
Abstract
An efficient analytical method based on molecularly imprinted solid-phase extraction (MISPE) coupled with high-performance liquid chromatography-diode array detection (HPLC-DAD) was established for the determination of iridoid glycosides (IGs) in Di-huang pills. As the solid-phase extraction medium, superhydrophilic molecularly imprinted polymers (MIPs) with high affinity and selectivity to IGs in water media were fabricated using divinyl galactose as a single cross-linking monomer. The structure, porosity, and hydrophilicity of MIPs were characterized. The properties involving dynamic adsorption, kinetic adsorption, and selectivity were evaluated. Under optimal conditions the MISPE-HPLC-DAD based method was applied for loganin, morroniside, cornin, and sweroside determination in three kinds of Di-huang pills. The limits of detection of four IGs were 0.002–0.003 mg g−1. Furthermore, the proposed method exhibited some merits including good linearity, excellent precision, and desirable accuracy. The established MISPE-HPLC-DAD method has great potential for the selective determination of IGs in Chinese patent drugs.

ᅟ
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Molecularly imprinted polymers (MIPs) can be synthesized by the polymerization of a mixture of functional monomers and cross linkers in the presence of template molecules dissolved in porogenic solvents [1]. As the artificial receptor, MIPs have received increasing interest for analytical applications, involving separation media, immunoassays, biosensors, and affinity supports for screening libraries of bioactive compounds [2]. MIPs can recognize the template molecule or its structural analogues based on specific molecular recognition cavities, which are complementary to the template molecule in shape, size, and spatial distribution [3,4,5,6,7,8,9,10]. In the process of MIPs synthesis, template molecules, cross linkers, and functional monomers are considered as the “three-elements of molecular imprinting.”
The optimization of MIPs formulation components has been relatively mature but complicated and time-consuming. A variety of variables should be determined via the preparation and evaluation of several polymers, such as how many and what type of functional monomers to use, how many and which cross linkers to use, the optimum ratio of templates/functional monomers, and the optimum ratio of cross linkers/functional monomers. The generalized process has limited the broader impact of molecularly imprinted technology (MIT) on society and inhibited the widespread use of MIT by the general scientific community. Molecular imprinting has not been made easy so far, except for some examples of the single cross linking monomer-based MIPs [11,12,13,14,15]. However, these MIPs have not been utilized for the imprinting of water-soluble compounds, most probably due to the incompatibility with water media.
Iridoid glycosides (IGs) are a series of typical water-soluble compounds which are found in many herbs [16, 17]. Due to the observably pharmacological and biological activities, the amounts of IGs have been utilized to evaluate the quality of some traditional Chinese patent drugs, such as Liu Wei Di Huang Wan (LWDHW), Dang Shao Di Huang Wan (DSDHW), Qi Ju Di Huang Wan (QJDHW), and so on [18,19,20].
High-performance liquid chromatography (HPLC) methods have been developed to analyze IGs [21,22,23,24,25,26,27]. However, the sample preparation of Chinese patent drugs presents an immense challenge because of interfering constituents within the matrix. Therefore, it is important to develop a rapid and selective sample preparation method for the analysis of active ingredients in Chinese patent drugs [28,29,30].
In this work, we aim to develop a water-compatible MIPs based on a single cross-linking monomer for the recognition of IGs in Di-huang pills. Superhydrophilic MIPs have been synthesized via the bulk polymerization in the presence of loganin as the template, divinyl galactose (DivG) as the single cross-linking monomer, 2,2-azobisisobutyronitrile (AIBN) as the initiator, and H2O as the porogenic solvent. The binding capacities of MIPs were investigated in water media. In addition, molecularly imprinted solid-phase extraction (MISPE) conditions were optimized in detail. Meanwhile, a method for determination of four IGs in Chinese patent drugs, including loganin, morroniside, cornin, and sweroside, was set up by combination of MISPE and HPLC. The recoveries of the developed method for selective determination of IGs are highly satisfactory.
Materials and methods
Materials
Loganin, morroniside, cornin, sweroside, arbutin, and genipin were provided by Yuanye biotechnology Co., Ltd. (Shanghai, China). Diisopropylidene galactose (DipG), acryloyl chloride, and allyl alcohol were obtained from Xiya Reagent (Chengdu, China). Methacrylic acid (MAA), N,N′-methylene diacrylamide (MBA), ethylene glycol dimethacrylate (EGDMA), methanol (MeOH, HPLC grade), and trifluoroacetic acid (TFA, HPLC grade) were obtained from J&K Scientific Ltd. (Beijing, China). Other analytical reagents were from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). The Chinese patent drugs, including LWDHW, DSDHW, and QJDHW were bought from the local drugstore in Jinan, China. Alkenyl glycosides glucose (AGG) was synthesized as the previously reported [31]. The chemical structures of typical compounds are shown in Fig. 1.

Chemical structures of typical compounds
Standards of IGs were dissolved individually in the deionized water to 2.0 mmol L−1 concentration for each analyte, and they were stored in darkness at 4 °C prior to tests. Working solutions were prepared by gradually diluting the corresponding stock standard solutions.
Instrument and analytical conditions
For HPLC, an Agilent 1120 system (Palo Alto, CA, USA) equipped with a diode-array detector (DAD) was used. A Waters C18 column (5 μm, 4.6 × 250 mm) was utilized for chromatographic separation. The mobile phase was MeOH-0.1% TFA in ultrapure water (3:7, v/v). The injection volume and flow rate were set at 10 μL and 1.0 mL min−1, respectively. The absorption wavelength was set at 235 nm.
The prepared polymers were characterized with Fourier transform infrared spectroscopy (FT-IR), Brunauer–Emmett–Teller (BET) N2 sorption isotherms, and water contact angle (CA). The detailed information about these instruments were shown in the supplementary material.
Synthesis of the single cross-linking monomer DivG
The synthetic route of DivG was shown in Scheme 1.

The synthetic route of DivG
Acryloyl DipG: DipG (2.6 g) and triethylamine (2.0 g) were suspended in dry CH2Cl2 (25 mL), and acryloyl chloride (2.0 g) was cautiously added at 0 °C. Then the reaction mixture was stirred for 6 h at 22 °C. After that, the reaction products were washed with saturated aqueous NaHCO3, diluted HCl (10%), and water, and then dried with MgSO4. After evaporating the solvent, residues were purified by the recrystallization in MeOH to yield white crystal (2.8 g, 89%).
DivG: Acryloyl DipG (2.0 g) was suspended in TFA (20 mL) and the mixture was refluxed for 4 h. After evaporating the solvent, acryloyl galactose was obtained.
Acetyl chloride (2.5 g) was added to allyl alcohol (20 g) at − 5 °C. After stirring for 30 min at − 5 °C, acryloyl galactose (2.0 g) was added and the reaction mixture was stirred for 1 h at 90 °C. The mixture was quenched with NaHCO3, filtered, and concentrated. The residue was purified by the recrystallization in methanol/ethyl acetate (9:1, v/v) at − 10 °C to yield DivG as white crystal (1.4 g, 82%). 1H NMR (CD3OD, 400 MHz) δ 5.84–5.88 (m, 1H), 5.21–5.26 (m, 1H), 5.06–5.09 (m, 1H), 4.76–4.80 (m, 1H), 3.95–4.15 (m, 3H), 3.59–4.15 (m, 6H), 3.21–3.25 (m, 1H), 3.21 (d, 1H, J = 1.2 Hz) ppm; 13C NMR (CD3OD, 100 MHz) δ 166.4, 131.8, 130.1, 128.4, 115.4, 104.2, 74.1, 73.9, 73.2, 71.9, 68.9, 63.1 ppm; HRMS (ESI) m/z calculated for C12H18O7 [M + Na]+ 297.0950, found: 297.0944.
Preparation of imprinted materials
MIPs and non-imprinted polymers (NIPs) were synthesized via the bulk polymerization by using DivG as the hydrophilic single cross-linking monomer (see supplementary material).
Adsorption text
Dynamic adsorption
Twenty milligrams of MIPs or NIPs were immersed into 2.5 mL of loganin solution at different concentrations (0.3–1.05 mmol L−1). After shaken for 3 h at 22 °C, the equilibrium concentrations of loganin in the filtrates were analyzed by HPLC-DAD. The equilibrium adsorption capacity (Qe, mg g−1) was calculated according to the Eq. (1) [32]:
In this equation, Ci (mmol L−1) represent the initial concentration of loganin, Ce (mmol L−1) is the equilibrium concentration of loganin, M represent the molar mass of loganin, m (mg) is the mass of polymers, and v (mL) is the volume of solution.
The data were further processed via the Langmuir Eq. (2) and Freundlich Eq. (3) [32, 33]:
where QmL (mg g−1) is Langmuir theoretical maximum adsorption capacity. KF and n are the Freundlich constants.
Kinetic adsorption
Twenty milligrams of MIPs or NIPs were immersed into 2.5 mL of loganin solution (0.45 mmol L−1). The mixture was shaken for a set time (5–150 min) at 22 °C and the free concentration of loganin in the filtrates at time t were analyzed by HPLC-DAD. The adsorption capacity (Qt, mg g−1) at time t was calculated according to the Eq. (4) [34]:
where Ct (mmol L−1) is the free concentration of loganin at time t.
To evaluate the adsorption kinetic mechanism, the kinetic data were calculated according to the pseudo-first-order (5) and pseudo-second-order (6) model [34]:
where Qe and Qt are the amounts of loganin adsorbed at equilibrium time and time t, respectively. Qm1 and Qm2 are the theoretical maximum adsorption capacity. k1 (min−1) is the rate of pseudo-first-order and k2 (g mg−1 min−1) indicates the rate of pseudo-second-order.
Selectivity study
The selectivity of MIPs and NIPs was evaluated using working standard solutions (0.45 mmol L−1) of loganin and five competitive compounds (morroniside, cornin, sweroside, arbutin, and genipin). Polymers (20 mg) were shaken with aliquots of each solution (2.5 mL) for 3 h. The free concentration of each analyte in the filtrates was analyzed by HPLC-DAD. The imprinting factor (IF = Qe, MIPs / Qe, NIPs) was utilized to evaluate the recognition capacity of MIPs.
Application of MISPE to samples
Each kind of Di-huang pills (0.1 g) was refluxed with the deionized water (100 mL) for 2 h. After filtered through a 0.45 μm PTFE membrane, the supernatant was accurately diluted with the deionized water to 100 mL for further experiments.
To prepare MISPE cartridges, MIPs (100 mg) were slurry-packed under vacuum into empty SPE cartridges (1 mL) and capped between two polyethylene disks. The MISPE cartridge was firstly wetted with deionized water (10 mL). Then, the extraction solution was flowed through the MISPE cartridges at a constant flow rate of 25.0 mL min−1. After washed with dioxane (2 mL), the MISPE cartridge was eluted with ammonia/ethanol (2 mL, pH = 8.5) at a constant flow rate of 20.0 mL min−1. The elution fraction was analyzed by HPLC-DAD.
Results and discussion
Optimization of MIPs preparation conditions
The structural skeleton of DivG assembles MAA and AGG which can possess more possible interaction sites. To investigate the adsorption capacity (Qe) and selectivity (IF) using DivG as the single cross-linking monomer for MIT, polymers were imprinted to loganin and compared with polymers imprinted with the traditional imprinting formulation MAA/EGDMA, MAA/MBA, AGG/EGDMA, and AGG/MBA [31, 35, 36]. As listed in Table 1, the values of Qe and IF indicated the imprinting property was better for MIPs using DivG versus the traditional formulation.
The molar ratio of the template-single cross-linking monomer is the critical factor that influences the imprinting property of MIPs. Different molar ratios of template-single cross-linking monomer were utilized to optimize the conditions for the synthesis of MIPs. As summarized in Table 1, the value of Qe increased with increasing the ratios of the single cross-linking monomer DivG, while that of the adsorption on NIPs were not drastically increasing. Considering the value of IF, the molecular ratio of 1:8 for the template:single cross-linking monomer was selected to prepare MIPs (Table 1, entry 9).
Characterization of polymers
The FT-IR spectrums of the synthesized polymers (see Electronic Supplementary Material (ESM) Fig. S1) show that there are some hydrophilic groups on MIPs and NIPs, such as hydroxyl groups (-OH), and the peak of 3440 cm−1 corresponds to the stretching vibration of O-H. The peaks showed at 2956 cm−1 indicate the presence of -CH2- groups. The strong peak at 1731 cm−1 can be ascribed to the stretching vibration of C=O. In addition, the peak at 1115 cm−1 is assigned to the stretching vibration of C-O-C. The same characteristic signals for both MIPs and NIPs demonstrate that the template compounds were totally eliminated in the synthesized products.
The porosity of MIPs-9 and NIPs-9 was analyzed by N2 adsorption-desorption at 77.3 K. Both MIPs-9 and NIPs-9 exhibited “type IV” N2 adsorption-desorption isotherm, associating with the capillary condensation taking place in the mesoporous material (Fig. 2) [35]. The BET surface area for MIPs-9 was 392 m2 g−1 with the total pore volume of 0.88 cm3 g−1. The BET surface area and pore volume for NIPs-9 were 386 m2 g−1 and 0.58 cm3 g−1, respectively. Similar pore size distributions for MIPs-9 and NIPs-9 were found, which consisted of a sharp peak at 9.0 nm (MIPs-9) and 6.0 nm (NIPs-9) (Fig. 2 inset). The total pore volume and pore size of MIPs-9 were both about 1.5 times of that of NIPs-9, which might be caused by the imprinted cavity. These results implied that the porosity of MIPs-9 was favorable to application as the SPE adsorbent.

N2 adsorption-desorption isotherms of MIPs (a) and NIPs (b)
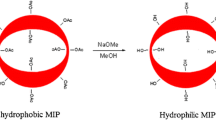
Video of CA measurement was recorded to evaluate the water-compatibility of polymers. One droplet of water was deposited on the surface of MIPs-9 slice, where it rapidly spread over the contact area (ESM Movie S1). As shown in Fig. 3, the water CA approached 0° within 1.14 s, which demonstrated the super-hydrophilicity of MIPs-9. The excellent water-compatibility of MIPs-9 was beneficial to bind analytes from aqueous matrices.

The profiles of a water drop on the films of MIPs
Adsorption text
Dynamic adsorption
The dynamic adsorption experiment was studied to evaluate the recognition ability of MIPs and NIPs toward loganin at 22 °C. As shown in Fig. 4a, the adsorption amounts of loganin on polymers were observably increased with increasing the initial concentration of loganin. MIPs exhibited higher affinity than NIPs and the adsorption reached saturation at the initial concentration of 0.95 mmol L−1. To study the adsorption mechanism, Langmuir and Freundlich models were utilized to fit the dynamic adsorption data. As shown in Figs. S2 and S3 in the ESM, the Freundlich model (R2MIPs = 0.9389 and R2NIPs = 0.9324) was more suitable for modeling the dynamic adsorption than the Langmuir model (R2MIPs = 0.6547 and R2NIPs = 0.3715). The value of n for MIPs was calculated to be 2.1 (n > 2), implying a good fitting with the Freundlich model and high heterogeneity [36, 37].

a The adsorption isotherms of loganin on MIPs and NIPs. b Dynamic adsorption isotherms of MIPs and NIPs for loganin
Kinetic adsorption
The adsorption kinetics were evaluated for MIPs and NIPs using loganin at the random concentration of 0.45 mmol L−1. Figure 4b showed the adsorption capacity for loganin on MIPs increased faster in the first 20 min than that of NIPs and approached equilibrium after that time. The faster binding of MIPs to the template molecules was mainly caused by imprinted cavities. Lagergren pseudo-first-order and pseudo-second-order equation were utilized to fit the kinetic adsorption data. As shown in Figs. S4 and S5 in the ESM, the pseudo-second-order equation (R2MIPs = 0.980 and R2NIPs = 0.911) was more suitable for modeling the kinetic adsorption than the pseudo-first-order equation (R2MIPs = 0.897 and R2NIPs = 0.903). According to the Lagergren theory, the value of Qm2 for MIPs was calculated to be 21.6 mg g−1, which was fitted well with the experimental data (19.3 mg g−1).
Selectivity study
Figure 5 illustrated the selectivity of MIPs and NIPs. The values of Qe for four IGs, loganin, morroniside, cornin, and sweroside, were > 15 mg g−1 for MIPs but < 5 mg g−1 for NIPs. The adsorption of arbutin and genipin onto MIPs was not noticeably different from adsorption onto NIPs. The values of IF were 4.58 (loganin), 4.41 (morroniside), 4.17 (cornin), 4.40 (sweroside), 1.92 (arbutin), and 1.67 (genipin), respectively, indicating the excellent selectivity of MIPs for IGs. The imprinting recognition between IGs and the polymeric matrix and the excellent compatibility of MIPs with aqueous media both contribute to the high selectivity.

Selectivity of MIPs and NIPs for different compounds
MISPE optimization
In our previous work, some MISPE-HPLC-DAD methods for the analysis of IGs have been developed [37,38,39,40]. Therefore, the same SPE protocol was utilized to evaluate the adsorbent. Using water as loading solvent, the recoveries of four IGs (0.3 mmol L−1 for each IG, including loganin, morroniside, cornin, and sweroside) were almost constant with increasing flow rate to 25.0 mL min−1. When the flow rate was unceasingly increased, the recoveries were decreased (ESM Fig. S6). Thus, the maximum loading flow rate of 25.0 mL min−1 was used for next experiments.
A suitable washing solvent should be utilized to both bind target analytes and elute the potential interferents from SPE cartridges. In this work, 2 mL of different washing solvents were evaluated, including n-hexane, ethyl acetate, toluene, acetone, n-butanol, and dioxane. Arbutin was used as the interferent. As shown in Fig. 6, only n-butanol and dioxane could elute the interferent arbutin from SPE cartridges. In view of higher recoveries, 2 mL of dioxane was chosen as the washing solvent.

Effect of washing solvents on the retention rate of IGs. a n-Hexane, b ethyl acetate, c toluene, d acetone, e n-butanol, and f dioxane. Peak identifications: 1, arbutin; 2, morroniside; 3, cornin; 4, sweroside; 5, loganin
Two milliliters of EtOH/Et3N (pH = 8.5) was studied to optimize the flow rate of elution solvent. As shown in (ESM Fig. S7), the recoveries were decreased when the flow rate was increased from 20.0 to 25.0 mL min−1. In order to obtain high recovery, the maximum elution flow rate of 20.0 mL min−1 was adopted.
Regeneration capability
The regeneration capability is an important factor for the application of sorbents. As shown in ESM Fig. S8, MISPE cartridges could be reused for five 30 times without an obvious loss of recoveries for 4 IGs. The decrease of recoveries after 30 times might be caused by the reduction of the imprinting sites in MIPs during the adsorption-elution cycles. Overall, MISPE cartridges showed excellent repeatability and reproducibility and were, therefore, suitable for the practical application.
Validation of MISPE-HPLC-DAD method
The MISPE-HPLC-DAD method was validated by the evaluation of linearity, limits of detection (LOD), limits of quantification (LOQ), and precision. The spiked samples ranging from 0.01 to 50 mg g−1 were extracted with MISPE cartridges. Three replicates were utilized for each concentration and calibration curves of the four IGs were obtained by plotting the peak area versus analyte concentration.
As listed in Table 2, satisfactory linearities (0.01–50 mg g−1) were obtained with corresponding correlation coefficients (r) 0.9977, 0.9968, 0.9971, and 0.9962 for loganin, morroniside, cornin, and sweroside, respectively. The calculated LODs and LOQs were 0.002–0.003 and 0.008–0.01 mg g−1, which were defined as the concentrations of analytes at the signal-to-noise ratios of 3 and 10, respectively. The precision of the MISPE-HPLC-DAD method was investigated by using replicates of the spiked samples on the same day (intra-day) and for three consecutive days (inter-day). As summarized in Table 2, the intra-day and inter-day precisions expressed as the relative standard deviation (RSD) were in the range of 3.4–4.7 and 5.2–6.9%, respectively. All these results illustrated that the established MISPE-HPLC-DAD method for four IGs can satisfy the analysis of real samples, since which has the good linearity and excellent precision.
Analysis of IGs in Chinese patent drug
The established MISPE method was utilized for the IGs analysis in three kinds of Dihuang pills, including LWDHW, DSDHW, and QJDHW. After accurately diluted with deionized water, the extract solution was loaded onto MISPE cartridges at the flow rate of 25 mL min−1. The cartridges were washed with dioxane (2 mL) and eluted with EtOH/Et3N (2 mL, pH = 8.5) at the flow rate of 20 mL min−1. The eluents were determined by HPLC-DAD.
As summarized in Table 3, the combined amounts of loganin and morroniside without spiking IGs were 1.04 mg g−1 for LWDHW, 0.84 mg g−1 for DSDHW, and 0.82 mg g−1 for QJDHW, which were higher than the level stipulated by the Chinese Pharmacopeia (0.9 mg g−1 for LWDHW, 0.64 mg g−1 for DSDHW, and 0.65 mg g−1 for QJDHW). Moreover, the recoveries for spiked drugs at two concentration level (0.1 and 1.0 mg g−1) were ranged from 88.9 to 102.0% with the RSD in the range from 3.1 to 5.8%.
The typical chromatograms of extracts from LWDHW were shown in Fig. 7. Without the enrichment, only morroniside chromatographic peak was visible since the concentrations of other IGs were too low (Fig. 7a). In the washing chromatogram for the MISPE cartridge, IGs peaks were very low, confirming the high retention of IGs in the MISPE cartridge (Fig. 7c). After the selective enrichment using the MISPE cartridge, the matrix interference was minimized and great interferences were eliminated efficiently (Fig. 7d). Although morroniside and loganin chromatogram peaks could be obtained through the C18 SPE cartridge, IGs peaks are obviously lower than peaks obtained from the MISPE cartridge (Fig. 7b vs d), which implied low recoveries for the C18 SPE cartridge. These results indicate that the proposed MISPE–HPLC-DAD method could be utilized to analyze IGs to evaluate the quality of Di-huang pills.

a Chromatogram of extract of LWDHW before percolating through SPE column. b Chromatogram of eluting solutions from C18 SPE column. c Chromatogram of washing solutions from MISPE column. d Chromatogram of eluting solutions from MISPE column. e standards of four IGs. Peak identifications: 1, morroniside; 2, cornin; 3, sweroside; 4, loganin
Conclusions
In this study, superhydrophilic MIPs were synthesized using DivG a single cross-linking monomer. MIPs were demonstrated by a series of experiments to be a promising and effective adsorbent with the selective recognition to IGs in aqueous media. The dynamic and kinetic adsorption data were well fitted by the Freundlich model and the pseudo-second-order model, respectively. The developed MISPE-HPLC-DAD method for the determination of IGs in Di-huang pills exhibited some merits including the good linearity, low LODs, excellent precision, and desirable accuracy. Accordingly, it indicates great potential in the analysis of water-soluble IGs to evaluate the quality of Chinese patent drugs.
References
Chen L, Xu S, Li J. Recent advances in molecular imprinting technology: current status, challenges and highlighted applications. Chem Soc Rev. 2011;40:2922–42.
Chen L, Wang X, Lu W, Wu X, Li J. Molecular imprinting: perspectives and applications. Chem Soc Rev. 2016;45:2137–211.
Liang S, Yan H, Cao J, Han Y, Shen S, Bai L. Molecularly imprinted phloroglucinol-formaldehyde-melamine resin prepared in a deep eutectic solvent for selective recognition of clorprenaline and bambuterol in urine. Anal Chim Acta. 2017;951:68–77.
Díaz-Álvarez M, Martín-Esteban A. Hollow fiber membrane-protected molecularly imprinted microspheres for micro solid-phase extraction and clean-up of thiabendazole in citrus samples. J Chromatogr A. 2018;1531:39–45.
Rossetti C, Ore OG, Sellergren B, Halvorsen TG, Reubsaet L. Exploring the peptide retention mechanism in molecularly imprinted polymers. Anal Bioanal Chem. 2017;409:5631–43.
Jia M, Yang J, Sun Y, Bai X, Wu T, Liu Z, et al. Improvement of imprinting effect of ionic liquid molecularly imprinted polymers by use of a molecular crowding agent. Anal Bioanal Chem. 2018;410:595–604.
Sun Y, Pang Y, Zhang J, Li Z, Liu J, Wang B. Application of molecularly imprinted polymers for the analysis of polycyclic aromatic hydrocarbons in lipid matrix-based biological samples. Anal Bioanal Chem. 2017;409:6851–60.
Sun Y, Zhong S. Molecularly imprinted polymers fabricated via Pickering emulsions stabilized solely by food-grade casein colloidal nanoparticles for selective protein recognition. Anal Bioanal Chem. 2018; https://doi.org/10.1007/s00216-018-1006-x.
Ye L. Molecularly imprinted polymers with multi-functionality. Anal Bioanal Chem. 2016;408:1727–33.
Guo P, Yuan X, Zhang J, Wang B, Sun X, Chen X, et al. Dummy-surface molecularly imprinted polymers as a sorbent of micro-solid-phase extraction combined with dispersive liquid–liquid microextraction for determination of five 2-phenylpropionic acid NSAIDs in aquatic environmental samples. Anal Bioanal Chem. 2018;410:373–89.
Sibrian-Vazquez M, Spivak DA. Molecular imprinting made easy. J Am Chem Soc. 2004;126:7827–33.
Sibrian-Vazquez M, Spivak DA. Enhanced enantioselectivity of imprinted polymers formulated with novel crosslinking monomers. Macromolecules. 2003;36:5105–13.
Wei Z, Wu X, Zhang B, Li R, Huang Y, Liu Z. Coatings of one monomer molecularly imprinted polymers for open tubular capillary electrochromatography. J Chromatogr A. 2011;1218:6498–504.
Sibrian-Vazquez M, Spivak DA. Improving the strategy and performance of molecularly imprinted polymers using cross-linking functional monomers. J Organomet Chem. 2003;68:9604–11.
Yoshimatsu K, LeJeune J, Spivak DA, Ye L. Peptide-imprinted polymer microspheres prepared by precipitation polymerization using a single bi-functional monomer. Analyst. 2009;134:719–24.
Boros CA, Stemrmitz FR. Iridoids. An updated review. Part I. J Nat Prod. 1990;53:1055–147.
Boros CA, Stemrmitz FR. Iridoids. An updated review. Part II. J Nat Prod. 1991;54:1173–246.
Chinese Pharmacopoeia Commission. Chinese pharmacopoeia, vol. I. Beijing: People’s Medical Publishing House; 2015. p. 704–−705.
Chinese Pharmacopoeia Commission. Chinese pharmacopoeia, vol. I. Beijing: People’s Medical Publishing House; 2015. p. 775–−777.
Chinese Pharmacopoeia Commission. Chinese pharmacopoeia, vol. I. Beijing: People’s Medical Publishing House; 2015. p. 935–−936.
Song Y, Li SL, Wu MH, Li HJ, Li P. Qualitative and quantitative analysis of iridoid glycosides in the flower buds of Lonicera species by capillary high performance liquid chromatography coupled with mass spectrometric detector. Anal Chim Acta. 2006;564:211–8.
Cao XY, Wang ZZ. Simultaneous determination of four iridoid and secoiridoid glycosides and comparative analysis of Radix Gentianae macrophyllae and their related substitutes by HPLC. Phytochem Anal. 2010;21:348–54.
Yang L, Wang Y, Wang L, Xiao H, Wang Z, Hu Z. Rapid quantification of iridoid glycosides analogues in the formulated Chinese medicine Longdan Xiegan decoction using high-performance liquid chromatography coupled with mass spectromentry. J Chromatogr A. 2009;1216:2098–103.
He ML, Cheng XW, Chen JK, Zhou TS. Simultaneous determination of five major biologically active ingredients in different parts of Gardenia jasminoides fruits by HPLC with diode-array detection. Chromatographia. 2006;64:713–7.
Zhao M, Tao J, Qian D, Liu P, Shang E, Jiang S, et al. Simultaneous determination of loganin, morroniside, catalpol and acteoside in normal and chronic kidney disease rat plasma by UPLC–MS for investigating the pharmacokinetics of Rehmannia glutinosa and Cornus officinalis Sieb drug pair extract. J Chromatogr B. 2016;1009–1010:122–9.
Li H, Li P, Ye W. Determination of five major iridoid glucosides in Flos Lonicerae by high-performance liquid chromatography coupled with evaporative light scattering detection. J Chromatogr A. 2003;1008:167–72.
Du W, Cai H, Wang M, Ding X, Yang H, Cai B. Simultaneous determination of six active components in crude and processed Fructus Corni by high performance liquid chromatography. J Pharm Biomed Anal. 2008;48:194–7.
Si W, Yang W, Guo D, Wu J, Zhang J, Qiu S, et al. Selective ion monitoring of quinochalcone C-glycoside markers for the simultaneous identification of Carthamus tinctorius L. in eleven Chinese patent medicines by UHPLC/QTOF MS. J Pharm Biomed Anal. 2016;117:510–21.
Yao C, Yang W, Wu W, Da J, Hou J, Zhang J, et al. Simultaneous quantitation of five Panax notoginseng saponins by multi heart-cutting two-dimensional liquid chromatography: method development and application to the quality control of eight Notoginseng containing Chinese patent medicines. J Chromatogr A. 2015;1402:71–81.
Zhao J, Ma S, Li S. Advanced strategies for quality control of Chinese medicines. J Pharm Biomed Anal. 2018;147:473–8.
Ji W, Zhang M, Wang D, Wang X, Liu J, Huang L. Superhydrophilic molecularly imprinted polymers based on a water-soluble functional monomer for the recognition of gastrodin in water media. J Chromatogr A. 2015;1425:88–96.
Kong XJ, Zheng C, Lan YH, Chi SS, Dong Q, Liu HL, et al. Synthesis of multirecognition magnetic molecularly imprinted polymer by atom transfer radical polymerization and its application in magnetic solid-phase extraction. Anal Bioanal Chem. 2018;410:247–57.
Gao SP, Zhang X, Zhang LS, Huang YP, Liu ZS. Molecularly imprinted polymer prepared with polyhedral oligomeric silsesquioxane through reversible addition–fragmentation chain transfer polymerization. Anal Bioanal Chem. 2017;409:3741–8.
Ji W, Sun R, Geng Y, Liu W, Wang X. Rapid, low temperature synthesis of molecularly imprinted covalent organic frameworks for the highly selective extraction of cyano pyrethroids from plant samples. Anal Chim Acta. 2018;1001:179–88.
Wang N, Wang YF, Omer AM, Ouyang X. Fabrication of novel surface-imprinted magnetic graphene oxide-grafted cellulose nanocrystals for selective extraction and fast adsorption of fluoroquinolones from water. Anal Bioanal Chem. 2017;409:6643–53.
Dima ŞO. Equilibrium and kinetic isotherms and parameters for molecularly imprinted with sclareol poly(acrylonitrile-co-acrylic acid) matrix. Polym Eng Sci. 2015;55:1152–62.
Umpleby RJ, Baxter SC, Chen Y, Shah RN, Shimizu KD. Characterization of molecularly imprinted polymers with the Langmuir-Freundlich isotherm. Anal Chem. 2001;73:4548–91.
Ji W, Wang T, Liu W, Liu F, Guo L, Geng Y, et al. Water-compatible micron-sized monodisperse molecularly imprinted beads for selective extraction of five iridoid glycosides from Cornus officinalis fructus. J Chromatogr A. 2017;1504:1–8.
Ji W, Zhang M, Liu W, Wang X, Duan W, Xie H, et al. Development of hydrophilic magnetic molecularly imprinted polymers by directly coating onto Fe3O4 with a water-miscible functional monomer and application in a solid-phase extraction procedure for iridoid glycosides. RSC Adv. 2016;6:50487–96.
Ji W, Zhang M, Gao Q, Cui L, Chen L, Wang X. Preparation of hydrophilic molecularly imprinted polymers via bulk polymerization combined with hydrolysis of ester groups for selective recognition of iridoid glycosides. Anal Bioanal Chem. 2016;408:5319–28.
Acknowledgments
Financial support from the National Natural Science Foundation of China (No. 81603286), the Key Science and Technology Program of Shandong (No. 2016GSF202033), the Natural Science Foundation of Shandong (ZR2016YL006), the Priority Research Program of the Shandong Academy of Sciences (Lanping Guo), and the Shandong Province Taishan Scholar Program (Lanping Guo) are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ji, W., Wang, R., Mu, Y. et al. Superhydrophilic molecularly imprinted polymers based on a single cross-linking monomer for the recognition of iridoid glycosides in Di-huang pills. Anal Bioanal Chem 410, 6539–6548 (2018). https://doi.org/10.1007/s00216-018-1257-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-018-1257-6