
Abstract
We describe a solid-phase extraction adsorbent based on molecularly imprinted polymers (MIPs), prepared with use of a mimic template. The MIPs were used for the selective extraction and determination of three chlorogenic acids as combined quality markers for Lonicera japonica and Lianhua qingwen granules. The morphologies and surface groups of the MIPs were assessed by scanning electron microscopy, Brunauer–Emmett–Teller surface area analysis, and Fourier transform infrared spectroscopy. The adsorption isotherms, kinetics, and selectivity of the MIPs were systematically compared with those of non-molecularly imprinted polymers. The MIPs showed high selectivity toward three structurally similar chlorogenic acids (chlorogenic acid, cryptochlorogenic acid, and neochlorogenic acid). A procedure using molecularly imprinted solid-phase extraction coupled with high-performance liquid chromatography was established for the determination of three chlorogenic acids from Lonicera japonica and Lianhua qingwen granules. The recoveries of the chlorogenic acids ranged from 93.1% to 101.4%. The limits of detection and limits of quantification for the three chlorogenic acids were 0.003 mg g−1 and 0.01 mg g−1, respectively. The newly developed method is thus a promising technique for the enrichment and determination of chlorogenic acids from herbal medicines.

Mimic molecularly imprinted polymers for the selective extraction of chlorogenic acids.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The increase in global demand for herbal medicines (HMs) means that quality control is becoming increasingly important in both clinical and business applications [1]. HMs, which contain unrefined mixtures of phytochemical compounds, are different from Western drugs, and the selection of appropriate compounds as quality markers presents a significant challenge [2]. Many bioactive components have been used as quality control markers for HMs [3, 4] but insufficient pharmacological and chemical evidence is usually available to justify the selection of a single molecule as a quality marker, resulting in poor biological relevance. The therapeutic benefits of HMs are widely acknowledged to result from the combined effects of multiple components on multiple targets [5]. Better quality control of HMs could, therefore, be achieved by use of combinations of compounds with congruent pharmacological activities.
Chlorogenic acid (CHA) and its structural analogues, cryptochlorogenic acid (CPCHA) and neochlorogenic acid (NCHA) (Fig. 1), have the congruent pharmacological activities 19.6967ptof scavenging free radicals, acting as antioxidants, and suppressing mutations [6]. Since chlorogenic acids (CGAs) are the main bioactive components of HMs such as Eucommia ulmodies, honeysuckle, and Lianhua qingwen granules, it is more appropriate to use a family of CGAs for the quality control of such HMs. However, the presence of related compounds, such as isochlorogenic acid A (ICHA-A) (Fig. 1), with similar chemical properties, means that the three CGAs of interest cannot be selectively extracted with organic solvents. The analysis of a family of CGAs in HMs is rendered more difficult by the fact that CPCHA and NCHA are often present in trace, or even ultratrace, amounts. Sample pretreatment steps are thus often required to increase the sensitivity and accuracy of methods used for the determination of these three CGAs.

The chemical structures of chlorogenic acids and the synthesis of the mimic template reduced chlorogenic acid
Molecularly imprinted solid-phase extraction (MISPE) is widely used for the selective recognition of target molecules [7], and solid-phase extraction (SPE) cartridges packed with molecularly imprinted polymers (MIPs) have been shown to have significantly increased selectivity [8,9,10,11,12,13,14]. MIPs can be prepared by copolymerization of cross-linkers and functional monomers in the presence of template molecules [15]. CHA-MIPs have been used for the enrichment and purification of CHA from samples of HMs, such as honeysuckle [16,17,18] and E. ulmodies [19,20,21,22]. Because of their good molecular recognition properties, MIPs provide both high selectivity and good recoveries for CHA. However, all previously reported CHA MIPs were synthesized with CHA as the template. The potential leakage of template molecules when MISPE is used, even after exhaustive washing steps, could seriously compromise the accuracy of the analytical method and cannot be ignored [23]. Template leakage could even render MISPE unsuitable for the simultaneous analysis of the whole family of CGAs. The best way to avoid template leakage is to use a structural analogue (a “mimic template”) of the target analyte as the template [24,25,26,27,28,29,30,31,32].
In the present work, we prepared MIPs using reduced CHA (R-CHA), which was synthesized by catalytic hydrogenation of CHA, as the mimic template. The influence of template leakage on quantitative analysis of CHA was eliminated by use of the mimic template. MIPs and non-molecularly imprinted polymers (NIPs) were characterized by systematic investigation of adsorption isotherms, kinetics, and selectivity. A new method, using MISPE coupled with high-performance liquid chromatography (HPLC), was developed to analyze three CGAs in the model traditional Chinese medicines Lonicera japonica Thunb. and Lianhua qingwen granules. Parameters such as linearity, limits of detection (LODs), limits of quantitation (LOQs), accuracy, and precision were used to validate the method.
Materials and methods
Materials
CHA, CPCHA, NCHA, and ICHA-A were purchased from Puxi Technology Co. (Beijing, China). Analytical grade CHA, which was used for the preparation of R-CHA, was obtained from Yuanye Biological Technology Co. (Shanghai, China). 4-Vinylpyridine (4-VP; functional monomer), divinylbenzene (DVB; cross-linker), and 2,2-azoisobutyronitrile (AIBN; initiator), and 10% Pd/C (hydrogenation catalyst) were purchased from J&K Scientific (Beijing, China). HPLC grade acetonitrile and methanol (MeOH) were purchased from Merck (Darmstadt, Germany). Analytical grade organic solvents were obtained from Sinopharm Chemical Reagent Co. (Shanghai, China).
Preparation of R-CHA
A suspension of CHA (3.54 g, 10 mmol) and 10% Pd/C (1.0 g) in MeOH (100 mL) was stirred at 20 °C under a H2 atmosphere for 4 h. After filtration through a pad of Celite, the filtrate was concentrated under reduced pressure. Flash column chromatography (silica gel, 20% MeOH–ethyl acetate) afforded R-CHA (3.38 g, 95%) as a white solid. 1H NMR (400 MHz, MeOD) δ (ppm) 6.57-6.54 (m, 2H), 6.42 (dd, 1H, J = 1.2 and 8.0 Hz), 5.15 (s, 1H), 4.07 (s, 1H), 3.49–3.39 (m, 1H), 2.68 (t, 2H, J = 7.6 Hz), 2.49 (t, 2H, J = 8.0 Hz), 2.05–1.99 (m, 3H), 1.90–1.81 (m, 1H). 13C NMR (100 MHz, MeOD) δ (ppm) 175.9, 172.2, 145.5, 143.9, 131.8, 119.2, 116.1, 115.9, 72.9, 71.6, 63.5, 56.5, 37.8, 36.2, 30.1, 19.0. Electrospray ionization mass spectrometry m/z = 355.1 [M – H+]. Anal calcd for C16H20O9: C, 53.93; H, 5.66. Found: C, 53.66; H, 5.81.
Calibration for potentiometric titration
The pH values of R-CHA solutions in MeOH–water (2:1, v/v) were determined with a PHS-3C pH meter (INESA, Shanghai, China), which had been standardized with aqueous buffer prepared from the standard sample. Subsequent calibration was conducted with the method described by Van Uitert and Haas [33], which is based on pH measurements of R-CHA in hydroalcoholic solution and in aqueous solution. The relation between the pH in MeOH–water (2:1, v/v) (pHw-ol) and the pH in pure water (pHw) was determined as follows:
Preparation of MIPs
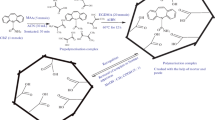
Preparation of the mimic imprinted polymers is shown schematically in Fig. 2. MIPs were synthesized by bulk polymerization with a noncovalent approach. The mimic template molecules (R-CHA, 1.0 mmol) and functional monomers (4-VP, 4.0 mmol) were mixed in MeOH–water (6 mL, 2:1, v/v) under magnetic stirring for 0.5 h to allow them to self-assemble by molecular interactions. After removal of the stirrer bar, cross-linker (DVB, 8.0 mmol) and initiator (AIBN, 0.05 g) were added to the solution of preassembled molecules. The mixture was thermopolymerized (55 °C) under a N2 atmosphere for 10 h. The rigid polymers obtained were crushed, ground, sieved (35–45 μm), sedimented in acetone, and dried under a vacuum. Because of the excellent solubility of R-CHA in water, the mimic template molecules were extracted with water–acetic acid (9:1, v/v), until no R-CHA was detected by HPLC in the extract. NIPs were simultaneously synthesized by the same method in the absence of the mimic template molecules. The composition of the prepolymerization mixtures is listed in Table S1.

The principle of the mimic imprinted polymers. AIBN 2,2-azoisobutyronitrile, DVB divinylbenzene, MIPs molecularly imprinted polymers, R-CHA reduced chlorogenic acid, 4-VP 4-vinylpyridine
Characterization
The functional groups of the synthesized particles were investigated by Fourier transform infrared spectroscopy (FT-IR; Nicolet iS10, Thermo Fisher Scientific, Waltham, MA, USA). Morphologies were determined by scanning electron microscopy (SWPRATM55, Carl Zeiss, Aalen, Germany). N2 adsorption isotherms were determined with an ASAP 2020 accelerated surface area and porosimetry system (Micromeritics Instrument, Norcross, GA, USA). Surface area and total pore volume were determined by the Brunauer–Emmett–Teller method [34].
Chromatographic conditions
A 1120 liquid chromatography system (Agilent Technologies, Santa Clara, CA, USA), equipped with an ultraviolet detector, was used to analyze the samples. A ZORBAX octadecylsilane analytical column (5 μm, 4.6 mm × 250 mm, Agilent Technologies, Palo Alto, USA) was used for the separation of analytes. The mobile phase was 0.2% formic acid in water (solvent A) and acetonitrile (solvent B). The gradient elution program was as follows: 0−10 min, 92% to 90% solvent A; 10−20 min, 90% to 85% solvent A; 20−30 min, 85% solvent A; 30−40 min, 85% to 75% solvent A; 40−50 min, 75% to 70% solvent A; 50−51 min, 70% to 92% solvent A. The flow rate was maintained at 1.0 mL min−1, and the column temperature was 30 °C. The injection volume and detection wavelength were 10 μL and 280 nm, respectively.
Binding characteristics
The adsorption isotherms were investigated by our mixing MIPs or NIPs (10 mg) with CHA solutions (5 mL) of different concentration (10−500 mg L−1). The mixtures were shaken for 100 min at 22 °C, and the sorbents were then collected by centrifugation. The quantity of CHA in the supernatants was analyzed by HPLC. The equilibrium adsorption capacity (Q e; mg g−1) of the MIPs was calculated with the following equation:
where C i and C e (mg L−1) are the initial and equilibrium concentration of CHA, respectively, m (mg) is the weight of sorbents, and v (mL) is the volume of the solution.
The adsorption isotherms were described by the Langmuir binding isotherm model as follows [35, 36]:
where K L and Q m are the Langmuir equilibrium constant and theoretical maximum adsorption capacity, respectively.
Adsorption kinetics were investigated by our mixing MIPs or NIPs (10 mg) with CHA solution (5 mL, 300 mg L−1) and shaking the mixture for different times (0−150 min). The binding quantity of CHA (Q t, mg g−1) was calculated with the following equation [37]:
where C t (mg L−1) represents the CHA concentration at time t.
ICHA-A was chosen as the reference compound to evaluate the selectivity of the MIPs toward the three CGAs. The reference compound is a phenylpropanoid that is widely present in L. japonica. The working solutions of ICHA-A and the three CGAs were prepared at an initial concentration of 300 mg L−1. The sorbent (10 mg) was shaken with working solution (5 mL) for 100 min at 22 °C. After removal of the sorbent, the quantity of analyte in the supernatant was analyzed by HPLC. The imprinting factor (IF) and selectivity coefficient (SC) were used to assess the selectivity of the MIPs:
Extraction of three CGAs from HM samples using MISPE
L. japonica or Lianhua qingwen granules (0.1 g) were refluxed with the deionized water (50 mL) for 1 h. After filtration through a 0.45-μm polytetrafluoroethylene membrane, the supernatant was accurately diluted with deionized water to 50 mL for further experiments.
MIPs (500 mg) were packed into empty SPE columns (5 mL). After wetting with deionized water (10 mL), extraction solution (50 mL) was passed through the MISPE columns (flow rate 10.0 mL min−1). The columns were then washed and the contents eluted with MeOH–acetic acid (9:1, v/v, 3 mL) and water–acetic acid (6:4, v/v, 3 mL). Finally, the amount of the three CGAs was analyzed by HPLC.
Results and discussion
Choice of template and functional monomer
The structural difference between CHA and R-CHA is shown in Fig. 1. R-CHA has the same functional groups as CHA but lacks the C=C double bond, suggesting that R-CHA would be a more suitable template. In support of this, the recovery of the template from different MIPs after free-radical bulk polymerization was significantly higher when R-CHA, rather than CHA, was used as the template (93.5% vs 69.1%, respectively). The functional groups of R-CHA MIPs (after and before removal of R-CHA), CHA MIPs (after and before removal of CHA), and NIPs were investigated by FT-IR spectroscopy (see Fig. S1). The peak around 1731 cm−1 indicated the presence of a C=O group, demonstrating the retention of CHA in CHA MIPs after removal of CHA. In sharp contrast, the FT-IR spectrum of R-CHA MIPs after removal of R-CHA was similar to that of NIPs, and showed no obvious peaks around 1731 cm−1. These results, taken together, demonstrate that R-CHA is more suitable as a template molecule and can avoid interference caused by template leakage during the chromatographic quantification of CHA.
Results from the binding experiments (Table S1) showed that MIPs synthesized with 4-VP had higher IFs than those synthesized with methacrylic acid (acidic monomer), likely because of favorable interactions between the basic 4-VP monomer and the acidic template. In addition to electrostatic interactions, π–π interactions between 4-VP and R-CHA are expected to contribute to the greater ability of MIPs to selectively recognize the acidic compound CHA, compared with those synthesized with methacrylic acid. These results are in agreement with previously reported results for CHA [19] and its analogues chicoric acid [38] and caffeic acid [39]. Therefore, 4-VP was chosen as the functional monomer to prepare CHA MIPs.
The amount of the functional monomer and the strength of its interaction with the template are two important factors in providing MIPs with effective recognition properties. Optimization of prepolymerization complexes is a crucial step in the preparation of MIPs [36]. Molar ratios of R-CHA to 4-VP were defined by pH titration studies so as to evaluate the effect of increasing the quantity of 4-VP on the interaction between the mimic template and the functional monomer. The pH values of R-CHA solutions (100 mL, 5 mmol L−1 in MeOH–water) were measured in the presence of 4-VP (0–10.0 mmol), with use of a PHS-3C pH meter. The pH of the solution increased from 3.1 to 6.9 as the concentration of 4-VP was increased from 0 to 1.9 mmol (Fig. S2), but further increases in the concentration of 4-VP caused no further change in pH. A molar ratio of 1:4 for template to functional monomer was thus used to prepare MIPs.
Choice of cross-linker
The cross-linker must provide an appropriate degree of flexibility for the MIPs, to allow both rapid achievement of binding equilibrium and good accessibility to the active sites. Polymers C in Table S1, which were synthesized with DVB as the cross-linker (template to functional monomer to cross-linker ratio 1:4:8), showed good IFs and rebinding results. DVB can form π–π interactions with R-CHA. On the basis studies of the described so far, the best MIPs were composed of R-CHA as the mimic template, 4-VP as the functional monomer, and DVB as the cross-linker, in a molar ratio of 1:4:8 (Table S1, MIPs C). MIPs C were used to further investigate binding properties in subsequent experiments.
Characterization of morphology
The morphologies of MIPs and the corresponding NIPs were determined by scanning electron microscopy (Fig. S3). Because bulk polymerization was used, the morphologies of the MIPs and NIPs were similar and the particles did not have a well-defined shape [40]. Particle sizes were in the range 30–50 μm, which is suitable for packing SPE columns. The MIPs had a more porous and rougher structure, which would contribute to the adsorption of compounds. The surface area and pore volume of the MIPs and NIPs were calculated by the Brunauer–Emmett–Teller method. Values for the MIPs (282.2 m2 g−1 and 0.453 m3 g−1, respectively) were significantly higher than those for the NIPs (132.2 m2 g−1 and 0.191 m3 g−1, respectively). The larger surface area and pore volume increase the adsorption capacity of MIPs, indicating that MIPs are very suitable for use as an adsorbent material.
Binding characteristics
Adsorption isotherms
Static adsorption experiments, with CHA as the model molecule, were conducted to evaluate the imprinting efficiency of the MIPs. As shown in Fig. 3, the amount of CHA adsorbed increased with increasing concentration of the working standard solution (10−500 mg L−1). The amounts of CHA adsorbed by the NIPs were much lower than those adsorbed by the MIPs, indicating that imprinted sites had been formed in the MIPs. The adsorption data were processed with the Langmuir equation (Eq. 3), whereby the ratio of free to bound CHA (C e/Q e) was plotted against the amount of free CHA (C e). The linear regression equations for MIPs and NIPs were y = 0.0038x + 0.5162 (R 2 = 0.905) and y = 0.0045x + 3.5984 (R 2 = 0.973), respectively (Fig. 4). The theoretical maximum adsorption amounts (Q m) were 222 mg g−1 and 263 mg g−1 for NIPs and MIPs, respectively, indicating that adsorption of CHA by MIPs is more favorable.

Adsorption isotherm curves for chlorogenic acid on molecularly imprinted polymers (MIPs) and non-molecularly imprinted polymers (NIPs)

Langmuir plots of molecularly imprinted polymers (MIPs) and non-molecularly imprinted polymers (NIPs).
Adsorption kinetics
Adsorption kinetics of MIPs and NIPs were studied over different periods, with a constant concentration of CHA (300 mg L−1). The adsorption capacity of MIPs increased rapidly during the first 12 min and then approached equilibrium (Fig. 5). In contrast, the adsorption capacity of the NIPs was lower and approached equilibrium only after 90 min. These results suggest that the imprinted cavities of the MIPs led to both faster adsorption and higher binding affinity for the analyte.

Kinetic adsorption curves for chlorogenic acid on molecularly imprinted polymers (MIPs) and non-molecularly imprinted polymers (NIPs)
Selectivity of adsorption
IFs and SCs were used to indicate the selective recognition by MIPs and NIPs of the three CGAs and the reference compound (ICHA-A). The results are summarized in Table 1. The IFs for CHA, CPCHA, and NCHA were 2.24, 2.42, and 2.90, respectively, suggesting that the adsorption capacity of MIPs for CGAs was much greater than that of NIPs. The SCs for CHA, CPCHA, and NCHA were 2.41, 2.60, and 3.12, respectively, indicating that MIPs showed excellent selectivity for CGAs compared with the competitor molecule, ICHA-A. Notably, Q e of ICHA-A on MIPs was similar to that on NIPs. This is because the structure of the mimic template is very different from that of ICHA-A, which means that the competitor molecule cannot be captured by the imprinted sites. The high adsorption capacity and excellent selectivity of MIPs toward CGAs can be attributed to the specific imprinting sites. MIPs could, therefore, be used for the selective enrichment of CGAs.
Optimization of the MISPE procedure
To eliminate interference and achieve satisfactory recoveries of the three CGAs, several parameters of the MISPE method were investigated. These included flow rate and types and volumes of loading, washing, and elution solvents.
Effect of sample loading solvents
Water (50 mL) spiked with different amounts of CGA standards (0.3, 0.6, 0.9, 1.2, and 1.5 mg for each CGA) was percolated through the cartridges to determine the mass capacity of the MISPE columns. Over the tested range, recoveries of more than 95% fort the CGAs were achieved. Recoveries of more than 85% were still achieved when the total loading amount was as high as 9.0 mg (3.0 mg for each CGA), indicating that the MISPE columns have good mass capacity for CGAs. Taking into consideration the concentration of CGAs in L. japonica and Lianhua qingwen granules, we used water (50 mL) spiked with 1.5 mg of each CGA (total 4.5 mg) to optimize other parameters.
Effect of flow rate
The effect of flow rates in the range 1.0–13.0 mL min−1 on the recoveries of CGAs was investigated. The recoveries of all three CGAs were more than 95% as the flow rate was increased from 1.0 to 10.0 mL min−1, but the recoveries decreased as the flow rate was increased from 10.0 to 13.0 mL min−1. A flow rate of 10.0 mL min−1 was thus selected for further studies.
Effect of washing solvent
The washing procedure plays an important role in reducing interference from impurities in the matrix, as well as in maximizing the specific interactions between MISPE columns and the analytes. After spiked water (50 mL) had been loaded onto the columns, water, acetonitrile, MeOH, MeOH–acetic acid (9:1, v/v), and water–acetic acid (9:1, v/v) (3 mL each) were evaluated as washing solvents. Water, acetonitrile, and MeOH provided high recoveries for both MISPE and non-molecularly imprinted SPE (NISPE) columns (Fig. S4), indicating that they were not appropriate as washing solvents. After washing with MeOH–acetic acid (9:1, v/v), the recoveries of all three CGAs for the MISPE columns were more than 90%, but the recoveries for the NISPE columns decreased to less than 70%, indicating that the selective interactions between CGAs and MISPE columns had been retained. CGAs could be sufficiently eluted from NISPE columns by water–acetic acid (9:1, v/v), whereas most of the CGAs were also removed from MISPE columns simultaneously. To reduce solvent consumption, different volumes of MeOH–acetic acid (9:1, v/v, 1–5 mL) were evaluated. As shown in Fig. S5, purification and recoveries were satisfactory with a volume of 3.0 mL, but recoveries of CGAs from MISPE columns significantly decreased with larger volumes. Therefore, 3 mL of MeOH–acetic acid (9:1, v/v) was used as the washing solution.
Effect of elution solvents
The elution solvent should sufficiently remove analytes from MISPE columns. Water (4 mL) spiked with different volumes of acetic acid was investigated as the elution solvent, and 6:4 (v/v) water–acetic acid was found to give the highest recoveries (Fig. 6). Different volumes (1–5 mL) of water–acetic acid (6:4, v/v) were then evaluated. The recoveries of CGAs increased as the volume of eluent increased from 1 to 3 mL, and were then almost constant. Thus, 3 mL of water–acetic acid (6:4, v/v) was used as the elution solvent.

Effect of different elution solvents on recoveries of three chlorogenic acids for molecularly imprinted solid-phase extraction and non-molecularly imprinted solid-phase extraction. CHA chlorogenic acid, CPCHA cryptochlorogenic acid, NCHA neochlorogenic acid
Validation of the MISPE–HPLC method
To validate the optimized MISPE method for extracting CGAs from real HM samples, the linearity, accuracy, precision, LODs, LOQs, and recoveries for the method were systematically established. Deionized water, which had been demonstrated to contain no CGAs, was spiked with the three CGAs to obtain real samples. Good linearity, with R 2 > 0.999, was obtained for the three CGAs over the concentration range 0.01–60 mg g−1 (Table 2). LODs and LOQs were calculated at three and ten times the signal-to-noise ratio. The LODs for CHA, CPCHA, and NCHA were 0.003, 0.003, and 0.003 mg g−1, respectively. The LOQs for CHA, CPCHA, and NCHA were 0.01, 0.01 and 0.01 mg g−1, respectively. The intraday and interday precision (relative standard deviations) were 3.5–4.3% and 5.9–7.1%, respectively, when evaluated with the method on the same day (n = 6) and on four consecutive days.
To evaluate the applicability of MISPE for recognizing CGAs in more complex HM matrixes, Salvia miltiorrhiza samples, which have been verified to contain no CGAs, were spiked with the CGAs at three concentrations (0.05, 1.0, and 20 mg g−1) to validate the recoveries. The recoveries of the three CGAs ranged from 93.1% to 101.4% with relative standard deviations less than 5.9% (Table 3), indicating that the newly developed MISPE method could be used for accurate and sensitive measurements.
Analysis of HM samples
The concentrations of the three CGAs in commercial L. japonica and Lianhua qingwen granules were analyzed by the newly developed MISPE–HPLC method. The concentrations of CHA in all samples of L japonica were greater than 15.0 mg g−1 (Table 3), in accordance with the quality standard established by the Chinese Pharmacopoeia [41]. Typical chromatograms for L. japonica and Lianhua qingwen granules are shown in Fig. 7. Only the CHA chromatographic peak was visible after 50 mL of extract had been concentrated to 3 mL (Fig. 7, chromatograms a), mainly because matrix interference was maximized by concentration of the sample and the impurity peaks became more obvious. Pretreatment by concentration of the sample was thus not suitable for the analysis of CGAs in HM samples. The chromatographic peaks obtained from the MISPE columns were markedly clearer than those obtained with three other adsorbents, C18, hydrophilic–lipophilic balance, and NISPE adsorbents (Fig. 7, chromatograms b–e). The peaks seen with MISPE were also noticeably bigger, implying higher recovery with MISPE. All of the results demonstrate that the MISPE method developed in this work has the ability to recognize a family of CGAs by specific imprinting. The MISPE–HPLC method could thus be used to analyze a family of CGAs as combined quality markers for the quality control of HMs.

Typical high-performance liquid chromatography chromatograms for the determination of chlorogenic acids in herbal medicine samples: concentrated extract before percolation through a solid-phase extraction column (a); eluting solutions from a C18 column (b); eluting solutions from a hydrophilic–lipophilic balance column (c); eluting solutions from a non-molecularly imprinted solid-phase extraction column; eluting solutions from a molecularly imprinted solid-phase extraction column (e). Peaks: 1 neochlorogenic acid, 2 chlorogenic acid, 3 cryptochlorogenic acid
Conclusions
In summary, an MISPE method has been developed for the selective extraction of CGAs (CHA, CPCHA, and NCHA) from L. japonica and Lianhua qingwen granules. MIPs, which were prepared with R-CHA as the mimic template, showed fast binding kinetics and a large adsorption capacity for CHA. The MISPE–HPLC method showed good recoveries, together with high repeatability and selectivity, in the determination of three CGAs in L. japonica and Lianhua qingwen granules. These excellent properties mean that MISPE is a promising candidate for other applications, especially for the analysis of CGAs in clinical samples based on SPE.
References
Li SP, Zhao J, Yang B. Strategies for quality control of Chinese medicines. J Pharm Biomed Anal. 2011;55:802–9.
Xie PS, Leung AY. Understanding the traditional aspect of Chinese medicine in order to achieve meaningful quality control of Chinese materia medica. J Chromatogr A. 2009;1216:1933–40.
Yang Y, Wang HJ, Yang J, Brantner AH, Lower-Nedza AD, Si N, et al. Chemical profiling and quantification of Chinese medicinal formula Huang-Lian-Jie-Du decoction, a systematic quality control strategy using ultra high performance liquid chromatography combined with hybrid quadrupole-orbitrap and triple quadrupole mass spectrometers. J Chromatogr A. 2013;1321:88–9.
Qi LW, Cao J, Li P, Wang YX. Rapid and sensitive quantitation of major constituents in Danggui Buxue Tang by ultra-fast HPLC–TOF/MS. J Pharm Biomed Anal. 2009;49:502–7.
Shi Z, Song D, Li R, Yang H, Qi L, Xin G, et al. Identification of effective combinatorial markers for quality standardization of herbal medicines. J Chromatogr A. 2014;1345:78–85.
Nakamura T, Nakazawa Y, Onizuka S. Antimutagenicity of Tochu tea (an aqueous extract of Eucommia ulmoides leaves): 1. The clastogen-suppressing effects of Tochu tea in CHO cells and mice. Mutat Res. 1997;338:7–20.
Chen L, Wang X, Lu W, Wu X, Li J. Molecular imprinting: perspectives and applications. Chem Soc Rev. 2016;45:2137–211.
Miranda LFC, Domingues DS, Queiroz MEC. Selective solid-phase extraction using molecularly imprinted polymers for analysis of venlafaxine, O-desmethylvenlafaxine, and N-desmethylvenlafaxine in plasma samples by liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2016;1458:46–53.
Cao J, Yan H, Shen S, Bai L, Liu H, Qiao F. Hydrophilic molecularly imprinted melamine-urea-formaldehyde monolithic resin prepared in water for selective recognition of plant growth regulators. Anal Chim Acta. 2016;943:136–45.
Ji W, Zhang M, Wang T, Wang X, Zheng Z, Gong J. Molecularly imprinted solid-phase extraction method based on SH-Au modified silica gel for the detection of three Sudan dyes in chili powder samples. Talanta. 2017;165:18–26.
Hernández-Mesa M, Cruces-Blanco C, García-Campaña AM. Capillary electrophoresis-tandem mass spectrometry combined with molecularly imprinted solid phase extraction as useful tool for the monitoring of 5-nitroimidazoles and their metabolites in urine samples. Talanta. 2017;163:111–20.
Zhao T, Guan X, Tang W, Ma Y, Zhang H. Preparation of temperature sensitive molecularly imprinted polymer for solid-phase microextraction coatings on stainless steel fiber to measure ofloxacin. Anal Chim Acta. 2015;853:668–75.
Urraca JL, Chamorro-Mendiluce R, Orellana G, Moreno-Bondi MC. Molecularly imprinted polymer beads for clean-up and preconcentration of β-lactamase-resistant penicillins in milk. Anal Bioanal Chem. 2016;408:1843–54.
Pichon V, Combès A. Selective tools for the solid-phase extraction of ochratoxin A from various complex samples: immunosorbents, oligosorbents, and molecularly imprinted polymers. Anal Bioanal Chem. 2016;408:6983–99.
Hu Y, Pan J, Zhang K, Lian H, Li G. Novel applications of molecularly-imprinted polymers in sample preparation. Trends Anal Chem. 2013;43:37–52.
Gu X, Xu R, Yuan G, Lu H, Gu B, Xie H. Preparation of chlorogenic acid surface-imprinted magnetic nanoparticles and their usage in separation of traditional Chinese medicine. Anal Chim Acta. 2010;675:64–70.
Golsefidi MA, Es’haghi Z, Sarafraz-Yazdi A. Design, synthesis and evaluation of a molecularly imprinted polymer for hollow fiber-solid phase microextraction of chlorogenic acid in medicinal plants. J Chromatogr A. 2012;1229:24–9.
Liu Q, Zhao Y, Pan J, Bruggen B, Shen J. A novel chitosan base molecularly imprinted membrane for selective separation of chlorogenic acid. Sep Purif Technol. 2016;164:70–80.
Li H, Li G, Li Z, Lu C, Li Y, Tan X. Surface imprinting on nano-TiO2 as sacrificial material for the preparation of hollow chlorogenic acid imprinted polymer and its recognition behavior. Appl Surf Sci. 2013;264:644–52.
Li H, Liu Y, Zhang Z, Liao H, Nie L, Yao S. Separation and purification of chlorogenic acid by molecularly imprinted polymer monolithic stationary phase. J Chromatogr A. 2005;1098:66–74.
Miura C, Matsunaga H, Haginaka J. Molecularly imprinted polymer for caffeic acid by precipitation polymerization and its application to extraction of caffeic acid and chlorogenic acid from Eucommia ulmodies leaves. J Pharm Biomed Anal. 2016;127:32–8.
Miura C, Li H, Matsunaga H, Haginaka J. Molecularly imprinted polymer for chlorogenic acid by modified precipitation polymerization and its application to extraction of chlorogenic acid from Eucommia ulmodies leaves. J Pharm Biomed Anal. 2015;114:139–44.
Sun X, Wang J, Li Y, Jin J, Yang J, Li F, et al. Highly class-selective solid-phase extraction of bisphenols in milk, sediment and human urine samples using well-designed dummy molecularly imprinted polymers. J Chromatogr A. 2014;1360:9–16.
You QP, Peng MJ, Zhang YP, Guo JF, Shi SY. Preparation of magnetic dummy molecularly imprinted polymers for selective extraction and analysis of salicylic acid in Actinidia chinensis. Anal Bioanal Chem. 2014;406:831–9.
Zhu F, Wang J, Zhu L, Tan L, Feng G, Liu S, et al. Preparation of molecularly imprinted polymers using theanine as dummy template and its application as SPE sorbent for the determination of eighteen amino acids in tobacco. Talanta. 2016;150:388–98.
Liu R, Feng F, Chen G, Liu Z, Xu Z. Barbell-shaped stir bar sorptive extraction using dummy template molecularly imprinted polymer coatings for analysis of bisphenol A in water. Anal Bioanal Chem. 2016;408:5329–35.
Wang XH, Zhang J, Peng C, Dong Q, Huang YP, Liu ZS. Comparison of multi-recognition molecularly imprinted polymers for recognition of melamine, cyromazine, triamterene, and trimethoprim. Anal Bioanal Chem. 2015;407:7145–55.
Mei XQ, He XP, Wang JT. Molecularly imprinted polymer as efficient sorbent of solid-phase extraction for determination of gonyautoxin 1,4 in seawater followed by high-performance liquid chromatography-fluorescence detection. Anal Bioanal Chem. 2016;408:5737–43.
Lian Z, Li HB, Wang J. Experimental and computational studies on molecularly imprinted solid-phase extraction for gonyautoxins 2,3 from dinoflagellate Alexandrium minutum. Anal Bioanal Chem. 2016;408:5527–35.
Wulandari M, Urraca JL, Descalzo AB, Amran MB, Moreno-Bondi MC. Molecularly imprinted polymers for cleanup and selective extraction of curcuminoids in medicinal herbal extracts. Anal Bioanal Chem. 2015;407:803–12.
Wang M, Chang X, Wu X, Yan H, Qiao F. Water-compatible dummy molecularly imprinted resin prepared in aqueous solution for green miniaturized solid-phase extraction of plant growth regulators. J Chromatogr A. 2016;1458:9–17.
Lenain P, Mavungu JDD, Dubruel P, Robbens J, Saeger SD. Development of suspension polymerized molecularly imprinted beads with metergoline as template and application in a solid-phase extraction procedure toward ergot alkaloids. Anal Chem. 2012;84:10411–8.
Van Uitert LG, Haas CG. Studies on coördination compounds. I. A method for determining thermodynamic equilibrium constants in mixed solvents. J Am Chem Soc. 1953;75:451–5.
Wang Y, Wang Y, Ouyang X, Yang L. Surface-imprinted magnetic carboxylated cellulose nanocrystals for the highly selective extraction of three fluoroquinolones from egg samples. ACS Appl Mater Interfaces. 2017;9:1759–69.
Ji W, Sun R, Duan W, Wang X, Wang T, Mu Y, et al. Selective solid phase extraction of chloroacetamide herbicides from environmental water samples by amphiphilic magnetic molecularly imprinted polymers. Talanta. 2017;170:111–8.
Ji W, Wang T, Liu W, Liu F, Guo L, Geng Y, et al. Water-compatible micron-sized monodisperse molecularly imprinted beads for selective extraction of five iridoid glycosides from Cornus officinalis fructus. J Chromatogr A. 2017;1504:1–8.
Xu X, Duhoranimana E, Zhang X. Preparation and characterization of magnetic molecularly imprinted polymers for the extraction of hexamethylenetetramine in milk samples. Talanta. 2017;163:31–8.
Saad EM, Madbouly A, Ayoub N, Nashar RME. Preparation and application of molecularly imprinted polymer for isolation of chicoric acid from Chicorium intybus L. medicinal plant. Anal Chim Acta. 2015;877:80–9.
Li N, Ng TB, Wong JH, Qiao JX, Zhang YN, Zhou R, et al. Separation and purification of the antioxidant compounds, caffeic acid phenethyl ester and caffeic acid from mushrooms by molecularly imprinted polymer. Food Chem. 2013;139:1161–7.
Pardo A, Mespouille L, Dubois P, Blankert B, Duez P. Molecularly imprinted polymers : compromise between flexibility and rigidity for improving capture of template analogues. Chem Eur J. 2014;20:3500–9.
Chinese Pharmacopoeia Commission. Chinese pharmacopoeia. Vol. I. People’s Medical Publishing House: Beijing, 2010; p 205−206.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (81473298, 81603286), the Key Science and Technology Program of Shandong (2014GSF119031), the Natural Science Foundation of Shandong (ZR2016YL006), the Shandong Province Taishan Scholar Program (Lanping Guo), and the Priority Research Program of the Shandong Academy of Sciences (Lanping Guo).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(PDF 517 kb)
Rights and permissions
About this article

Cite this article
Ji, W., Zhang, M., Yan, H. et al. Selective extraction and determination of chlorogenic acids as combined quality markers in herbal medicines using molecularly imprinted polymers based on a mimic template. Anal Bioanal Chem 409, 7087–7096 (2017). https://doi.org/10.1007/s00216-017-0667-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0667-1