
Abstract
With recent advances in ionization sources and instrumentation, ion mobility spectrometers (IMS) have transformed from a detector for chemical warfare agents and explosives to a widely used tool in analytical and bioanalytical applications. This increasing measurement task complexity requires higher and higher analytical performance and especially ultra-high resolution. In this review, we will discuss the currently used ion mobility spectrometers able to reach such ultra-high resolution, defined here as a resolving power greater than 200. These instruments are drift tube IMS, traveling wave IMS, trapped IMS, and field asymmetric or differential IMS. The basic operating principles and the resulting effects of experimental parameters on resolving power are explained and compared between the different instruments. This allows understanding the current limitations of resolving power and how ion mobility spectrometers may progress in the future.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ion mobility spectrometers (IMS) separate and analyze ions based on their motion through a neutral gas under the influence of an electric field. Ion separation in IMS occurs usually within milliseconds, and mostly based on the collision properties between ions and neutral gas molecules. This results in three major advantages of IMS that have led to their rising popularity in many applications.
-
IMS can be easily coupled to extremely efficient atmospheric pressure chemical ionization sources, allowing for limits of detection in the low pptv-range for substances amenable for chemical ionization.
-
IMS offer separation in a dimension different to gas chromatography, liquid chromatography, and mass spectrometry on a millisecond timescale. Thus, both fast stand-alone detection devices and hyphenated instruments for multidimensional separations are feasible.
-
IMS separation is based on the physical structure of ions, providing size and shape information.
The combination of the first two of these three strengths has led to its widespread use as a detector for chemical warfare agents [1, 2], explosives [2,3,4], drugs [5], and other hazardous compounds beginning in the 1970s. A detailed history of IMS can be found in the textbook on IMS by Eiceman, Karpas, and Hill [6]. Today, IMS can be found at most airports and in many military units [2]. More recently, often coupled with gas chromatography for pre-separation, IMS have been used in the analysis of more complex samples, for example in the quality control of food [7,8,9] and pharmaceuticals [10, 11], process control [12], or exhaled breath gas analysis [13, 14].
However, it has been the combination of the latter two of these three strengths that has led to its rise in more analytical tasks, especially bioanalytical applications [15,16,17,18,19,20,21,22,23,24]. On the one hand, operating on a millisecond to second timescale, IMS perfectly combines both with chromatographic separation such as gas chromatography (GC), liquid chromatography (LC), or supercritical fluid chromatography (SFC) as well as with mass spectrometry (MS) [25]. This allows three-dimensional, or, when using 2D GC or 2D LC, even four-dimensional separation [26, 27], enabling the selectivity needed for extremely complex samples. On the other hand, IMS provides not only another dimension of separation but also structural information about the ions [15, 16, 20, 22, 28]. This allows distinguishing between many isomers and differently folded structures of biomolecules.
Just as mass spectrometers divide into different instruments, e.g., time-of-flight, sector, quadrupole, and ion trap devices, IMS can be also grouped into different instruments. Recent reviews list as many as eight different main operating principles [29, 30]. However, only a few of them are able to reach ultra-high resolution. Thus, two aspects should be discussed first in this review—the very basics of ion mobility spectrometry and how to define ultra-high resolution.
Basics of ion mobility spectrometry
Generally, ion mobility spectrometry separates and analyzes ions based on their motion through a neutral buffer gas under the influence of an electric field. The ion mobility K is then defined as the proportionality factor between the ion’s drift velocity vd and the electric field strength E according to Eq. 1.
Ions are constantly accelerated in the direction of the electric field, but collide with neutrals due to their thermal motion, and decelerate. These processes quickly reach an equilibrium, leading to a constant drift velocity. As heavier ions accelerate slower, but also lose less energy during a collision, the likelihood of a collision becomes the most important parameter. Thus, ion mobility spectrometry is mostly a separation based on the ion-neutral collision properties. At extremely low collision energies, the ion mobility does also not depend on the structure of the ion, as repulsion due to polarization already occurs at large distances. Therefore, this is called the “polarization limit.” As given by Eq. 2 [31, 32], the ion mobility only depends on the ion mass m, the neutral mass M, the dipole polarizability of the neutral αd, and the number density of the neutrals N.
However, this case is only applicable in cryogenic systems, as the thermal energy of the ions at room temperature is sufficient to leave this realm. At these thermal energies, the ion mobility will depend on the collision cross section (CCS) between ion and neutral Ω as given by Eq. 3. Thus, the ion mobility is related to the size and shape of an ion. Other variables are the charge state z, the elementary charge e, the Boltzmann constant kB, and the absolute temperature T. It is noteworthy that a certain correlation between ion mass m and ion-neutral collision cross section Ω exists, as molecules containing many atoms grow both heavier and larger.
It is important to note that Eq. 3 was derived under the assumption of a negligibly low electric field. As soon as an electric field is applied, correction factors would be required to account for ion energies above the thermal energy of the ions and directional bias of collisions [33]. This increase in ion energy is often referred to as “ion heating,” as the effects are identical to those of increasing thermal energy [31]. Several correction approaches exist, most typical the two-temperature theory [34, 35], the three-temperature theory [36, 37], and the momentum-transfer theory [33], each with different adjustments and with different quality of correction depending on the ions studied [38]. However, these correction factors can typically be neglected at low electric field strengths. An estimation for what might be considered a low electric field strength is given by Eq. 4 [39]. Here, N0 is Loschmidt’s constant, which is the number density at standard conditions, and K0 is the ion mobility at standard conditions, known as the reduced ion mobility. It is noteworthy that the limit is given as the ratio between the electric field E and the number density of the neutral N, as either doubling E or halving N would have the same effect on the energy upon collision. E/N is called the reduced field strength and given in Townsend (Td). Furthermore, it is important to note that not only the static drift field needs to be considered but also AC fields for example for ion focusing [40].
The drift velocity can also be calculated directly from the reduced ion mobility K0 and the reduced field strength E/N as given by Eq. 5. Thus, ions move with the same drift velocity regardless of pressure when adjusting E to the same reduced field strength E/N.
At much higher energies, even more structural information can be revealed about the ions. This is shown for isotopomers, which have the same mass and the same collision cross section, but can nevertheless be separated at high reduced field strengths [41, 42]. Thus, the different mass distribution within the ion must also affect these high-energy collisions. Due to the complexity and limitations of the corrections mentioned above for more complex ion-molecule systems, the ion mobility at high electric field strengths is often empirically described through the alpha function α(E/N) as given by Eq. 6. As the alpha function may not depend on the direction of the electric field, it is typically represented through an even power series [31, 43].
The alpha function and therefore also K(E/N) contain various effects, as the increased ion energy may not only effect Eq. 3 but also cause changes to the collision cross section itself, for example, through declustering ion-molecule complexes, through changing conformations, or simply due to the collision cross section being energy dependent. The alpha function can be measured through two different approaches, either by using a drift tube ion mobility spectrometer able to reach such high ratios of E/N [44,45,46] or by using an electric field alternating between high and low E/N, known from differential ion mobility spectrometry (DMS) or field asymmetric ion mobility spectrometry (FAIMS) [47, 48]. While a high reduced field strength drift tube IMS measures K(E/N) directly, differential or field asymmetric IMS can only obtain information on the alpha function, but not on the ion mobility. Furthermore, due to the dynamic field, measurements from differential or field asymmetric IMS may be perturbed by dynamic effects and also require a complex calculation to obtain the alpha function from measurement data.
Thus, ion mobility spectrometry includes measurements of the true low-field ion mobility, of an ion mobility perturbed by added energy, of the ion mobility at a defined increased energy, and of the alpha function.
Definition of ultra-high-resolution IMS
Following the terms of the IUPAC definition for chromatography, resolution in ion mobility spectrometry is defined as the separation between two peaks [49]. In practice, comparing resolution between two IMS would require to measure the same substances. Thus, the resolving power, defined as the ratio between the position of a peak and its full width at half maximum (FWHM), is usually used instead. As long as the relative positions of peaks remain the same, the resolution is proportional to the resolving power. Measurement results from a drift tube IMS may be reported in terms of the drift time td, the inverse ion mobility 1/K, or the collision cross section Ω. The inverse ion mobility is often used instead of the ion mobility as it is proportional to the other two quantities. No matter which of the three scales is chosen, resolving power, resolution, and peak capacity remain the same for a drift tube IMS, as all three scales are proportional to each other. However, for IMS with nonlinear ion motion, such as traveling wave IMS, this is not the case as shown in Fig. 1. Despite the fact that the separation and thus resolution are exactly the same for both IMS, the resolving power of the traveling wave IMS appears to be worse in the time domain due to the different ion motion mechanism. Only after conversion to a common scale such as the inverse ion mobility 1/K or the collision cross section Ω, the resolving powers of the two devices may be compared.

Illustration of the observed resolving powers in the time and CCS domains of a drift tube IMS with linear ion motion and a traveling wave IMS with nonlinear ion motion. Both devices show exactly the same degree of separation between the two peaks despite the fact that their resolving power appears different in the time domain due to different time scales
It has been suggested to use the resolving power in the scale of the collision cross section Ω for comparison [50] and we will follow this suggestion throughout the paper. Furthermore, it needs to be noticed that multiply charged ions are easier to separate and thus comparisons between instruments should be done using ions of the same charge state.
This way, all IMS measurements directly related to the collision cross section can be compared with each other. Only the alpha function is not directly related to the collision cross section and thus the resolving power of FAIMS cannot be compared directly with other IMS. At constant resolving power, ion separation by FAIMS may be better or worse compared with other IMS depending on the substance [51]. Furthermore, resolving powers can obviously not be compared directly between IMS and gas or liquid chromatography due to their different separation space.
Following a previous definition [52], one can attribute high resolution to IMS with a resolving power above 80, which is the upper end for most commercial devices. A resolving power above 200 is considered ultra-high resolution, which is sufficient to resolve two peaks equal in height with a 1% difference in collision cross section with a valley of 12.5% of their peak height [53]. Furthermore, sharper peaks may also ease determining the peak position exactly and may even, in the case of a constant number of ions and thus a constant peak area, be higher, improving the signal-to-noise ratio [54]. Currently, only five IMS technologies have reached ultra-high resolution: drift tube ion mobility spectrometers (DT-IMS), the ion cyclotron mobility spectrometer, traveling wave ion mobility spectrometers with extended path lengths (cyclic-TW-IMS and SLIM-TW-IMS), trapped ion mobility spectrometers (TIMS), and differential or field asymmetric ion mobility spectrometers (DMS/FAIMS). Thus, this review will focus on these instruments. The different operational principles are illustrated in Figs. 2 and 3. An overview comparing the main parameters is given at the end of the review in Table 1.

Different applications of the electric field to the drift region in the different ion mobility spectrometers. (A) Constant field strength in the drift tube IMS. (B) Switched segments in the ion cyclotron mobility spectrometer. (C) Moving wave in traveling wave IMS. (D) Field gradient trapping the ions against the gas flow in trapped IMS

Application of the electric field in a field asymmetric IMS
Drift tube IMS
The drift tube ion mobility spectrometer is the original embodiment of an ion mobility spectrometer. The original measurements in the 1930s by Tyndall and Powell [66] as well as Bradbury and Nielsen [67] used this setup, as well as the first military trace gas detectors such as the chemical agents monitor (CAM) [2, 6] and the first analytical IMS(-MS) instruments [68, 69]. In order to measure the ion mobility, a small packet of ions is injected by an ion shutter into a drift tube with a constant electric field and the drift time required to reach the detector is measured. Drift tubes can be manufactured in a wide variety of sizes and from a wide variety of materials such as resistive glass tubes [70, 71], low temperature co-fired ceramics (LTCC) [72], printed circuit boards (PCB) [73, 74], or even 3D printing [75]. The expected drift time of an ion with ion mobility K through a drift tube of length L with the electric field E is given by Eq. 8.
Drift tube IMS can be considered a “jack of all trades” instrument, being applicable to most measurement tasks. The full ion mobility spectrum is acquired within a single measurement of a few to a few 10 ms and can be averaged to increase the signal-to-noise ratio and thus lower the limits of detection. However, the short time frame of spectra requires fast mass spectrometers for fully nested operation [76, 77]. Ion yield can be vastly increased by using multiplexing [78,79,80,81,82], a field switching shutter for compatible ion sources [83, 84], or injection from an ion trap when operating at reduced pressure [85]. There is no inherent limit to the ion mobility range of a drift tube IMS—when measuring negative ions in gases not capturing electrons such as nitrogen, even an electron peak can be observed [86, 87], while the upper limit is only given by the ability to bring ions into the gas phase. However, it should be noted that measuring very slow and very fast ions in a single spectrum requires an ion shutter with extremely low discrimination of slow ions, meaning that the initial packet width should not depend on the ion mobility [46, 88, 89]. Furthermore, as there is a direct relationship between drift time and ion mobility, ion mobility and collision cross section can be directly obtained from a measurement without calibration through Eqs. 8 and 3. Specialized high-accuracy drift tube IMS are able to measure the ion mobility within ± 0.1% [90,91,92] and serve as the reference standard for other ion mobility measurements.
Generally, the resolving power of any separation by ion mobility strongly depends on the diffusion during the ion drift. The diffusion-limited resolving power Rp,Diff [93, 94] can be calculated from drift time, diffusion, and velocity of the ion motion [95] as given by Eq. 9. The diffusion-limited resolving power just depends on three parameters—the absolute temperature T, the charge state of the ions z, and the drift voltage U. As mentioned above, higher charge states and lower temperatures improve IMS separation power, which needs to be kept in mind when comparing different IMS. Furthermore, as these constants will appear in many more equations, we define a combined constant C as given by Eq. 10 to simplify notations.
However, analytical modeling of drift tube IMS has progressed much further over the years to include additional effects such as initial ion packet width and amplifier distortion, which in turn also create a dependence on ion mobility and drift length. Every drift tube has an optimum drift voltage that maximizes its resolving power by striking a balance between the diffusion during long drift times and the increasing effect of the additional peak width caused by ion injection and detection at short drift times [93, 94, 96,97,98]. The maximum resolving power can be given as a function of the optimum drift voltage [54, 55, 96] and is \( \sqrt{2/3} \) of the diffusion-limited resolving power at the same drift voltage [54]. This of course requires an IMS designed to operate at this optimum drift voltage [96]. Increasing the drift voltage above the optimum results in a slight loss of resolving power, but universally increases the signal-to-noise ratio [99] due to sharper peaks, less ion losses, and more averages due to the reduced time frame of the spectra. However, we will use the simple model in Eq. 9 in this review, as models for other IMS concepts just consider ion motion, and thus the different models will be comparable.
Atmospheric pressure drift tube IMS (AP-DT-IMS)
As most other ultra-high-resolution IMS concepts are tailored to be coupled with mass spectrometers and therefore operated at reduced pressures, we will consider atmospheric pressure drift tube IMS and low pressure drift tube IMS separately for easier comparison. Beside IMS aiming to reach high reduced field strengths [44,45,46], stand-alone instruments are practically always operated at atmospheric pressure. Furthermore, some IMS-MS also use drift tubes at atmospheric pressure due to their superior separation performance [57, 100,101,102].
The expected resolving power of a drift tube IMS is derived by inserting Eq. 10 into Eq. 9 and given by Eq. 11. As drift tube IMS employ a constant and uniform field as shown in Fig. 2(A), the drift voltage U and drift field strength E are directly related through the length L.
The main advantages of operating IMS at atmospheric pressure are easier coupling to highly effective atmospheric pressure chemical ionization sources for maximum ion yield and sensitivity and low E/N even at high voltages. Therefore, high drift voltages can be applied to short drift tubes to maximize resolving power as long as the electronics for ion injection and detection are sufficiently fast. The highest resolving drift tube IMS reported achieves a resolving power of 250 to 260 for several small, single charged ions such as DMMP, benzene, toluene, and acetone by applying a drift voltage of 25 kV across a 15-cm drift tube at 1000 mbar [53, 55]. Another drift tube IMS with a length of 63 cm operated at 325 mbar with 10 kV has been reported to reach a resolving power of 172 for single charged C60 clusters [103] and 240 for the minus four charge state of CH3(SO2NHSO2(CH2)6)5SO2NHSO2CH3 [56] as well as 260 for an unidentified peak [56]. Using a drift region at atmospheric pressure with a length of 13 cm and a drift voltage of only 3.6 kV, a resolving power of 216 was obtained for the plus 11 charge state of cytochrome c [57]. This extremely high resolving power at low drift voltage again emphasizes the need to keep the charge state in mind when comparing resolving powers.
In summary, the available maximum drift voltage is the resource limiting both the resolving power and the signal-to-noise ratio and therefore the ultimate limit of drift tube IMS performance. Future improvements in atmospheric pressure drift tube IMS will thus most likely proceed together with improvements in compact high voltage power supplies, power, and data isolation as either the ion source or the detector is referenced to this high voltage and improved systems design to handle high voltages in small enclosures without breakdown. Furthermore, increasing drift voltage at the same drift length requires faster electronics for ion injection and detection.
Low pressure drift tube IMS (LP-DT-IMS)
When coupling IMS with MS, a reduced pressure in the drift tube simplifies the ion transfer and gives the opportunity to store or mass-select ions prior to injection. However, below a certain pressure, the low-field limit given by Eq. 4 must be considered due to decreasing neutral density N. At this point, it is no longer possible to apply arbitrary drift voltages to a drift tube without heating the ions, leaving two possible scenarios. If the low-field mobility is the quantity of interest, the drift field is now fixed to a maximum Emax and increasing the resolving power is only possible by increasing the drift length as shown by Eq. 12.
This has given rise to drift tubes several meters long. An IMS-IMS-IMS-MS instrument with a drift tube as long as 3 m has been reported [104]; however, to our knowledge, no resolving power when using the whole drift tube as one has been reported. For a 2-m-long drift tube operating between 15 and 20 mbar, a resolving power of 109 was measured for the plus two charge state of angiotensin II using a drift voltage of 5 kV [58]. Moving back to higher pressure would be a possible remedy. Halving the length and doubling the pressure would result in both the same resolving power and reduced field strength, but in a more compact instrument and, due to the constant drift velocity given by Eq. 5, also in halved measurement times. On the other hand, high reduced field strengths can also be used to purposefully increase the ion energy and thus measure not only K(0) but also K(E/N), adding another dimension of ion separation and characterization [44, 45]. This also leads to higher resolving powers, as high voltages in short drift tubes are now possible again. For example, by applying a drift voltage of 18 kV across a drift tube of 30 cm at a pressure of 20 mbar, which equals 120 Td and is close to the breakdown of air, a resolving power of 140 has been achieved for single charged methyl salicylate [46]. Still, the breakdown of the drift gas creates an ultimate limit for the achievable field strength. Thus, although low pressure drift tube IMS offer a number of advantages, achieving ultra-high resolution is difficult.
Extending separation time
As the achievable performance of an accordingly designed drift tube IMS is limited by the maximum drift voltage available, while the maximum drift field strength Emax is limited at reduced pressure, an increase in resolving power would only be possible through longer drift tubes. Thus, several quite different approaches have been developed to overcome this limitation. They all share one common defining feature—they are able to trade measurement time for resolving power by prolonging the measurement and thus “reusing” the same drift voltage. This is possible as all such IMS operate at reduced pressure and can thus employ radial focusing. Nevertheless, as can be seen from Fig. 2, they all retain the same principle of ion motion along a drift region, but with differently applied electric fields. Often, ions move several cycles through the instrument and the voltage is only applied to segments of the drift region. In this review, we will unanimously use n for the number of cycles and m for the number of segments. While this differs from the nomenclature used in some of the references, it ensures that the notation is consistent throughout this work.
It should be kept in mind that the ion focusing may affect the measurement through ion heating. Furthermore, these techniques are only applicable for ions that can actually be focused, leading to a limitation of the accessible mobility range and mass range [105, 106]. However, for most applications not related to trace gas analysis, this limitation is typically not of practical relevance. Additionally, extending the separation time also increases the diffusion, meaning that more averages and thus even more measurement time are typically necessary to maintain the same signal-to-noise ratio. Generally, one would expect four times the measurement time to double resolving power and another four times to maintain the signal-to-noise ratio. However, these devices are relatively new and theories regarding the ion motion and separation do not consider the signal-to-noise ratio yet.
Ion cyclotron mobility spectrometer
The first such device was the ion cyclotron mobility spectrometer or cyclical drift tube IMS originally published in 2009 [107] followed by two improved versions in 2010 [108] and 2013 [59]. It consists of four curved quarter-circle drift tube segments with ion funnels in between to re-focus the ions, together forming a circular drift tube with a length of 181 cm. Thus, possible operating pressure is limited to a few millibars by the ion funnel operating range. The drift field is switched periodically to keep the ions moving around the circle as shown in Fig. 2(B) for n cycles, leading to an n times longer drift tube that only requires the drift voltage for two of its m segments to be switched on at the same time. Thus, the resolving power and required voltage are given by Eqs. 13 and 14. The drift time is simply what would be expected of a drift tube prolonged to this length.
However, this is only strictly true during the first few cycles. As the spectrum spreads out around the ring, ions not moving at the drift field switching frequency are eliminated. Faster ions move ahead of the drift field and are discharged at its front, while slower ions lag behind the drift field and are discharged there. This turns the device into an ion filter as known from overtone ion mobility spectrometry (OMS) [109,110,111]. Here, the ion current passing the device at a certain drift field application frequency is measured. The resolving power equation for OMS is rather complex [110] and depends both on the resolving power expected from the effective drift length and on the number of segments passed along that length. However, we will not go into detail here, as the resolving power expected from the effective length seems to be sufficient for comparison.
Using n = 100 cycles to achieve a drift length of about 180 m results in resolving powers above 1000 [59], here measured for the z = 3 charge states of substance P at a pressure of 3 mbar. This was published in 2013 and the first reported resolving power above 1000 in the history of IMS. Furthermore, the measured mobilities agree well with those measured by a conventional drift tube IMS [108], meaning that no additional calibration would be required. However, possible ion heating effects have not been studied, although the good agreement with drift tube values suggests that they are low.
Despite the possibility to filter four ion packets simultaneously as four segments exist [59], the device is ultimately limited by the significant loss of ions at long drift times of more than a second. Combined with the need to scan the drift field application frequency to record the full ion mobility spectrum, extremely long measurement times are required. This may presumably be the reason why no further research concerning the ion cyclotron mobility spectrometer has been reported despite the outstanding separation capability.
Traveling wave ion mobility spectrometry
Traveling wave ion mobility spectrometry was first published in 2004 [112, 113], with a second-generation instrument being published in 2011 [114]. Again, the drift field is not applied across the whole drift tube, but only across small segments. These segments are not switched on and off as a whole, but move along the drift tube ring by ring as shown in Fig. 2(C), forming the namesake traveling waves that push the ions through the drift tube. Furthermore, unlike a moving drift field, the waves move faster than the ions and thus cause them to roll over the waves instead of moving through the IMS with constant drift velocity. Normally, ions would be pushed towards the drift tube wall at the front and at the end of a traveling wave and eliminated. However, an additional RF potential is applied between adjacent rings similar to an ion funnel [105, 115, 116], focusing the ions towards the center of the drift tube throughout the whole length and minimizing ion discharge at the walls. Again, operating pressures are limited to a few millibars by the operating range of the ion focusing.
Due to the effect of ions rolling over the moving wave, the drift velocity becomes a nonlinear function of the ion mobility, depending approximately on its square assuming an idealized triangular waveform [117]. For all other waveforms, the dependence is significantly more complex and depends on the distribution of the electric field strengths inside the wave [117]. Thus, calibration with suitable standard ions becomes necessary in order to extract ion mobilities and collision cross sections [118, 119]. Furthermore, heating of ions by the RF confinement needs to be considered [120,121,122], especially when choosing the standard ions [123,124,125], as these may experience heating effects too. Assuming an idealized triangular waveform and selecting the wave velocity to maintain rollover for the ions with the highest mobility Kmax, the drift time can be estimated by Eq. 15 for comparison with other IMS instruments.
Calculating the theoretical collision cross section resolving power of a traveling wave ion mobility spectrometry (TW-IMS) [117], again assuming the ideal triangular waveform, results in Eq. 16. This is derived from equations 27 and 34 from reference [117]. The nonlinear ion motion in traveling wave IMS increases the ion mobility separation and therefore the collision cross section resolving power by a factor of \( \sqrt{4} \). It should be noted that this is not observable on the nonlinear drift time scale, underlining the mentioned need to use a common scale for resolving power comparisons. However, resolving power decreases for ion mobilities lower than the maximum ion mobility Kmax, diminishing this advantage. As given by Eq. 17, the required voltage is reduced by twice the number of waves m in the traveling wave IMS compared with a drift tube IMS. The factor 2 stems from the electric potential decreasing with the maximum field strength Emax in both directions from its peak at the center of each wave.
By setting L/m constant, which means fixing the width b of a traveling wave, the required voltage UTW becomes independent of the drift length of the IMS. This is the main advantage of traveling wave IMS—while its resolving power is approximately the same compared with drift tube IMS using the same length and field strength, it can be built at arbitrary lengths without requiring additional voltage.
Generally, the resolving power reported for traveling wave IMS is moderate for the first- and second-generation instruments, ranging from 10 [113] to 40 [114]. However, in recent years, two approaches have emerged that make full use of the advantages of the traveling wave approach by vastly extending the drift lengths. At a fixed maximum electric field strength, this is the only possibility for resolving power improvement. The first approach is a cyclic multi-pass arrangement first presented in 2014 [126, 127] with a second-generation instrument reported in 2017 [61]. This traveling wave IMS is simply arranged in a circle with a port for transferring ions in and out. Further modifications are not necessary, as the measurement principle already allows for infinitely long drift tubes. The length is thus multiplied by the number of cycles n in the instrument as given by Eq. 18, resulting in a longer effective drift length, while the required voltage still remains the same.
As expected, the resolving power grows with the square root of the number of cycles. It reaches about 550 for 50 cycles equivalent to an effective drift length of 50 m [61], measured for the single charged peptides SDGRG and GRGDS at a pressure of 1.8 mbar. The total drift time was about 90 ms. Due to the constant focusing, ion transmission was relatively high. An ion loss of 35% was reported for six cycles, which corresponds to an effective drift length of 6 m.
It should be noted that, unlike in the ion cyclotron mobility spectrometer, ions are not eliminated by principle during the measurement. Thus, it is necessary either to distinguish between ions having traveled different numbers of cycles or to eliminate parts of the spectrum to obtain only data from ions all having the same cycle number. It was shown that the former is actually possible in IMS-MS systems due to the typical mass-mobility correlation [127]. However, multiplexing as in drift tube IMS is most likely impossible due to the overlap that would result between different injections having traveled different cycles.
The second approach is based on structures for lossless ion manipulations (SLIM) as focusing devices instead of an ion guide. SLIM are planar structures etched on the surfaces of printed circuit boards (PCB), meaning that extremely long devices can be manufactured with low additional effort. They consist of two parallel boards with many parallel stripe electrodes, creating an arrangement similar to the rings in the drift tube shown in Fig. 2. Again, a RF field is applied between adjacent stripes, focusing the ions towards the center between the two boards [128]. However, as now only two parallel stripes exist instead of a full ring, additional DC guard electrodes are placed bordering the stripe electrodes to prevent the ions from moving sideways. Similar to an ion funnel, operating pressures for SLIM are in the millibar range. SLIM have been first introduced in 2014 [128, 129] and it was quickly shown that both drift tube IMS [130, 131] and traveling wave IMS [132] can be built that way. Apart from the additional focusing electrodes, ion motion remains the same as shown in Fig. 2(C). Exhibiting both the ability to store stable ions for hours [133] and lossless transmission across distances as long as 1 km [60], extremely long drift tubes become feasible. Furthermore, additional manipulation structures have been reported such as 90° turns [134], switches between multiple paths on one level [131, 134], and even switching between several levels [135].
By combining these building blocks, it is possible to create folded, serpentine drift tubes to reach extremely long drift lengths without a prohibitively large instrument size. Starting with a length of 44 cm [136], the device was later prolonged to a drift length of 13 m [137] on a 45.9-cm × 32.5-cm printed circuit board. Currently, a 3D instrument stacking several layers for even longer drift length is being developed. Longer paths have the advantage that extremely long drift lengths with less overlap of different parts of the spectrum can be achieved. Nevertheless, a routing system to allow multiple cycles along the 13-m-long path has also been implemented, reaching effective drift lengths as long as 1 km [60]. Again, resolving powers grow with the square root of the number of cycles, reaching a value of 1860 for the Agilent Tune Mix m/z 622 and 922 ions for 40 cycles, equaling an effective drift length of 540 m [60]. This is to our knowledge the highest resolving power ever reported in ion mobility spectrometry.
The separation with 40 cycles also takes over 13 s, as the drift velocity remains constant since the electric field strength cannot be increased any further. This time frame is significantly longer than conventional ion mobility separations, although still extremely fast compared with chromatographic methods. Thus, despite the high resolving power, diffusion for the prolonged time results in peak widths of tens of milliseconds—as long as the time frame of a full ion mobility spectrum in many drift tube IMS. While these timescales allow simpler coupling to mass spectrometers, they are the main challenges in cyclic or SLIM traveling wave IMS. On the one hand, as the number of ions determines the peak area, a wider peak is necessarily lower at the same number of ions. Therefore, even at perfectly lossless transmission, a much higher number of ions is required to maintain signal intensity. To this end, ion introduction through a flat SLIM funnel [138], trapping inside the SLIM structures [139], and compression of the ion packet inside the SLIM structure [139, 140] have been used to increase ion population. On the other hand, ion lifetime can become a limitation depending on the ions studied. While the ions in the presented studies have been shown to be stable for hours [133], others ions are known to have lifetimes of only milliseconds [141,142,143,144,145].
Trapped ion mobility spectrometry (TIMS)
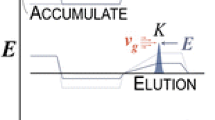
The abovementioned separation techniques prolong the effective drift length of the ions through folded drift tubes and through flying along these drift tubes several times. Another possibility for increasing the effective drift length is using a fast counter flow of drift gas, pushing the ions back [146]. Trapped ion mobility spectrometry combines this concept with trapping the ions and was first published in 2011 [147, 148]. A short review of theory and hardware advances can be found in [149]. Generally, a drift gas flow with the velocity vg pushes the ions towards the detector, while a position-dependent electric field E pushes them back as shown in Fig. 2(D). This way, ions advance up to the position where the drag from the drift gas and the drag from the electric field are in balance, that is, vg = KE [64]. Then, ions are eluted towards the detector or mass spectrometer by slowly lowering the maximum of the electric field, allowing the gas flow to push ions with sufficiently low ion mobility across the electric field plateau. From the point in time where the electric field was no longer able to trap the ion, its ion mobility can be determined. Unlike the other methods, no ion packet injection is necessary; instead, the trapping region can simply be filled by a continuous ion current. Again, radial ion focusing is employed to minimize ion losses, limiting the operating pressure to the millibar range. In the case of trapped IMS, the drift rings are split into four segments to create a quadrupole field for focusing.
It can be shown that under typical conditions the effective drift length is the displacement caused by the gas velocity vg during the time required for crossing the electric field plateau tp [64], resulting in the resolving power given by Eq. 19. Ee is the electric field strength on the plateau at the moment of elution. A recent derivation arrives at a slightly different result where the effective drift length is additionally multiplied by the ratio between the ion velocity due to the electric field and the gas velocity [150]. However, this factor is expected to be close to unity under conditions for high resolving power.
In principle, the actual physical length of the instrument does not define the effective drift length. Instead, gas velocity vg and time to pass the electric field plateau tp need to be maximized together with the electric field strength on the plateau at the moment of elution Ee. The latter follows directly from the elution condition, vg = KEe. The time to pass the electric field plateau depends not only on its length L and the ion mobility K but also on the scanning rate of the electric field β, since the electric field continues to change while the ions cross the electric field plateau. Combining these effects results in Eq. 20 [64, 150], which equals Equation 22 in reference [64] and Equations 17 and 18 in reference [150]. It is especially noteworthy that slower ions separate much better, as they require a higher electric field to elute and spend a longer time on the plateau. This dependence is more pronounced than in any other type of IMS, making trapped IMS especially efficient for large molecules, such as in bioanalytical applications.
The time to record an ion mobility spectrum, as given by Eq. 21, can be calculated by using the equation for the expected elution time from reference [64] and calculating the difference between the most extreme ion mobility values of interest. This is also an advantage compared with the other presented types of IMS, as it is possible to measure only the range between the highest and lowest ion mobilities of interest. In drift tube and traveling wave IMS, the delay between ion injection and arrival of the most mobile ions of interest passes without providing any analytical information.
However, the above equations are only approximations as the exact time spent in the trapped IMS contains additional nonlinear terms [64, 151]. Thus, while there have been attempts to directly calculate mobilities and collision cross section values [151], trapped IMS usually require calibration to extract both values [152, 153]. The effects of ion heating have also been studied [141, 154], showing that they need to be considered.
As shown by Eq. 20, there are three tuning parameters to improve the resolving power of trapped IMS. The first is increasing the length of the analyzer, which would also require higher voltage. However, this is less efficient than in the other types of IMS. Nevertheless, as current analyzers are only as long as 4.6 cm, increasing the length remains a possible option. The second is increasing the gas flow velocity, which strongly increases the separation performance; however, complex fluid dynamics need to be considered [150, 155] and non-idealities such as a non-uniform flow profile or the pressure drop across the analyzer gain influence. Furthermore, the ability to still trap the slowest ions using the maximum possible field strength limits the maximum possible gas flow velocity [156]. In order to compare with other types of IMS also limited by the maximum possible field strength, we will substitute vg by KminEmax in Eq. 20, leading to Eq. 22.
The only remaining and most common possibility is decreasing the scanning rate β in order to increase the effective drift length but also measurement time. Like increasing the physical length, decreasing β is less efficient than in other IMS; however, as it is simply the slew rate of a voltage, it can be set to any desired value. Just as increasing the effective drift length in other IMS, losses in signal-to-noise ratio go along with longer measurement times [64]. To mitigate these effects, ions can be stored in a second electric field gradient during analysis to increase the duty cycle to 100% [157] and nonlinear scans can be employed to shorten the measurement time by only resolving the ion mobility range of interest [158, 159]. Such scans of a limited ion mobility range can also be used to couple trapped IMS to slower mass spectrometers such as FT-ICR [160].
The highest reported resolving powers range from 320 to 400 for a set of single charged polybrominated diphenyl ether metabolites using a scanning rate of β = 579 V m−1 s−1 (10 V in 500 ms) [62]. Using a scanning rate of β = 3536 V m−1 s−1 (122 V/s for 900 ms), a resolving power of 295 was obtained for the plus seven charge state of ubiquitin [63]. Using a reported scanning rate of β = 2691 V m−1 s−1 in a 900-ms scan, a resolving power of 228 was obtained for the single charged m/z = 1822 ion of an ESI tune mix [64]. It should be noted that often the voltage difference and scan time are reported instead of β. We converted these quantities into β using the potential gradients shown in Figure 3 of reference [151]. As expected, the highest resolving powers are achieved at the lowest β and for higher charge states.
Differential or field asymmetric ion mobility spectrometry (DMS/FAIMS)
Differential mobility spectrometry (DMS) or field asymmetric ion mobility spectrometry (FAIMS) differs considerably from the techniques discussed before that a dedicated textbook exists [47]. Often, however not always in an identical fashion, the two names have been used to distinguish between cylindrical and planar devices. As only planar devices have been demonstrated to achieve ultra-high resolution, we will only consider them in this review.
Field asymmetric IMS were originally developed in the USSR [161] and first published in 1991 [43, 162]. While the separation principle differs from other IMS, it can be directly explained from Eq. 6. A gas stream pushes ions along two parallel plates, the ion filter region, between which a time-varying separation voltage is applied as shown in Fig. 3. It generates low E/N for a longer time and high E/N for a shorter time and in opposite direction. The integrals are identical so that ions with α(E/N) = 0 would experience no net displacement along the electrical field axis. All other ions are deflected towards one of the plates depending on the shape of their alpha function. A small, constant compensation voltage is applied between the two plates and scanned to allow different ions to pass the filter region, obtaining a spectrum. Ion heating is obviously unavoidable in field asymmetric IMS, as it is the measurement principle [163,164,165]. Operation is possible under a wide range of pressures [166, 167].
Due to its operation at high E/N, measurement results of field asymmetric IMS can only be compared with measurements of high-field drift tube IMS [168, 169]. However, this orthogonality to low-field IMS also enables multidimensional IMS such as FAIMS-IMS setups [170, 171]. Furthermore, the alpha function shows less correlation with the ion mass than the collision cross section, possessing slightly better orthogonality to mass spectrometry than to low-field IMS [47]. As field asymmetric IMS act as a filter, they pass a continuous stream of selected ions and thus achieve 100% duty cycle if only a single ion species is being monitored. This is especially useful when coupling field asymmetric IMS to slow mass spectrometers. If multiple ions need to be monitored, the duty cycle drops accordingly and becomes one over the number of points in a spectrum.
The peak width of a field asymmetric IMS accounting for the filtering effect and diffusion can be estimated according to Eq. 23 [172]. Expressing the diffusion coefficient D through the ion mobility K by using the Nernst-Einstein-Townsend relation is in this case only of limited validity, as ion heating will increase diffusion disproportionally [31, 173]. Nevertheless, doing so as an approximation allows for vastly simplifying the equation to a form similar to those for other IMS. Increasing the time the ions spend in the filter tfilt or the ion mobility K narrows the peaks as ions not passing the filter move further towards the plates at the same compensation voltage.
The reduced compensation field strength Ec/N defining the peak position in the spectrum depends in a complex way on the shape of the alpha function, the shape of the applied separation voltage waveform, and the magnitude of the reduced separation field strength ED/N [43, 172, 174]. A rough approximation ignoring all terms of higher order is given by Eq. 24. α2 is the first term of the series expansion shown in Eq. 6, while f3 is the average of the cube of the separation waveform. Increasing any of the three coefficients in Eq. 24 will move the peaks further apart from each other, improving resolving power at a constant peak width. It is interesting to note that the peak width depends on the absolute ion mobility, but the peak position on the alpha function. Therefore, resolving power can sometimes be a misleading quantity in field asymmetric IMS, as under certain conditions, groups of peaks may shift simultaneously to lower or higher compensation voltages. By carefully tuning experimental parameters to showcase such effects, it is for example possible to increase the apparent resolving power from around 20 to 7900 without any increase in resolution between the peaks [175].
Being a nonlinear separation method, more possibilities for improving separation performance exist compared with other types of IMS as shown by Eqs. 23 and 24. First, the filter time tfilt can be increased as needed by lowering the gas velocity. However, this will require larger gaps between the plates to still maintain acceptable ion transmission [172], as no focusing exists in planar field asymmetric IMS. Furthermore, the total measurement time already increases with increasing resolving power due to being an ion filter, as more points are needed to maintain the same number of points per peak [176] and can already be several minutes for a single high resolution spectrum. Thus, longer filter times are often not a feasible way to higher resolving power. Second, the ion mobility K can be increased by using high fractions of light gases such as helium or hydrogen in the drift gas, also increasing the energy uptake of the ions [176,177,178,179]. This has vastly helped to increase resolving power of field asymmetric IMS; however, there is no possibility to surpass the ion mobility in pure hydrogen or helium for further improvement. Furthermore, the increased risk of electrical breakdown has to be kept in mind. Third, the magnitude of the alpha function can be increased through modifiers which cluster with the target ions during the low-field period [180,181,182]. This can be an ion-specific process and thus add selectivity, increasing the spread between different peaks even further. Fourth, changing the shape of the separation voltage waveform to better approximate a rectangular shape will increase f3 at the cost of more complex high-voltage electronics and higher power consumption due to high charging currents [183, 184]. This optimization approach is obviously limited when having reached a rectangular shape. Fifth, increasing the reduced dispersion field is limited by breakdown based on Paschen’s curve [173, 179] and ion losses due to fragmentation at extremely high fields [185]. While most of these parameters have already been explored to their limit individually, the various possible combinations allow tuning field asymmetric IMS for the measurement task at hand. For example, it has recently been shown that maintaining resolving power is possible when replacing helium with nitrogen, thus widening the peaks, but increasing the dispersion field as possible by the higher electrical breakdown of nitrogen, thus increasing the peak shift [186].
Using a mixture with 14% nitrogen and 86% hydrogen in combination with an extremely stable compensation voltage generator and filter times of 200 ms, a resolving power of 440 to 460 has been obtained for four times charged Syntide 2 [65]. Similar resolving powers were obtained in a 50/50 mixture with 700-ms filter time. At these resolving powers and filter times, the total measurement time for scanning a single peak already exceeds 1 min [65]. It was suggested that with better stability of experimental parameters during these timescales, resolving power might be significantly above 500. It is important to remember once again that FAIMS operates in a different separation space and thus, values for resolving power cannot be easily compared with other IMS. Nevertheless, ultra-high resolving power in field asymmetric IMS translates to many separations not possible in low-field IMS, for example, distinguishing between isotopomers [41, 42].
Conclusion
In this review, we have analyzed the main principles of ion mobility spectrometers with respect to ultra-high resolution and their limitations to further improvements. While drift tube IMS have the major advantage of providing a direct measurement of the ion mobility and collision cross section, they have most likely reached their maximum possible resolving power at values between 100 and 140 when operated at low pressure. As higher fields are not permissible due to electrical breakdown and ion heating, the only way to increase resolving power at constant pressure is to increase the drift length. However, with instruments as long as 3 m, the practical limit for most applications has most likely been reached or even surpassed. Thus, higher resolving power in drift tube IMS is only possible when moving to higher pressures. Atmospheric pressure drift tube IMS provide, at the same low level of ion heating, much higher field strengths and thus faster analysis time in a smaller device. Here, increases in resolving power are still possible as long as the required drift voltages can be managed, allowing for full spectra to be recorded with ultra-high resolution in milliseconds. Current resolving powers are as high as 250 in a 15-cm-long drift tube.
Staying at reduced pressure while also circumventing the need for high voltage, increasing the effective drift length is another way to ultra-high resolving power. This can be done either by traveling a long folded drift tube, possibly even several times, such as in cyclic or SLIM traveling wave IMS, or by pushing against a gas stream as in trapped IMS. At the cost of increased measurement time and requiring calibration to obtain collision cross sections, even higher resolving powers than possible with ultra-high-resolution atmospheric pressure drift tube IMS can be obtained. Generally, the traveling wave variants reach the highest resolving powers with values of 550 and 1860, as it scales with the square root of additional measurement time. Resolving power in trapped IMS only scales with the fourth root of additional measurement time; however, measurement time can be set regardless of the physical drift length through the scanning rate and it is possible to scan only the ion mobility range of interest with high resolving power. This way, resolving powers of up to 400 have been achieved.
Furthermore, differential or field asymmetric IMS offer an ion separation orthogonal to the abovementioned instruments. While no measurement of ion mobility or collision cross sections is possible and measurement times are extremely long for ultra-high resolving power, they offer additional possibilities for tuning the ion separation and, as an ion filter, deliver a continuous stream of ions for further analysis. This enables better coupling to slow mass spectrometers and even multidimensional IMS systems, such as FAIMS-IMS.
References
Puton J, Namieśnik J. Ion mobility spectrometry - current status and application for chemical warfare agents detection. TrAC Trends Anal Chem. 2016. https://doi.org/10.1016/j.trac.2016.06.002.
Eiceman GA, Stone JA. Peer reviewed: ion mobility spectrometers in national defense. Anal Chem. 2004. https://doi.org/10.1021/ac041665c.
Mäkinen M, Nousiainen M, Sillanpää M. Ion spectrometric detection technologies for ultra-traces of explosives: a review. Mass Spectrom Rev. 2011. https://doi.org/10.1002/mas.20308.
Buryakov IA. Detection of explosives by ion mobility spectrometry. J Anal Chem. 2011. https://doi.org/10.1134/S1061934811080077.
Borsdorf H, Eiceman GA. Ion mobility spectrometry: principles and applications. Appl Spectrosc Rev. 2006. https://doi.org/10.1080/05704920600663469.
Eiceman GA, Karpas Z, Hill HH. Ion mobility spectrometry. 3rd ed. Boca Raton: CRC Press; 2013.
Vautz W, Zimmermann D, Hartmann M, Baumbach JI, Nolte J, Jung J. Ion mobility spectrometry for food quality and safety. Food Addit Contam. 2006. https://doi.org/10.1080/02652030600889590.
Karpas Z. Applications of ion mobility spectrometry (IMS) in the field of foodomics. Food Res Int. 2013. https://doi.org/10.1016/j.foodres.2012.11.029.
Hernández-Mesa M, Escourrou A, Monteau F, Le Bizec B, Dervilly-Pinel G. Current applications and perspectives of ion mobility spectrometry to answer chemical food safety issues. TrAC Trends Anal Chem. 2017. https://doi.org/10.1016/j.trac.2017.07.006.
O’Donnell RM, Sun X, Harrington P d B. Pharmaceutical applications of ion mobility spectrometry. TrAC Trends Anal Chem. 2008. https://doi.org/10.1016/j.trac.2007.10.014.
Campuzano ID, Lippens JL. Ion mobility in the pharmaceutical industry - an established biophysical technique or still niche? Curr Opin Chem Biol. 2018. https://doi.org/10.1016/j.cbpa.2017.11.008.
Baumbach JI. Process analysis using ion mobility spectrometry. Anal Bioanal Chem. 2006. https://doi.org/10.1007/s00216-005-3397-8.
Baumbach JI. Ion mobility spectrometry coupled with multi-capillary columns for metabolic profiling of human breath. J Breath Res. 2009. https://doi.org/10.1088/1752-7155/3/3/034001.
Fink T, Baumbach JI, Kreuer S. Ion mobility spectrometry in breath research. J Breath Res. 2014. https://doi.org/10.1088/1752-7155/8/2/027104.
Clemmer DE, Jarrold MF. Ion mobility measurements and their applications to clusters and biomolecules. J Mass Spectrom. 1997. https://doi.org/10.1002/(SICI)1096-9888(199706)32:6<577:AID-JMS530>3.0.CO;2-4.
Creaser CS, Griffiths JR, Bramwell CJ, Noreen S, Hill CA, Thomas CL. Paul. Ion mobility spectrometry: a review. Part 1. Structural analysis by mobility measurement. Analyst. 2004. https://doi.org/10.1039/b404531a.
Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE. Biomolecule analysis by ion mobility spectrometry. Annu Rev Anal Chem (Palo Alto, Calif.). 2008. https://doi.org/10.1146/annurev.anchem.1.031207.113001.
Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck, Albert JR. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc Rev. 2010. https://doi.org/10.1039/b914002f.
Kliman M, May JC, McLean JA. Lipid analysis and lipidomics by structurally selective ion mobility-mass spectrometry. Biochim Biophys Acta. 2011. https://doi.org/10.1016/j.bbalip.2011.05.016.
Williams DM, Pukala TL. Novel insights into protein misfolding diseases revealed by ion mobility-mass spectrometry. Mass Spectrom Rev. 2013. https://doi.org/10.1002/mas.21358.
Konijnenberg A, Butterer A, Sobott F. Native ion mobility-mass spectrometry and related methods in structural biology. Biochim Biophys Acta. 2013. https://doi.org/10.1016/j.bbapap.2012.11.013.
Zhong Y, Hyung S-J, Ruotolo BT. Ion mobility-mass spectrometry for structural proteomics. Expert Rev Proteomics. 2012. https://doi.org/10.1586/epr.11.75.
Gray CJ, Thomas B, Upton R, Migas LG, Eyers CE, Barran PE, et al. Applications of ion mobility mass spectrometry for high throughput, high resolution glycan analysis. Biochim Biophys Acta. 2016. https://doi.org/10.1016/j.bbagen.2016.02.003.
Paglia G, Kliman M, Claude E, Geromanos S, Astarita G. Applications of ion-mobility mass spectrometry for lipid analysis. Anal Bioanal Chem. 2015. https://doi.org/10.1007/s00216-015-8664-8.
Zheng X, Wojcik R, Zhang X, Ibrahim YM, Burnum-Johnson KE, Orton DJ, et al. Coupling front-end separations, ion mobility spectrometry, and mass spectrometry for enhanced multidimensional biological and environmental analyses. Annu Rev Anal Chem (Palo Alto, Calif.). 2017. https://doi.org/10.1146/annurev-anchem-061516-045212.
Stephan S, Jakob C, Hippler J, Schmitz OJ. A novel four-dimensional analytical approach for analysis of complex samples. Anal Bioanal Chem. 2016. https://doi.org/10.1007/s00216-016-9460-9.
Lipok C, Hippler J, Schmitz OJ. A four dimensional separation method based on continuous heart-cutting gas chromatography with ion mobility and high resolution mass spectrometry. J Chromatogr A. 2018. https://doi.org/10.1016/j.chroma.2017.07.013.
Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014. https://doi.org/10.1038/nchem.1889.
Cumeras R, Figueras E, Davis CE, Baumbach JI, Gràcia I. Review on ion mobility spectrometry. Part 1: current instrumentation. Analyst. 2014. https://doi.org/10.1039/c4an01100g.
Ewing MA, Glover MS, Clemmer DE. Hybrid ion mobility and mass spectrometry as a separation tool. J Chrom A. 2016. https://doi.org/10.1016/j.chroma.2015.10.080.
Mason EA, McDaniel EW. Transport properties of ions in gases. Weinheim, FRG: Wiley-VCH Verlag GmbH & Co. KGaA; 1988.
Heiche G, Mason EA. Ion mobilities with charge exchange. J Chem Phys. 1970. https://doi.org/10.1063/1.1673997.
Siems WF, Viehland LA, Hill HH. Improved momentum-transfer theory for ion mobility. 1. Derivation of the fundamental equation. Anal Chem. 2012. https://doi.org/10.1021/ac301779s.
Viehland LA, Mason EA. Gaseous lon mobility in electric fields of arbitrary strength. Ann Phys. 1975. https://doi.org/10.1016/0003-4916(75)90233-X.
Viehland LA, Mason EA. Gaseous ion mobility and diffusion in electric fields of arbitrary strength. Ann Phys. 1978. https://doi.org/10.1016/0003-4916(78)90034-9.
Lin SL, Viehland LA, Mason EA. Three-temperature theory of gaseous ion transport. Chem Phys. 1979. https://doi.org/10.1016/0301-0104(79)85040-5.
Viehland LA, Lin SL. Application of the three-temperatue theory of gaseous ion transport. Chem Phys. 1979. https://doi.org/10.1016/0301-0104(79)80112-3.
Siems WF, Viehland LA, Hill HH. Correcting the fundamental ion mobility equation for field effects. Analyst. 2016. https://doi.org/10.1039/c6an01353h.
Yousef A, Shrestha S, Viehland LA, Lee EPF, Gray BR, Ayles VL, et al. Interaction potentials and transport properties of coinage metal cations in rare gases. J Chem Phys. 2007. https://doi.org/10.1063/1.2774977.
Barber S, Blake RS, White IR, Monks PS, Reich F, Mullock S, et al. Increased sensitivity in proton transfer reaction mass spectrometry by incorporation of a radio frequency ion funnel. Anal Chem. 2012. https://doi.org/10.1021/ac300894t.
Shvartsburg AA, Clemmer DE, Smith RD. Isotopic effect on ion mobility and separation of isotopomers by high-field ion mobility spectrometry. Anal Chem. 2010. https://doi.org/10.1021/ac101992d.
Kaszycki JL, Bowman AP, Shvartsburg AA. Ion mobility separation of peptide isotopomers. J Am Soc Mass Spectrom. 2016. https://doi.org/10.1007/s13361-016-1367-3.
Buryakov IA, Krylov EV, Nazarov EG, Rasulev UK. A new method of separation of multi-atomic ions by mobility at atmospheric pressure using a high-frequency amplitude-asymmetric strong electric field. Int J Mass Spectrom Ion Process. 1993. https://doi.org/10.1016/0168-1176(93)87062-W.
Langejuergen J, Allers M, Oermann J, Kirk AT, Zimmermann S. High kinetic energy ion mobility spectrometer: quantitative analysis of gas mixtures with ion mobility spectrometry. Anal Chem. 2014. https://doi.org/10.1021/ac5011662.
Langejuergen J, Allers M, Oermann J, Kirk AT, Zimmermann S. Quantitative detection of benzene in toluene- and xylene-rich atmospheres using high-kinetic-energy ion mobility spectrometry (IMS). Anal Chem. 2014. https://doi.org/10.1021/ac5034243.
Kirk AT, Grube D, Kobelt T, Wendt C, Zimmermann S. A high resolution high kinetic energy ion mobility spectrometer based on a low-discrimination tri-state ion shutter. Anal Chem. 2018. https://doi.org/10.1021/acs.analchem.7b04586.
Shvartsburg AA. Differential mobility spectrometry - FAIMS and beyond Boca Raton, Fla. London: CRC; Taylor & Francis [distributor]; 2008.
Schneider BB, Nazarov EG, Londry F, Vouros P, Covey TR. Differential mobility spectrometry/mass spectrometry history, theory, design optimization, simulations, and applications. Mass Spectrometry Review. 2015. https://doi.org/10.1002/mas.21453.
Nič M, Jirát J, Košata B, Jenkins A, McNaught A. IUPAC Compendium of Chemical Terminology Research Triagle Park. Zurich: NC: IUPAC; 2009.
Dodds JN, May JC, McLean JA. Correlating resolving power, resolution, and collision cross section - unifying cross-platform assessment of separation efficiency in ion mobility spectrometry. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b02827.
Ungethüm B, Walte A, Münchmeyer W, Matz G. Comparative measurements of toxic industrial compounds with a differential mobility spectrometer and a time of flight ion mobility spectrometer. Int J Ion Mobil Spec. 2009. https://doi.org/10.1007/s12127-009-0028-7.
Shvartsburg AA, Smith RD. Ultrahigh-resolution differential ion mobility spectrometry using extended separation times. 2011. https://doi.org/10.1021/ac102689p.
Kirk AT, Raddatz C-R, Zimmermann S. Separation of isotopologues in ultra-high-resolution ion mobility spectrometry. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.6b03300.
Kirk AT, Bakes K, Zimmermann S. A universal relationship between optimum drift voltage and resolving power. Int J Ion Mobil Spec. 2017. https://doi.org/10.1007/s12127-017-0219-6.
Kirk AT, Zimmermann S. Pushing a compact 15 cm long ultra-high resolution drift tube ion mobility spectrometer with R = 250 to R = 425 using peak deconvolution. Int. J. Ion Mobil. Spec. 2015. https://doi.org/10.1007/s12127-015-0166-z.
Srebalus CA, Li J, Marshall WS, Clemmer DE. Gas-phase separations of electrosprayed peptide libraries. Anal Chem. 1999. https://doi.org/10.1021/ac9903757.
Wu C, Siems WF, Asbury GR, Hill HH. Electrospray ionization high-resolution ion mobility spectrometry-mass spectrometry. 1998. https://doi.org/10.1021/ac980414z.
Kemper PR, Dupuis NF, Bowers MT. A new, higher resolution, ion mobility mass spectrometer. Int J Mass Spectrom. 2009. https://doi.org/10.1016/j.ijms.2009.01.012.
Glaskin RS, Ewing MA, Clemmer DE. Ion trapping for ion mobility spectrometry measurements in a cyclical drift tube. Anal Chem. 2013. https://doi.org/10.1021/ac4015066.
Deng L, Webb IK, Garimella SVB, Hamid AM, Zheng X, Norheim RV, et al. Serpentine ultra-long path with extended routing (SUPER) high resolution traveling wave ion mobility-MS using structures for lossless ion manipulations. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b00185.
Giles K, Ujma J, Wildgoose JL, Green M, Richardson K, Langridge D, et al. Design and Performance of a second-generation cyclic ion mobility enabled Q-TOF. In: ASMS Conference on Mass Spectrometry and Allied Topics; 2017.
Adams KJ, Montero D, Aga D, Fernandez-Lima F. Isomer separation of polybrominated diphenyl ether metabolites using nanoESI-TIMS-MS. Int. J. Ion Mobil. Spec. 2016. https://doi.org/10.1007/s12127-016-0198-z.
Ridgeway ME, Silveira JA, Meier JE, Park MA. Microheterogeneity within conformational states of ubiquitin revealed by high resolution trapped ion mobility spectrometry. Analyst. 2015. https://doi.org/10.1039/C5AN00841G.
Michelmann K, Silveira JA, Ridgeway ME, Park MA. Fundamentals of trapped ion mobility spectrometry. J Am Soc Mass Spectrom. 2015. https://doi.org/10.1007/s13361-014-0999-4.
Shvartsburg AA, Seim TA, Danielson WF, Norheim R, Moore RJ, Anderson GA, et al. High-definition differential ion mobility spectrometry with resolving power up to 500. J Am Soc Mass Spectrom. 2013. https://doi.org/10.1007/s13361-012-0517-5.
Tyndall AM, Powell CF. The mobility of ions in pure gases. Proc R Soc A. 1930. https://doi.org/10.1098/rspa.1930.0149.
Bradbury NE, Nielsen RA. Absolute values of the electron mobility in hydrogen. Phys Rev. 1936. https://doi.org/10.1103/PhysRev.49.388.
Cohen MJ, Karasek FW. Plasma chromatography --a new dimension for gas chromatography and mass spectrometry. J Chromatogr Sci. 1970. https://doi.org/10.1093/chromsci/8.6.330.
Karasek FW, Cohen MJ, Carroll DI. Trace studies of alcohols in the plasma chromatograph--mass spectrometer. J Chromatogr Sci. 1971. https://doi.org/10.1093/chromsci/9.7.390.
Kwasnik M, Fuhrer K, Gonin M, Barbeau K, Fernandez FM. Performance, resolving power, and radial ion distributions of a prototype nanoelectrospray ionization resistive glass atmospheric pressure ion mobility spectrometer. Anal Chem. 2007. https://doi.org/10.1021/ac071226o.
Kaplan K, Graf S, Tanner C, Gonin M, Fuhrer K, Knochenmuss R, et al. Resistive glass IM-TOFMS. Anal Chem. 2010. https://doi.org/10.1021/ac1017259.
Pfeifer KB, Rohde SB, Peterson KA, Rumpf AN. Development of rolled miniature drift tubes using low temperature co-fired ceramics (LTCC). Int J Ion Mobil Spec. 2004;7(2):52–8.
Bohnhorst A, Kirk AT, Zimmermann S. Simulation aided design of a low cost ion mobility spectrometer based on printed circuit boards. Int J Ion Mobil Spec. 2016. https://doi.org/10.1007/s12127-016-0202-7.
Reinecke T, Clowers BH. Implementation of a flexible, open-source platform for ion mobility spectrometry. HardwareX. 2018. https://doi.org/10.1016/j.ohx.2018.e00030.
Hollerbach A, Baird Z, Cooks RG. Ion separation in air using a 3D printed ion mobility spectrometer. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b00469.
Hoaglund CS, Valentine SJ, Sporleder CR, Reilly JP, Clemmer DE. Three-dimensional ion mobility/TOFMS analysis of electrosprayed biomolecules. Anal Chem. 1998. https://doi.org/10.1021/ac980059c.
Henderson SC, Valentine SJ, Counterman AE, Clemmer DE. ESI/ion trap/ion mobility/time-of-flight mass spectrometry for rapid and sensitive analysis of biomolecular mixtures. Anal Chem. 1999. https://doi.org/10.1021/ac9809175.
Knorr FJ, Eatherton RL, Siems WF, Hill HH. Fourier transform ion mobility spectrometry. Anal Chem. 1985. https://doi.org/10.1021/ac50001a018.
Szumlas AW, Ray SJ, Hieftje GM. Hadamard transform ion mobility spectrometry. Anal Chem. 2006. https://doi.org/10.1021/ac051743b.
Clowers BH, Siems WF, Hill HH, Massick SM. Hadamard transform ion mobility spectrometry. Anal Chem. 2006. https://doi.org/10.1021/ac050615k.
Clowers BH, Siems WF, Yu Z, Davis AL. A two-phase approach to fourier transform ion mobility time-of-flight mass spectrometry. Analyst. 2015. https://doi.org/10.1039/c5an00941c.
Davis AL, Liu W, Siems WF, Clowers BH. Correlation ion mobility spectrometry. Analyst. 2017. https://doi.org/10.1039/C6AN02249A.
McGann WJ, Jenkins A, Ribiero K, Napoli J. New high-efficiency ion trap mobility detection system for narcotics and explosives. SPIE Proc. 1993. https://doi.org/10.1117/12.171280.
Kirk AT, Zimmermann S. Bradbury-Nielsen vs. field switching shutters for high resolution drift tube ion mobility spectrometers. Int J Ion Mobil Spec. 2014. https://doi.org/10.1007/s12127-014-0153-9.
Clowers BH, Ibrahim YM, Prior DC, Danielson WF, Belov ME, Smith RD. Enhanced ion utilization efficiency using an electrodynamic ion funnel trap as an injection mechanism for ion mobility spectrometry. Anal Chem. 2008. https://doi.org/10.1021/ac701648p.
Karasek FW, Kane DM. Effect of oxygen on response of the electron-capture detector. Anal Chem. 1973. https://doi.org/10.1021/ac60325a016.
Jafari MT. Low-temperature plasma ionization ion mobility spectrometry. Anal Chem. 2011. https://doi.org/10.1021/ac1022937.
Tabrizchi M, Shamlouei HR. Relative transmission of different ions through shutter grid. Int J Mass Spectrom. 2010. https://doi.org/10.1016/j.ijms.2010.01.011.
Chen C, Chen H, Li H. Pushing the resolving power of Tyndall-Powell gate ion mobility spectrometry over 100 with no sensitivity loss for multiple ion species. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b03629.
Crawford CL, Hauck BC, Tufariello JA, Harden CS, McHugh V, Siems WF, et al. Accurate and reproducible ion mobility measurements for chemical standard evaluation. Talanta. 2012. https://doi.org/10.1016/j.talanta.2012.09.003.
Hauck BC, Siems WF, Harden CS, McHugh VM, Hill HH. Construction and evaluation of a hermetically sealed accurate ion mobility instrument. Int J Ion Mobil Spec. 2017. https://doi.org/10.1007/s12127-017-0224-9.
Hauck BC, Siems WF, Harden CS, McHugh VM, Hill HH. High accuracy ion mobility spectrometry for instrument calibration. Anal Chem. 2018. https://doi.org/10.1021/acs.analchem.7b04987.
Rokushika S, Hatano H, Baim MA, Hill HH. Resolution measurement for ion mobility spectrometry. Anal Chem. 1985. https://doi.org/10.1021/ac00286a023.
Watts P, Wilders A. On the resolution obtainable in practical ion mobility systems. Int J Mass Spectrom Ion Process. 1992. https://doi.org/10.1016/0168-1176(92)80003-J.
Revercomb HE, Mason EA. Theory of plasma chromatography/gaseous electrophoresis. Review Anal Chem. 1975. https://doi.org/10.1021/ac60357a043.
Kirk AT, Allers M, Cochems P, Langejuergen J, Zimmermann S. A compact high resolution ion mobility spectrometer for fast trace gas analysis. Analyst. 2013. https://doi.org/10.1039/c3an00231d.
Siems WF, Wu C, Tarver EE, Hill, Herbert H Jr, Larsen PR, et al. Measuring the resolving power of ion mobility spectrometers. Anal Chem. 1994. https://doi.org/10.1021/ac00095a014.
Kanu AB, Gribb MM, Hill HH. Predicting optimal resolving power for ambient pressure ion mobility spectrometry. Anal Chem. 2008. https://doi.org/10.1021/ac8008143.
Kirk AT, Zimmermann S. An analytical model for the optimum drift voltage of drift tube ion mobility spectrometers with respect to resolving power and detection limits. Int. J. Ion Mobil. Spec. 2015. https://doi.org/10.1007/s12127-015-0176-x.
Steiner WE, Clowers BH, Fuhrer K, Gonin M, Matz LM, Siems WF, et al. Electrospray ionization with ambient pressure ion mobility separation and mass analysis by orthogonal time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2001. https://doi.org/10.1002/rcm.495.
Sabo M, Páleník J, Kučera M, Han H, Wang H, Chu Y, et al. Atmospheric pressure corona discharge ionisation and ion mobility spectrometry/mass spectrometry study of the negative corona discharge in high purity oxygen and oxygen/nitrogen mixtures. Int J Mass Spectrom. 2010. https://doi.org/10.1016/j.ijms.2010.03.004.
Allers M, Timoumi L, Kirk AT, Schlottmann F, Zimmermann S. Coupling of a high-resolution ambient pressure drift tube ion mobility spectrometer to a commercial time-of-flight mass spectrometer. J Am Soc Mass Spectrom. 2018. https://doi.org/10.1007/s13361-018-2045-4.
Dugourd P, Hudgins RR, Clemmer DE, Jarrold MF. High-resolution ion mobility measurements. Rev. Sci. Instrum. 1997. https://doi.org/10.1063/1.1147873.
Merenbloom SI, Koeniger SL, Valentine SJ, Plasencia MD, Clemmer DE. IMS-IMS and IMS-IMS-IMS/MS for separating peptide and protein fragment ions. Anal Chem. 2006. https://doi.org/10.1021/ac052208e.
Tolmachev AV, Kim T, Udseth HR, Smith RD, Bailey TH, Futrell JH. Simulation-based optimization of the electrodynamic ion funnel for high sensitivity electrospray ionization mass spectrometry. Int J Mass Spectrom. 2000. https://doi.org/10.1016/S1387-3806(00)00265-7.
Page JS, Tolmachev AV, Tang K, Smith RD. Theoretical and experimental evaluation of the low m/z transmission of an electrodynamic ion funnel. J Am Soc Mass Spectrom. 2006. https://doi.org/10.1016/j.jasms.2005.12.013.
Merenbloom SI, Glaskin RS, Henson ZB, Clemmer DE. High-resolution ion cyclotron mobility spectrometry. 2009. https://doi.org/10.1021/ac801880a.
Glaskin RS, Valentine SJ, Clemmer DE. A scanning frequency mode for ion cyclotron mobility spectrometry. 2010. https://doi.org/10.1021/ac1017474.
Kurulugama RT, Nachtigall FM, Lee S, Valentine SJ, Clemmer DE. Overtone mobility spectrometry: part 1. Experimental observations. J Am Soc Mass Spectrom. 2009. https://doi.org/10.1016/j.jasms.2008.11.022.
Valentine SJ, Stokes ST, Kurulugama RT, Nachtigall FM, Clemmer DE. Overtone mobility spectrometry: part 2. Theoretical considerations of resolving power. J Am Soc Mass Spectrom. 2009. https://doi.org/10.1016/j.jasms.2009.01.001.
Valentine SJ, Kurulugama RT, Clemmer DE. Overtone mobility spectrometry: part 3. On the origin of peaks. J Am Soc Mass Spectrom. 2011. https://doi.org/10.1007/s13361-011-0087-y.
Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Applications of a travelling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom: RCM. 2004. https://doi.org/10.1002/rcm.1641.
Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, et al. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int J Mass Spectrom. 2007. https://doi.org/10.1016/j.ijms.2006.07.021.
Giles K, Williams JP, Campuzano I. Enhancements in travelling wave ion mobility resolution. Rapid Commun Mass Spectrom: RCM. 2011. https://doi.org/10.1002/rcm.5013.
Gerlich D. Inhomogeneous RF fields: a versatile tool for the study of processes with slow ions. In: Ng CY, Baer M, editors. State-selected and state-to-state ion-molecule reaction dynamics. New York: J. Wiley; 1992.
Shaffer SA, Tolmachev A, Prior DC, Anderson GA, Udseth HR, Smith RD. Characterization of an improved electrodynamic ion funnel interface for electrospray ionization mass spectrometry. Anal Chem. 1999. https://doi.org/10.1021/ac990346w.
Shvartsburg AA, Smith RD. Fundamentals of traveling wave ion mobility spectrometry. Anal Chem. 2008. https://doi.org/10.1021/ac8016295.
Gelb AS, Jarratt RE, Huang Y, Dodds ED. A study of calibrant selection in measurement of carbohydrate and peptide ion-neutral collision cross sections by traveling wave ion mobility spectrometry. Anal Chem. 2014. https://doi.org/10.1021/ac503379e.
Hines KM, May JC, McLean JA, Xu L. Evaluation of collision cross section calibrants for structural analysis of lipids by traveling wave ion mobility-mass spectrometry. Anal Chem. 2016. https://doi.org/10.1021/acs.analchem.6b01728.
Morsa D, Gabelica V, de Pauw E. Effective temperature of ions in traveling wave ion mobility spectrometry. Anal Chem. 2011. https://doi.org/10.1021/ac201509p.
Merenbloom SI, Flick TG, Williams ER. How hot are your ions in TWAVE ion mobility spectrometry? J Am Soc Mass Spectrom. 2012. https://doi.org/10.1007/s13361-011-0313-7.
Morsa D, Gabelica V, de Pauw E. Fragmentation and isomerization due to field heating in traveling wave ion mobility spectrometry. J Am Soc Mass Spectrom. 2014. https://doi.org/10.1007/s13361-014-0909-9.
Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal Chem. 2010. https://doi.org/10.1021/ac1022953.
Michaelevski I, Eisenstein M, Sharon M. Gas-phase compaction and unfolding of protein structures. Anal Chem. 2010. https://doi.org/10.1021/ac1021419.
Zhong Y, Hyung S-J, Ruotolo BT. Characterizing the resolution and accuracy of a second-generation traveling-wave ion mobility separator for biomolecular ions. Analyst. 2011. https://doi.org/10.1039/c0an00987c.
Giles K, Wildgoose JL, Pringle SD, Garside J, Carney P, Nixon P, et al. Design and utility of a multi-pass cyclic ion mobility separator. In: ASMS Conference on Mass Spectrometry and Allied Topics; 2014.
Giles K, Wildgoose JL, Pringle SD, Langridge DJ, Nixon P, Garside J, et al. Characterising a T-wave enabled multi-pass cyclic ion mobility separator. In: ASMS Conference on Mass Spectrometry and Allied Topics; 2015.
Tolmachev AV, Webb IK, Ibrahim YM, Garimella, Sandilya VB, Zhang X, et al. Characterization of ion dynamics in structures for lossless ion manipulations. Anal Chem. 2014. https://doi.org/10.1021/ac502054p.
Garimella SVB, Ibrahim YM, Webb IK, Tolmachev AV, Zhang X, Prost SA, et al. Simulation of electric potentials and ion motion in planar electrode structures for lossless ion manipulations (SLIM). J Am Soc Mass Spectrom. 2014. https://doi.org/10.1007/s13361-014-0976-y.
Webb IK, Garimella, Sandilya VB, Tolmachev AV, Chen T-C, Zhang X, et al. Experimental evaluation and optimization of structures for lossless ion manipulations for ion mobility spectrometry with time-of-flight mass spectrometry. Anal Chem. 2014. https://doi.org/10.1021/ac502055e.
Webb IK, Garimella, Sandilya VB, Tolmachev AV, Chen T-C, Zhang X, et al. Mobility-resolved ion selection in uniform drift field ion mobility spectrometry/mass spectrometry: dynamic switching in structures for lossless ion manipulations. Anal Chem. 2014. https://doi.org/10.1021/ac502139e.
Hamid AM, Ibrahim YM, Garimella, Sandilya VB, Webb IK, Deng L, et al. Characterization of traveling wave ion mobility separations in structures for lossless ion manipulations. Anal Chem. 2015. https://doi.org/10.1021/acs.analchem.5b02481.
Zhang X, Garimella, Sandilya VB, Prost SA, Webb IK, Chen T-C, et al. Ion trapping, storage, and ejection in structures for lossless ion manipulations. Anal Chem. 2015. https://doi.org/10.1021/acs.analchem.5b00214.
Garimella SVB, Ibrahim YM, Webb IK, Ipsen AB, Chen T-C, Tolmachev AV, et al. Ion manipulations in structures for lossless ion manipulations (SLIM): computational evaluation of a 90° turn and a switch. Analyst. 2015. https://doi.org/10.1039/c5an00844a.
Ibrahim YM, Hamid AM, Cox JT, Garimella SVB, Smith RD. Ion elevators and escalators in multi-level structures for lossless ion manipulations (SLIM). Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.6b04500.
Hamid AM, Garimella SVB, Ibrahim YM, Deng L, Zheng X, Webb IK, et al. Achieving high resolution ion mobility separations using traveling waves in compact multiturn structures for lossless ion manipulations. Anal Chem. 2016. https://doi.org/10.1021/acs.analchem.6b01914.
Deng L, Ibrahim YM, Hamid AM, Garimella SVB, Webb IK, Zheng X, et al. Ultra-high resolution ion mobility separations utilizing traveling waves in a 13 m serpentine path length structures for lossless ion manipulations module. Anal Chem. 2016. https://doi.org/10.1021/acs.analchem.6b01915.
Deng L, Ibrahim YM, Garimella SVB, Webb IK, Hamid AM, Norheim RV, et al. Greatly increasing trapped ion populations for mobility separations using traveling waves in structures for lossless ion manipulations. Anal Chem. 2016. https://doi.org/10.1021/acs.analchem.6b02678.
Deng L, Garimella SVB, Hamid AM, Webb IK, Attah IK, Norheim RV, et al. Compression ratio ion mobility programming (CRIMP) accumulation and compression of billions of ions for ion mobility-mass spectrometry using traveling waves in structures for lossless ion manipulations (SLIM). Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b00189.
Garimella SVB, Hamid AM, Deng L, Ibrahim YM, Webb IK, Baker ES, et al. Squeezing of ion populations and peaks in traveling wave ion mobility separations and structures for lossless ion manipulations using compression ratio ion mobility programming. Anal Chem. 2016. https://doi.org/10.1021/acs.analchem.6b03660.
McKenzie-Coe A, Debord JD, Ridgeway M, Park M, Eiceman GA, Fernandez-Lima F. Lifetimes and stabilities of familiar explosive molecular adduct complexes during ion mobility measurements. Analyst. 2015. https://doi.org/10.1039/c5an00527b.
Jurado-Campos N, Garrido-Delgado R, Martínez-Haya B, Eiceman GA, Arce L. Stability of proton-bound clusters of alkyl alcohols, aldehydes and ketones in ion mobility spectrometry. Talanta. 2018. https://doi.org/10.1016/j.talanta.2018.03.030.
Ewing RG, Eiceman GA, Harden CS, Stone JA. The kinetics of the decompositions of the proton bound dimers of 1,4-dimethylpyridine and dimethyl methylphosphonate from atmospheric pressure ion mobility spectra. Int J Mass Spectrom. 2006. https://doi.org/10.1016/j.ijms.2006.04.003.
Rajapakse MY, Stone JA, Eiceman GA. Decomposition kinetics of nitroglycerine·Cl(-)(g) in air at ambient pressure with a tandem ion mobility spectrometer. J Phys Chem A. 2014. https://doi.org/10.1021/jp412444b.
Rajapakse RMMY, Stone JA, Eiceman GA. An ion mobility and theoretical study of the thermal decomposition of the adduct formed between ethylene glycol dinitrate and chloride. Int J Mass Spectrom. 2014. https://doi.org/10.1016/j.ijms.2014.07.039.
Loboda A. Novel ion mobility setup combined with collision cell and time-of-flight mass spectrometer. J Am Soc Mass Spectrom. 2006. https://doi.org/10.1016/j.jasms.2006.01.013.
Fernandez-Lima F, Kaplan DA, Suetering J, Park MA. Gas-phase separation using a trapped ion mobility spectrometer. Int J Ion Mobil Spectrom. 2011. https://doi.org/10.1007/s12127-011-0067-8.
Fernandez-Lima FA, Kaplan DA, Park MA. Note: Integration of trapped ion mobility spectrometry with mass spectrometry. Rev Sci Instrum. 2011. https://doi.org/10.1063/1.3665933.
Ridgeway ME, Lubeck M, Jordens J, Mann M, Park MA. Trapped ion mobility spectrometry - a short review. Int J Mass Spectrom. 2018. https://doi.org/10.1016/j.ijms.2018.01.006.
Larriba-Andaluz C, Chen X, Nahin M, Wu T, Fukushima N. Analysis of ion motion and diffusion confinement in inverted drift tubes and trapped ion mobility spectrometry devices. Anal Chem. 2018. https://doi.org/10.1021/acs.analchem.8b03930.
Bleiholder C. Towards measuring ion mobilities in non-stationary gases and non-uniform and dynamic electric fields (I). Transport equation. Int J Mass Spectrom. 2016. https://doi.org/10.1016/j.ijms.2016.01.003.
Chai M, Young MN, Liu FC, Bleiholder C. A transferable, sample-independent calibration procedure for trapped ion mobility spectrometry (TIMS). Anal Chem. 2018. https://doi.org/10.1021/acs.analchem.8b01326.
Haler JRN, Kune C, Massonnet P, Comby-Zerbino C, Jordens J, Honing M, et al. Comprehensive ion mobility calibration: poly(ethylene oxide) polymer calibrants and general strategies. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b02564.
Liu FC, Kirk SR, Bleiholder C. On the structural denaturation of biological analytes in trapped ion mobility spectrometry – mass spectrometry. Analyst. 2016. https://doi.org/10.1039/C5AN02399H.
Silveira JA, Michelmann K, Ridgeway ME, Park MA. Fundamentals of trapped ion mobility spectrometry part II - fluid dynamics. J Am Soc Mass Spectrom. 2016. https://doi.org/10.1007/s13361-015-1310-z.
Benigni P, Marin R, Molano-Arevalo JC, Garabedian A, Wolff JJ, Ridgeway ME, et al. Towards the analysis of high molecular weight proteins and protein complexes using TIMS-MS. Int J Ion Mobil Spec. 2016. https://doi.org/10.1007/s12127-016-0201-8.
Silveira JA, Ridgeway ME, Laukien F, Mann M, Park MA. Parallel accumulation for 100% duty cycle trapped ion mobility-mass spectrometry. Int J Mass Spectrom. 2016. https://doi.org/10.1016/j.ijms.2016.03.004.
Silveira JA, Danielson W, Ridgeway ME, Park MA. Altering the mobility-time continuum - nonlinear scan functions for targeted high resolution trapped ion mobility-mass spectrometry. Int J Ion Mobil Spec. 2016. https://doi.org/10.1007/s12127-016-0196-1.
Benigni P, Porter J, Ridgeway ME, Park MA, Fernandez-Lima F. Increasing analytical separation and duty cycle with nonlinear analytical mobility scan functions in TIMS-FT-ICR MS. Anal Chem. 2018. https://doi.org/10.1021/acs.analchem.7b04053.
Benigni P, Fernandez-Lima F. Oversampling selective accumulation trapped ion mobility spectrometry coupled to FT-ICR MS: fundamentals and applications. Anal Chem. 2016. https://doi.org/10.1021/acs.analchem.6b01946.
Nazarov EG. A journey into DMS/FAIMS technology. Int. J. Ion Mobil. Spec. 2012. https://doi.org/10.1007/s12127-012-0112-2.
Buryakov IA, Krylov EV, Makas AL, Nazarov EG, Pervukhin VV, Rasulev UK. Separation of ions according to their mobility in a strong alternating current electric field. Sov Tech Phys Lett. 1991;17(6):446–7.
Shvartsburg AA, Li F, Tang K, Smith RD. Distortion of ion structures by field asymmetric waveform ion mobility spectrometry. Anal Chem. 2007. https://doi.org/10.1021/ac061306c.
Robinson EW, Shvartsburg AA, Tang K, Smith RD. Control of ion distortion in field asymmetric waveform ion mobility spectrometry via variation of dispersion field and gas temperature. Anal Chem. 2008. https://doi.org/10.1021/ac800655d.
Krylov EV, Coy SL, Nazarov EG. Temperature effects in differential mobility spectrometry. Int J Mass Spectrom. 2009. https://doi.org/10.1016/j.ijms.2008.10.025.
Nazarov EG, Coy SL, Krylov EV, Miller RA, Eiceman GA. Pressure effects in differential mobility spectrometry. 2006. https://doi.org/10.1021/ac061092z.
Shvartsburg AA, Haris A, Andrzejewski R, Entwistle A, Giles R. Differential ion mobility separations in the low-pressure regime. Anal Chem. 2017. https://doi.org/10.1021/acs.analchem.7b03925.
Viehland LA, Guevremont R, Purves RW, Barnett DA. Comparison of high-field ion mobility obtained from drift tubes and a FAIMS apparatus. Int J Mass Spectrom. 2000. https://doi.org/10.1016/S1387-3806(99)00224-9.
Nazarov EG, Langejuergen J, Kirk AT, Allers M, Oermann J, Zimmermann S. Comparison of ion alpha parameters obtained from DMS and HiKE-IMS data. In: International Conference on Ion Mobility Spectrometry; 2015.
Nazarov EG, Anderson AG, Krylov EV, Coy SL, Miller RA, Burchfield D, et al. Miniature DMS-IMS detector for enhanced revolving power. In: Proceedings of 16th International Conference on Ion Mobility Spectrometry; 2007.
Shvartsburg AA, Tang K, Smith RD. Two-dimensional ion mobility analyses of proteins and peptides. Methods Mol Biol (Clifton, N.J.). 2009. https://doi.org/10.1007/978-1-59745-493-3_26.
Krylov EV, Nazarov EG, Miller RA. Differential mobility spectrometer: model of operation. Int J Mass Spectrom. 2007. https://doi.org/10.1016/j.ijms.2007.07.003.
Shvartsburg AA, Smith RD, Wilks A, Koehl A, Ruiz-Alonso D, Boyle B. Ultrafast differential ion mobility spectrometry at extreme electric fields in multichannel microchips. 2009. https://doi.org/10.1021/ac900892u.
Krylov E, Nazarov EG, Miller RA, Tadjikov B, Eiceman GA. Field dependence of mobilities for gas-phase-protonated monomers and proton-bound dimers of ketones by planar field asymmetric waveform ion mobility spectrometer (PFAIMS). J Phys Chem A. 2002. https://doi.org/10.1021/jp020009i.
Santiago BG, Harris RA, Isenberg SL, Glish GL. Resolving powers of >7900 using linked scans - how well does resolving power describe the separation capability of differential ion mobility spectrometry. Analyst. 2015. https://doi.org/10.1039/C5AN00845J.
Shvartsburg AA, Smith RD. Accelerated high-resolution differential ion mobility separations using hydrogen. Anal Chem. 2011. https://doi.org/10.1021/ac202386w.
Shvartsburg AA, Danielson WF, Smith RD. High-resolution differential ion mobility separations using helium-rich gases. Anal Chem. 2010. https://doi.org/10.1021/ac902852a.
Shvartsburg AA, Smith RD. Separation of protein conformers by differential ion mobility in hydrogen-rich gases. Anal Chem. 2013. https://doi.org/10.1021/ac4015963.
Shvartsburg AA, Ibrahim YM, Smith RD. Differential ion mobility separations in up to 100% helium using microchips. J Am Soc Mass Spectrom. 2014. https://doi.org/10.1007/s13361-013-0797-4.
Krylova N, Krylov E, Eiceman GA, Stone JA. Effect of moisture on the field dependence of mobility for gas-phase ions of organophosphorus compounds at atmospheric pressure with field asymmetric ion mobility spectrometry. J Phys Chem A. 2003. https://doi.org/10.1021/jp0221136.
Schneider BB, Covey TR, Coy SL, Krylov EV, Nazarov EG. Chemical effects in the separation process of a differential mobility/mass spectrometer system. 2010. https://doi.org/10.1021/ac902571u.
Blagojevic V, Chramow A, Schneider BB, Covey TR, Bohme DK. Differential mobility spectrometry of isomeric protonated dipeptides: modifier and field effects on ion mobility and stability. Anal Chem. 2011. https://doi.org/10.1021/ac200100s.
Shvartsburg AA, Smith RD. Optimum waveforms for differential ion mobility spectrometry (FAIMS). J Am Soc Mass Spectrom. 2008. https://doi.org/10.1016/j.jasms.2008.05.008.
Krylov EV, Coy SL, Vandermey J, Schneider BB, Covey TR, Nazarov EG. Selection and generation of waveforms for differential mobility spectrometry. Rev Sci Instrum. 2010. https://doi.org/10.1063/1.3284507.
Schneider BB, Londry F, Nazarov EG, Kang Y, Covey TR. Maximizing ion transmission in differential mobility spectrometry. J Am Soc Mass Spectrom. 2017. https://doi.org/10.1007/s13361-017-1727-7.
Baird MA, Anderson GA, Shliaha PV, Jensen ON, Shvartsburg AA. Differential ion mobility separations/mass spectrometry with high resolution in both dimensions. Anal Chem. 2018. https://doi.org/10.1021/acs.analchem.8b04518.
Funding
This work is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—ZI 1288/4–1 and ZI 1288/7-1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Published in the topical collection Close-Up of Current Developments in Ion Mobility Spectrometry with guest editor Gérard Hopfgartner.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kirk, A.T., Bohnhorst, A., Raddatz, CR. et al. Ultra-high-resolution ion mobility spectrometry—current instrumentation, limitations, and future developments. Anal Bioanal Chem 411, 6229–6246 (2019). https://doi.org/10.1007/s00216-019-01807-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-019-01807-0