genes standard solutions into 200 µL of the “Y” shaped aptasensor. After that, 210 μL of the mixture was incubated at 37 ℃ for 80 min and the fluorescence spectra were measured. The fluorescence intensity of the solution gradually increased with the increase of AFB1 and aflD gene concentrations (Fig. 4A). For the single detection of AFB1, the fluorescence intensity measured exhibited a good linear relationship. The linear regression equation for AFB1 was y = 868.68x + 4470.2, R2 = 0.9913 with a linear range of 0.5–500 ng·mL−1. Based on 3σ/k, the limit of detection (LOD) of AFB1 was determined to be 0.64 ng·mL−1 (Fig. 4B). For the detection of aflD genes, the linear equation was fitted as y = 1315.1x + 6661.7 and its correlation coefficient was R2 = 0.9905 with a linear range of 0.5–500 nM (Fig. 4C). The LOD of aflD genes was estimated to be as low as 0.75 nM [41].

A Linear curve of fluorescence recovery after adding target; linearity diagram of fluorescence detection obtained with different AFB1 B and aflD gene C concentrations
The analytical performance of the proposed aptasensor has been compared with the currently available single mycotoxins or aflD gene detection methods. The detection method, the range of linearity, and the LOD are summarized in Table 1. Although there are reports that the detection limit of AFB1 or aflD genes is very low, it can only be used to detect a single target. The results show that this method have wider linear ranges while detecting dual targets. And the sensitivity of this method is much higher than most reported methods.
Specificity analysis
In view of investigating the selectivity of the proposed fit-sensitive aptasensor, four possible interferences were applied, including DON, FB1, OTA, and ZEN. In addition to this, the mismatched DNA sequences were added to the prepared probes solution and the fluorescence intensity was measured. The selectivity of the ligand sensor was assessed by comparisons of the change in fluorescence intensity against other fungal toxins under the same conditions. There was little recovery of fluorescence in the presence of other fungal toxins except AFB1 which was mainly attributed to the combination of high affinity between the aptamer and target (Fig. 5A). As demonstrated in Fig. 5B, the degree of fluorescence recovery decreased with increasing amounts of mismatched bases. The fluorescence after the reaction of random DNA sequences was hardly recovered, and the fluorescence intensity was close to the blank group. The results show that the sensor has good specificity and selectivity.

Selectivity of sensing system: A the fluorescent response intensity of different numbers of different mycotoxins; B the fluorescent response intensity of different numbers of mismatched bases
Determination and recovery assay of AFB1 and aflD genes in rice flour and corn flour samples
In practical applications, fluorescent biosensors were used to determine the concentrations of AFB1 and aflD genes in rice flour and corn flour samples. On the basis of linear equations, the concentrations of AFB1 and aflD genes were observed to be in proximity to the spiked amounts. Recovery rates were calculated by the comparison of known and found amounts of AFB1 which ranged from 88.4 to approximately 115.32% in the rice flour samples and 90.7 ~ 102.58% in the corn flour samples. The recoveries of aflD genes ranged from 84.32 to approximately 109.3% in the rice flour samples and 89.48 ~ 100.99% in the corn flour samples (Table 2).
Conclusions
In summary, a “Y” shaped fluorescent biosensor was constructed based on Si-CDs and CdTe QDs conjugate with ssDNA to construct fluorescent probes. The probes were complemented with BHQ1-cDNA-BHQ3 to construct a “Y” shaped fluorescent aptasensor for simultaneous detection of AFB1 and aflD genes. The high affinity of the aptamer for its target led to the separation of the QDs-aptamer from the “Y” shaped DNA nanostructures. The fluorescent biosensor displayed the high sensitivity and selectivity for AFB1 and aflD genes. In addition, the adequate determination of AFB1 and aflD genes in rice flour and corn flour samples confirmed the applicability of this strategy. The design of the fluorescent aptasensor provided a prospective approach for the simultaneous detection of several mycotoxins and facilitated the use of aptamers in biosensors.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Kabak B, Dobson ADW, Var I. Strategies to prevent mycotoxin contamination of food and animal feed: a review. Crit Rev Food Sci Nutr. 2006;46:593–619. https://doi.org/10.1080/10408390500436185.
Xiong ZW, Wang Q, Xie YJ, Li N, Yun W, Yang LZ. Simultaneous detection of aflatoxin B1 and ochratoxin A in food samples by dual DNA tweezers nanomachine. Food Chem. 2021;338: 128122. https://doi.org/10.1016/j.foodchem.2020.128122.
Zhou SY, Xu LG, Kuang H, Xiao J, Xu CL. Immunoassays for rapid mycotoxin detection: state of the art. Analyst. 2020;145:7088–102. https://doi.org/10.1039/d0an01408g.
Fan Y, Li J, Amin K, Yu H, Yang HH, Guo ZJ, Liu JS. Advances in aptamers, and application of mycotoxins detection: a review. Food Res Int. 2023;170: 113022. https://doi.org/10.1016/j.foodres.2023.113022.
De Boevre M, Di Mavungu JD, Landschoot S, Audenaert K, Eeckhout M, Maene P, Haesaert G, De Saeger S. Natural occurrence of mycotoxins and their masked forms in food and feed products. World Mycotoxin J. 2012;5:207–19. https://doi.org/10.3920/wmj2012.1410.
Pereira VL, Fernandes JO, Cunha SC. Mycotoxins in cereals and related foodstuffs: a review on occurrence and recent methods of analysis. Trends Food Sci Tech. 2014;36:96–136. https://doi.org/10.1016/j.tifs.2014.01.005.
Ostry V, Malir F, Toman J, Grosse Y. Mycotoxins as human carcinogens—the IARC Monographs classification. Mycotoxin Res. 2016;33:65–73. https://doi.org/10.1007/s12550-016-0265-7.
Freire L, Sant’Ana AS, Modified mycotoxins: an updated review on their formation, detection, occurrence, and toxic effects. Food Chem Toxicol. 2018;111:189-205. https://doi.org/10.1016/j.fct.2017.11.021.
Abia WA, Warth B, Sulyok M, Krska R, Tchana AN, Njobeh PB, Dutton MF, Moundipa PF. Determination of multi-mycotoxin occurrence in cereals, nuts and their products in Cameroon by liquid chromatography tandem mass spectrometry (LC-MS/MS). Food Control. 2013;31:438–53. https://doi.org/10.1016/j.foodcont.2012.10.006.
Var I, Kabak B, Gök F. Survey of aflatoxin B1 in helva, a traditional Turkish food, by TLC. Food Control. 2007;18:59–62. https://doi.org/10.1016/j.foodcont.2005.08.008.
Pietri A, Fortunati P, Mulazzi A, Bertuzzi T. Enzyme-assisted extraction for the HPLC determination of aflatoxin M1 in cheese. Food Chem. 2016;192:235–41. https://doi.org/10.1016/j.foodchem.2015.07.006.
da Luz SR, Pazdiora PC, Dallagnol LJ, Dors GC, Chaves FC. Mycotoxin and fungicide residues in wheat grains from fungicide-treated plants measured by a validated LC-MS method. Food Chem. 2017;220:510–6. https://doi.org/10.1016/j.foodchem.2016.09.180.
Qian J, Ren CC, Wang CQ, Chen W, Lu XT, Li HN, Liu Q, Hao N, Li HM, Wang K. Magnetically controlled fluorescence aptasensor for simultaneous determination of ochratoxin A and aflatoxin B1. Anal Chim Acta. 2018;1019:119–27. https://doi.org/10.1016/j.aca.2018.02.063.
Pehlivan ZS, Torabfam M, Kurt H, Ow-Yang C, Hildebrandt N, Yüce M. Aptamer and nanomaterial based FRET biosensors: a review on recent advances (2014–2019). Microchim Acta. 2019;186:1–22. https://doi.org/10.1007/s00604-019-3659-3.
Zhang WQ, Ling J, Wen D, Cheng ZJ, Wang SP, Ding YJ. Simultaneous detection of acute myocardial infarction -related miR-199a and miR-499 based on a dual-emission CdTe fluorescent probe and T7 exonuclease-assisted signal amplification. Sens Actuators B Chem. 2022;371: 132484. https://doi.org/10.1016/j.snb.2022.132484.
Suo ZG, Liang XJ, Jin HL, He BS, Wei M. A signal-enhancement fluorescent aptasensor based on the stable dual cross DNA nanostructure for simultaneous detection of OTA and AFB1. Anal Bioanal Chem. 2021;413:7587–95. https://doi.org/10.1007/s00216-021-03723-8.
Kesharwani P, Ma RY, Sang L, Fatima M, Sheikh A, Abourehab MAS, Gupta N, Chen Z-S, Zhou Y. Gold nanoparticles and gold nanorods in the landscape of cancer therapy. Mol Cancer. 2023;22:98–128. https://doi.org/10.1186/s12943-023-01798-8.
Liu Z, Tao J, Zhu Z, Zhang Y, Wang H, Pang P, Wang H, Yang W. A sensitive electrochemical assay for T4 polynucleotide kinase activity based on Fe3O4@TiO2 and gold nanoparticles hybrid probe modified magnetic electrode. J Electrochem Soc. 2022;169: 027504. https://doi.org/10.1149/1945-7111/ac4f6c.
Maiti P, Sarkar S, Singha T, Dutta Roy S, Mahato M, Karmakar P, Paul S, Paul PK. Enhancement of fluorescence mediated by silver nanoparticles: implications for cell imaging. Langmuir. 2023;39:6713–29. https://doi.org/10.1021/acs.langmuir.3c00204.
Luo YS, Liu F, Li EZ, Fang Y, Zhao G, Dai X, Li JJ, Wang B, Xu MY, Liao B, Sun GP. FRET-based fluorescent nanoprobe platform for sorting of active microorganisms by functional properties. Biosens Bioelectron. 2020;148: 111832. https://doi.org/10.1016/j.bios.2019.111832.
Qian J, Cui HN, Lu XT, Wang CQ, An KQ, Hao N, Wang K. Bi-color FRET from two nano-donors to a single nano-acceptor: a universal aptasensing platform for simultaneous determination of dual targets. Chem Eng J. 2020;401: 126017. https://doi.org/10.1016/j.cej.2020.126017.
Li JZ, Zhao XD, Wang Y, Li S, Qin YK, Han T, Gao ZX, Liu H. A highly sensitive immunofluorescence sensor based on bicolor upconversion and magnetic separation for simultaneous detection of fumonisin B1 and zearalenone. Analyst. 2021;146:3328–35. https://doi.org/10.1039/d1an00004g.
Qin YK, Li S, Wang Y, Peng Y, Han DP, Zhou HY, Bai JL, Ren SY, Li S, Chen RP, Han T, Gao ZX. A highly sensitive fluorometric biosensor for Fumonisin B1 detection based on upconversion nanoparticles-graphene oxide and catalytic hairpin assembly. Anal Chim Acta. 2022;1207: 339811. https://doi.org/10.1016/j.aca.2022.339811.
Liang M, Lei ZL, Li YL, Xiao Y. A simple strategy to enhance the luminescence of metal nanoclusters and its application for turn-on detection of 2-thiouracil and hyaluronidase. Talanta. 2022;236: 122876. https://doi.org/10.1016/j.talanta.2021.122876.
Wang H-B, Tao B-B, Wu N-N, Zhang H-D, Liu Y-M. Glutathione-stabilized copper nanoclusters mediated-inner filter effect for sensitive and selective determination of p-nitrophenol and alkaline phosphatase activity. Spectrochim Acta A Mol Biomol Spectrosc. 2022;271: 120948. https://doi.org/10.1016/j.saa.2022.120948.
Ruscito A, DeRosa MC. Small-molecule binding aptamers: selection strategies, characterization, and applications. Front Chem. 2016;4:14–27. https://doi.org/10.3389/fchem.2016.00014.
Dong ZY, Zhang Q, Chen B-Y, Hong JM. Oxidation of bisphenol A by persulfate via Fe3O4-α-MnO2 nanoflower-like catalyst: mechanism and efficiency. Chem Eng J. 2019;357:337–47. https://doi.org/10.1016/j.cej.2018.09.179.
Zhuo ZJ, Yu YY, Wang ML, Li J, Zhang ZK, Liu J, Wu XH, Lu AP, Zhang G, Zhang BT. Recent advances in SELEX technology and aptamer applications in biomedicine. Int J Mol Sci. 2017;18:2142. https://doi.org/10.3390/ijms18102142.
Khan ZG, Patil PO. A comprehensive review on carbon dots and graphene quantum dots based fluorescent sensor for biothiols. Microchem J. 2020;157: 105011. https://doi.org/10.1016/j.microc.2020.105011.
Shao XL, Zhu LJ, Feng YX, Zhang YZ, Luo YB, Huang KL, Xu WT. Detachable nanoladders: a new method for signal identification and their application in the detection of ochratoxin A (OTA). Anal Chim Acta. 2019;1087:113–20. https://doi.org/10.1016/j.aca.2019.08.057.
Dunn MR, Jimenez RM. Chaput JC, Analysis of aptamer discovery and technology. Nat Rev Chem. 2017;1:0076. https://doi.org/10.1038/s41570017-0076.
Zhu WT, Zhou YS, Liu S, Luo M, Du J, Fan JP, Xiong H, Peng HL. A novel magnetic fluorescent molecularly imprinted sensor for highly selective and sensitive detection of 4-nitrophenol in food samples through a dual-recognition mechanism. Food Chem. 2021;348: 129126. https://doi.org/10.1016/j.foodchem.2021.129126.
Gao JW, Chen ZY, Mao LB, Zhang W, Wen W, Zhang XH, Wang SF. Electrochemiluminescent aptasensor based on resonance energy transfer system between CdTe quantum dots and cyanine dyes for the sensitive detection of ochratoxin A. Talanta. 2019;199:178–83. https://doi.org/10.1016/j.talanta.2019.02.044.
Lu ZS, Chen XJ, Wang Y, Zheng XT, Li CM. Aptamer based fluorescence recovery assay for aflatoxin B1 using a quencher system composed of quantum dots and graphene oxide. Microchim Acta. 2014;182:571–8. https://doi.org/10.1007/s00604-014-1360-0.
Deng HP, Liu QW, Wang X, Huang R, Liu HX, Lin QM, Zhou XM, Xing D. Quantum dots-labeled strip biosensor for rapid and sensitive detection of microRNA based on target-recycled nonenzymatic amplification strategy. Biosens Bioelectron. 2017;87:931–40. https://doi.org/10.1016/j.bios.2016.09.043.
Kumar YVVA, Renuka RM, Achuth J, Mudili V, Poda S. Development of a FRET-based fluorescence aptasensor for the detection of aflatoxin B1 in contaminated food grain samples. RSC Adv. 2018;8:10465-10473. https://doi.org/10.1039/c8ra00317c.
Zhu J, Wang SN, Li JJ, Zhao JW. The effect of core size on the fluorescence emission properties of CdTe@CdS core@shell quantum dots. J Lumin. 2018;199:216–24. https://doi.org/10.1016/j.jlumin.2018.03.047.
Wang YF, Wang K, Han ZX, Yin ZM, Zhou CJ, Du FL, Zhou SY, Chen P, Xie Z. High color rendering index trichromatic white and red LEDs prepared from silane-functionalized carbon dots. J Mater Chem C. 2017;5:9629–37. https://doi.org/10.1039/c7tc02297b.
Liu T, Fu LX, Yin CH, Wu M, Chen LG, Niu N. Design of smartphone platform by ratiometric fluorescent for visual detection of silver ions. Microchem J. 2022;174: 107016. https://doi.org/10.1016/j.microc.2021.107016.
Ma Q, Nie DQ, Sun XY, Xu YL, He JX, Yang L, Yang LZ. A versatile Y shaped DNA nanostructure for simple, rapid and one-step detection of mycotoxins. Spectrochim Acta A Mol Biomol Spectrosc. 2022;281: 121634. https://doi.org/10.1016/j.saa.2022.121634.
Hu O, Li ZY, Tong YL, Wang QY, Chen ZG. DNA functionalized double quantum dots-based fluorescence biosensor for one-step simultaneous detection of multiple microRNAs. Talanta. 2021;235: 122763. https://doi.org/10.1016/j.talanta.2021.122763.
Guo P, Yang W, Hu H, Wang Y, Li P. Rapid detection of aflatoxin B1 by dummy template molecularly imprinted polymer capped CdTe quantum dots. Anal Bioanal Chem. 2019;411:2607–17. https://doi.org/10.1007/s00216-019-01708-2.
Chen L, Wen F, Li M, Guo X, Li S, Zheng N, Wang J. A simple aptamer-based fluorescent assay for the detection of aflatoxin B1 in infant rice cereal. Food Chem. 2017;215:377–82. https://doi.org/10.1016/j.foodchem.2016.07.148.
Wang C, Yu H, Zhao Q. A simple structure-switch aptasensor using label-free aptamer for fluorescence detection of aflatoxin B1. Molecules. 2022;27:1–9. https://doi.org/10.3390/molecules27134257.
Sedighi-Khavidak S, Mazloum-Ardakani M, Rabbani Khorasgani M, Emtiazi G, Hosseinzadeh L. Detection of aflD gene in contaminated pistachio withAspergillus flavusby DNA based electrochemical biosensor. Int J Food Prop. 2017;20:S119–30. https://doi.org/10.1080/10942912.2017.1291675.
Li Y, Yu T, Li J, Kong D, Shi Q, Liu C, Dong C. A novel fluorescent FRET hairpin probe switch for aflD gene detection in real fermented soybean paste. Food Anal Methods. 2021;14:2469–77. https://doi.org/10.1007/s12161-021-02080-7.
Yu T, Peng S, Sun Q, Kong D, Liu C, Shi Q, Li Y, Chen Y, Fluorescence resonance energy transfer biosensor based on nitrogen doped carbon quantum dots and hairpin probe to sensitively detect aflatoxin biosynthesis-related genes aflD in rice. Res Sq 2022;1–15. https://doi.org/10.21203/rs.3.rs-1814775/v1.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31901799, 32001804), the China Postdoctoral Science Foundation (No. 2021M692370), the JUST Emerging Science and Technology Innovation Team Grant (No. 1182921902), the Jiangsu Dual Innovation Talent Program (No. 1184902001), and the Jiangsu University of Science and Technology Research Start-up Fund (No. 1182932001, 1182922001).
Author information
Authors and Affiliations
Contributions
Yaqi Li: funding acquisition, conceptualization, writing, review and editing. Qingyue Sun: conceptualization, investigation, writing—original draft. Xin Chen: the main strengths in the stage of article revision including synthesis and preparation new quantum dot probes, data analysis, and literature searching and organization. Shuangfeng Peng: data curation, formal analysis. Dezhao Kong: supervision. Chang Liu: validation. Qi Zhang and Qiaoqiao Shi: methodology. Yong Chen: supervision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Y., Sun, Q., Chen, X. et al. Simultaneous detection of AFB1 and aflD gene by “Y” shaped aptamer fluorescent biosensor based on double quantum dots. Anal Bioanal Chem 416, 883–893 (2024). https://doi.org/10.1007/s00216-023-05074-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-05074-y