
Abstract
In this article, some recent trends and developments in ambient desorption/ionization mass spectrometry (ADI-MS) are reviewed, with a special focus on quantitative analyses with direct, open-air sampling. Accurate quantification with ADI-MS is still not routinely performed, but this aspect is considered of utmost importance for the advancement of the field. In fact, several research groups are devoted to the development of novel and optimized ADI-MS approaches. Some key trends include novel sample introduction strategies for improved reproducibility, tailored sample preparation protocols for removing the matrix and matrix effects, and multimode ionization sources. In addition, there is significant interest in quantitative mass spectrometry imaging.

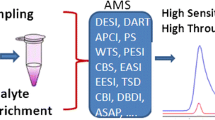
Conceptual diagram of the ambient desorption/ionization mass spectrometry approach with different desorption/ionization probes
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ambient desorption/ionization mass spectrometry (ADI-MS), also referred to as ‘ambient mass spectrometry,’ involves the direct sampling and ionization of analytes from samples under ambient conditions and requires minimal or no sample pretreatment. While the ionization process in mass-spectrometric analyses was traditionally performed in vacuo inside the mass spectrometer, the advent of differentially pumped interfaces and atmospheric pressure ionization (API) methods (e.g., electrospray ionization (ESI) [1], atmospheric pressure chemical ionization (APCI) [2], and atmospheric pressure photoionization (APPI) [3, 4]) allowed the ionization step to be performed at atmospheric pressure, greatly simplifying sample introduction. In late 2004, the introduction of desorption electrospray ionization (DESI) [5] by Cooks and coworkers, followed by that of direct analysis in real time (DART) [6] by Cody et al. in early 2005, further simplified ionization for MS by moving into the open-air environment, where the samples are present in native forms. Since the introduction of these methods, there has been a rapid boom in the development of these types of ionization techniques such that there are now more than 40 different ambient ionization techniques, which have been compiled in a recent review article [7].
Thirteen years since the first description of DESI and DART, a multitude of papers have been published on ADI-MS methods and applications spanning a wide range of scientific disciplines. In fact, as of December 2017, more than 1000 manuscripts involving ADI-MS approaches appear on the ISI Web of Knowledge. However, the vast majority of papers utilize ADI-MS methods to obtain qualitative information, while only a small selection of papers detail quantitative aspects of ADI-MS. Is the lack of quantitative analyses due to inherent limitations of ADI-MS techniques? If so, can these pitfalls be addressed or circumvented? To shed some light on these questions, this article highlights trends and developments in ADI-MS with a special focus on quantification and emerging applications. Progress in the field of plasma-based and non-plasma-based ADI sources is reviewed based on research papers published from 2014 and 2011, respectively, to early 2017 to avoid overlap with earlier reviews [8,9,10].
Concept of ambient desorption/ionization mass spectrometry
In order to judge the strengths and weaknesses of ADI-MS with respect to quantitative capabilities, it is necessary to give a brief introduction to the technique. The compelling feature of ADI-MS is that it allows mass spectrometric analysis without sample preparation or sample pretreatment by utilizing the mass spectrometer as the analyte separator as well as the detector. Furthermore, the term ‘ambient’ not only refers to the operation of the ionization source at atmospheric pressure (as is the case with APCI, APPI, and ESI), but also the concept of a freely accessible open space, i.e., a large sampling area, in front of the atmospheric pressure interface of the instrument (see, for example, [11]).
The primary purpose of ADI-MS is to enable the analysis of samples in their native state without sample preparation. This approach enables rapid screening with analysis times of much less than one minute per sample, which is significantly faster than chromatography-based MS techniques (e.g., gas chromatography, GC-MS, or liquid chromatography, LC-MS), and offers higher throughput than flow-injection analysis–mass spectrometry (FIA-MS) methods [12].
In a traditional ADI-MS experiment, analytes are desorbed from a solid or liquid sample in the open air with the effluent from a specially designed desorption/ionization source (cf. Fig. 1). The gas-phase analytes become charged through ionization processes common to established ionization sources, and are pulled into the reduced-pressure environment of the mass spectrometer, where the analyte ions are separated based on their mass-to-charge ratio (m/z) and detected. The desorption/ionization source is therefore the most important part of this setup as it is responsible for many roles, including sample collection, sample preparation (e.g., dissolution, digestion, solid/liquid extraction), analyte separation/selection (e.g., chromatographic or similar methods), analyte vaporization, ionization, and transport into the mass spectrometer (Fig. 1).

Conceptual diagram of the ambient desorption/ionization mass spectrometry approach with different desorption/ionization probes. Reagent ions from the ion-generation region are carried to the untreated sample with a sweep gas, aerosol, and/or liquid. This desorption/ionization beam releases analytes into the gas phase and subsequently ionizes them. Ions are drawn into the mass spectrometer inlet, separated by m/z, and detected. ASAP atmospheric solids analysis probe, DART direct analysis in real time, FAPA flowing atmospheric pressure afterglow, LTP low-temperature plasma, DBDI dielectric barrier desorption ionization, PADI plasma-assisted desorption ionization, DESI desorption electrospray ionization, LAESI laser ablation electrospray ionization, LMJ-SSP liquid microjunction surface-sampling probe
Analyte separation is mainly based on the mass analyzer and, to a lesser extent, the desorption/ionization source. Therefore, high-resolution MS and/or MS/MS are used to increase selectivity. MS/MS techniques work best with intact molecules and little fragmentation and, thus, soft ionization is required. Fortunately, this potential issue is usually mitigated by the significant amount of collisional cooling that occurs at atmospheric pressure after the ionization step. Therefore, mass spectra generated by ADI-MS should be simple and easy to interpret, but that is not always the case in practice. Usually, screening ADI-MS experiments are performed as qualitative analyses of the surface of solid samples or in solutions with low matrix content. However, quantitative analysis of complex samples by ADI-MS is quite challenging due to the aforementioned analytical load placed on the desorption/ionization source. To make ADI-MS techniques more quantitative and analytically useful, the following aspects must be carefully and fully addressed:
-
Sample introduction
-
Reproducibility (in each of the sampling/sample introduction, desorption, ionization, and mass-transport steps)
-
Sensitivity
-
Selectivity (in the desorption and/or ionization steps)
-
Matrix effects
-
Linearity, precision, accuracy
-
Method validation
Not all aspects can be addressed here due to page limitations, but they are certainly important for the advancement of the field. The key challenges associated with different aspects of quantitative ADI-MS are outlined in Table 1 and will be discussed in the following sections.
Sample introduction
Sample introduction and the subsequent desorption/ionization processes have a great influence on the quantitative capabilities of ADI-MS techniques. For example, changes in the type of pseudomolecular ions formed (e.g., sodiated vs. protonated) caused by variations in cations from the source or sample may affect reproducibility [46]. Of greater concern is poor reproducibility in analyses caused by sample heterogeneity and ion-collection location. Sample-surface properties including hardness, shape, roughness, etc. also greatly affect the analyte signal [47]. In the case of plasma-based sources, the distribution of reagent ions, ionization chemistries, and temperature vary significantly in the region between the ionization sources and the inlet of the mass spectrometer [26, 48,49,50]. As such, it is important to have reproducible sample introduction to ensure reproducible desorption and consistent analyte-ion formation [26, 37, 51, 52]. Additionally, the sampling geometry has a strong influence on signal levels and detectability, particularly for DESI. The initial designs of DESI sources were highly flexible with more than ten adjustable parameters. However, it was shown early on that analyte signal was heavily impacted by each of these variables, which include various geometrical settings and distance relationships between the spray tip, sample surface, and mass spectrometer orifice. Consequently, geometry-independent DESI [13] was introduced, which significantly improved reproducibility as well as the simplicity of operation.
In another attempt, transmission-mode methods were developed, where samples are placed on a transmissive mesh and the source effluent passes through this mesh. In transmission-mode DESI [14], for example, the emitter is oriented perpendicular to a sample-containing mesh, while the electrospray droplets dissolve, desorb, and ionize analytes from the mesh as they pass through. This configuration works well for liquid samples in particular, and needs less optimization of angles and distances than standard DESI [11]. The reproducible signals offered by transmission-mode sample introduction coupled with DART ionization were successful enough that it led to a commercial product in 2011 from IonSense, Inc., the DART ID-CUBE [15]. For the DART ID-CUBE, samples are placed on the narrow waist of a metal mesh held within a cardboard frame. An external power supply is used to resistively heat the mesh to volatilize analytes, effectively separating the desorption and ionization steps, which provides an additional means of analyte separation during analysis. In the future, automated sample dosing onto the device could be one way to further improve the reproducibility of the overall approach.
Further improvement in sampling through selective preconcentration has been achieved by coating the transmission mesh with polydimethylsiloxane (PDMS) [16]. The PDMS layer serves as a solid-phase microextraction medium for the extraction and preconcentration of semivolatile species in aqueous samples (solid-phase mesh-enhanced sorption from headspace, SPMESH) [16], which could then be analyzed by DART-MS. Through the use of DART-MS/MS and isotopically labeled internal standards, a detection limit of 71 μg/L for linalool in water was achieved. However, the accuracy and precision for linalool in grape macerate was compromised by isobaric interferences present at the monitored MS/MS transition for the unlabeled form, demonstrating the difficulty in performing quantitative analyses with ADI-MS. While transmission-mode approaches have greatly improved the reproducibility of sample introduction and the quantitative capabilities of these methods, it is somewhat removed from the original concept of ADI-MS in that solid samples are not directly probed in their native environment.
Paper spray [53] is an ambient desorption/ionization method which has shown great promise for quantitative analyses for a number of applications, usually with the aid of isotopically labeled standards [43, 53,54,55,56]. For instance, Manicke et al. [43] demonstrated paper spray MS for the quantification of pharmaceuticals in dried blood spots. The relative standard deviation of replicate analyses was as low as 8% when an isotopically labeled internal standard was added to the paper either before or after sample deposition. The variability worsened to 16% when the internal standard was added to the solvent eluent. In a subsequent study, 15 different therapeutic drugs in dried-blood-spot samples were detected with LODs as low as 1 ng/mL [44]. Quantitative information was achieved over approximately three orders of magnitude with accuracies within 10% of the actual concentration and variability of 10% when an internal standard was deposited prior to the application of the blood spots. While paper spray-MS has been shown to be very useful for quantitative analyses with internal standards, significant variation in absolute signals has been observed with different types of filter papers or even within the same batch of a certain type of paper; the reasons for this variability are not yet fully understood [57].
Coupling laser-sampling approaches with ADI sources, such as laser-ablation electrospray ionization (LAESI) [45], matrix-assisted laser desorption electrospray ionization (MALDESI) [58], and laser-ablation flowing atmospheric pressure afterglow (LA-FAPA) [17], has also improved the reproducibility of sample introduction, while simultaneously providing localized sample information that can be used to generate chemical images. With regard to the aforementioned approaches, LODs of 8 and 25 fmol for verapamil and reserpine, respectively, have been achieved with LAESI-MS with four orders of linear dynamic range for quantification [45]. Infrared MALDESI was used for quantitative bioimaging of the antiretroviral drug emtricitabine in incubated human cervical tissue [18]. For quantification, stable isotope-labeled emtricitabine was used, which was spotted on the cryosectioned tissue slice by a modified microliter syringe. One hundred nanoliters of each standard solution were used for each calibration spot, which covered an area approximately 1 mm in diameter on the tissue. For LA-FAPA-MS, a spatial resolution of ~20 μm with a LOD of 5 fmol was achieved for caffeine [17]. While the use of robotics, automated motion control, and lasers can improve sampling precision, these approaches are significantly more complex and expensive.
Another approach that has been employed to improve the reproducibility of sampling makes use of a liquid microjunction surface sampling probe (LMJ-SSP) [19, 20] interfaced with an atmospheric-pressure spray-based ionization source. With the LMJ-SSP, a liquid microjunction is formed between the probe and the sample surface to perform in situ microextraction. The LMJ-SSP can be applied to virtually all species that can be dissolved and transferred into the probe. Detection limits in the low nanogram range were reported for mixtures of dyes on TLC plates [20]. After several years of development, a commercial product is now available from Prosolia, Inc. (Flowprobe™) that can be used for spot sampling and imaging of cells and tissues. Though not yet demonstrated, it should be feasible to also integrate sample preconcentration and cleanup into the workflow of LMJ-SSP in the future.
Sensitivity
One of the greatest challenges in analytical chemistry is to develop techniques that are sensitive enough to detect trace levels of many different analytes in a short amount of time. While ADI-MS certainly reduces the analysis time compared to LC-MS, that speed can come at the expense of compromised sensitivity [21]. Matrix effects can also occur during the desorption (or volatilization) step, further complicating quantitative analyses. These desorption and ionization matrix effects can lead to poor sensitivity for analytes in certain matrices. In addition, ionization of nonpolar analytes is a challenge in ADI-MS analysis.
In plasma-based techniques, the type and density of reagent-ion production is highly dependent on the source operating parameters, such as the discharge voltage/current, gas flow rate, gas composition, and electrode configuration or spacing [22, 26, 27, 48, 59, 60]. Furthermore, discharge type (e.g., corona discharge, direct current (DC) glow discharge, radiofrequency (RF) glow discharge, dielectric-barrier discharge (DBD), etc.) and the associated ion-formation processes also influence the type, distribution, and density of the reagent species. For instance, the FAPA is based on a DC atmospheric-pressure glow discharge (APGD) [61] operated at ca. 10 W of power, which leads to a larger population of reagent ions and, in turn, better sensitivity as compared to the corona-to-glow discharge of the DART source and the dielectric-barrier discharge of the low-temperature plasma (LTP) probe [62].
Some studies have been performed with plasma-based sources to improve the ionization efficiency of nonpolar analytes. In one example, desorption atmospheric pressure chemical ionization (DAPCI) was optimized to successfully ionize and detect petroleum constituents such as hydronaphthalenes, thiophenes, alkyl-substituted benzenes, pyridines, fluorenes, and polycyclic aromatic hydrocarbons (PAHs) [63]. In another example, operating parameters of the FAPA source, such as the discharge current and gas flow rate, were tuned to enhance sensitivity for nonpolar analytes by increasing the density of charge-transfer reagents [26]. In the charge-transfer mode, which occurs at high discharge currents and low gas flow rates, the molecular-ion signal for the nonpolar analyte 2,2′-dichloroquaterphenyl improved by more than two orders of magnitude over the more conventional proton-transfer mode. Enhanced molecular-ion signals for nonpolar analytes with DART-MS were also observed by Cody [36] by adjusting several operating parameters. Namely, high grid potentials, close distances between the DART source and mass spectrometer inlet, and high gas-heater temperatures led to the formation of molecular ions for nonpolar aliphatic compounds. Desorption atmospheric pressure photoionization (DAPPI) [64] was also found to be equally or more effective for the determination of nonpolar analytes compared to DESI with detection limits in the range of 56–670 fmol.
Since the introduction of these ADI sources, several technological developments have been employed to improve analytical performance. Yu et al. [21] coupled the DART source with a triple-quadrupole MS with additional pumping attached to the inlet of MS to compensate for the increased vacuum loading from the high flow rate of helium. This arrangement, which is similar to a jet separator in gas chromatography MS, enhanced sensitivity between 10 and 100 times. The original design of the FAPA source, which utilized a hole in a plate anode to form the flowing afterglow, produced a substantial amount of chemical background across a broad mass range and resulted in significant oxidation of aromatic analytes. These issues complicated analyte detection and identification and compromised the analytical performance of the source [23]. A redesigned FAPA source with a pin-to-capillary geometry was devised, which led to a drastic decrease in chemical background levels in both positive- and negative-ionization modes, as well as less oxidation of aromatic analytes. A detection limit of 4 amol was found for the direct determination of the agrochemical ametryn with the pin-to-capillary FAPA source in neat solvent.
Because ADI sources are typically coupled to mass spectrometers that are designed to sample ions from ESI/APCI, optimization of the ion sampling efficiency/transfer is mostly done empirically, if at all. Moving forward, computer modeling of the ion sampling/transport and MS interface/ion optics is considered a valuable tool that should be used more in the future to improve the overall sensitivity of ADI-MS.
Selectivity
Sufficient selectivity is a prerequisite not only for analyte identification and the prevention of false positives, but also for reliable quantification. Selectivity can be improved through the use of instrumental means, such as tandem mass spectrometry (e.g., MS/MS or MSn) [24, 25], high-resolution mass spectrometry (HR-MS) [28, 65], coupling with ion-mobility spectrometry (IMS) [29, 30], physical sample-preparation approaches (e.g., derivatization or selective extraction/preconcentration) [28, 66], or by altering the desorption and/or ionization chemistry of the source [67].
The ability to perform high-resolution exact mass measurements, such as with an orbitrap, Fourier-transform ion cyclotron resonance (FT-ICR), or a time-of-flight mass analyzer (TOF-MS), offers additional selectivity over a large mass range. Accurate mass measurements can reduce isobaric interferences in the spectra. Additionally, with the resolving powers offered by current FT-ICR and orbitrap instruments, chemical noise levels can be quite low, thus improving sensitivity and LODs. For instance, Jillian et al. [16] observed a one order of magnitude improvement in the LOD for linalool with DART-MS when high-resolution orbitrap mass spectra were acquired as compared to those from a low-resolution triple-quadrupole instrument. Accurate mass measurements alone, though, cannot be used to separate or differentiate between isomeric species commonly encountered in natural systems [31, 34, 68], which can complicate nontargeted analyses.
In ADI-MS, the mass spectrometer itself serves as a separation device as well as a detection tool. Therefore, in the analysis of extremely complex samples, additional selectivity in the form of separating analyte-ion signals becomes quite important. Tandem mass spectrometry methods provide an additional means of selectivity via isolation and fragmentation of the analyte ions. The masses of the resultant fragment ions, along with the mass of the intact molecule, can be used to confirm identity through known fragmentation transitions in the case of targeted analyses [69], or can be compared to libraries of tandem mass spectra in the case of nontargeted analyses [70, 71]. Tandem mass spectra also provide lower chemical noise, which further improves the sensitivity of ADI-MS analyses [72]. However, current MSn approaches are performed serially in time and, as such, cannot be reasonably applied to more than a few analyte ions, let alone the entire mass range.
Yang et al. applied DAPCI for melamine detection in milk products [25]. For improved selectivity, they performed MS3 experiments in a linear ion trap MS (cf. Fig. 2). To shed light on the fragmentation behavior, additional experiments were carried out with deuterated melamine. Melamine-d6 contains three deuterated amine groups that are susceptible to proton–deuterium exchange. Therefore, the measurements were presumably carried out in deuterated solvent, as a mass shift of seven (m/z 127 vs 134) was observed. For MS quantification, the usage of isotopically labeled 15N- or 13C-melamine is recommended, because these isotopes do not exchange in solvent.

Mass spectra of melamine recorded by DAPCI-MS. a Mass spectrum of authentic melamine (1 ng) on filter paper surface. b MS/MS product-ion spectrum of protonated melamine at m/z 127. c MS/MS product-ion spectrum of deuterated melamine at m/z 134. d MS3 spectrum of the ionic fragments (m/z 85) produced from protonated melamine. e MS3 spectrum of the ionic fragment at m/z 90 produced from deuterated melamine. f Mass spectrum of powdered milk on a filter paper surface; the signal detected at m/z 127 yielded the same MS/MS spectrum as that of protonated authentic melamine (shown in Fig. 2b). Reprinted with permission from [25]
Additional rapid orthogonality to ADI-MS analyses, and improved selectivity, can be obtained with the temperature-programmed vaporization of analytes. Programmed thermal desorption with external heating leads to the separation of sample components in time as a result of their different vapor pressures. In the DIP-APCI-MS determination of coumarin in woodruff-flavored liquor [73], the analyte signal at m/z 147.0441 exhibited a delayed rise as compared to the chronogram of an isotopically labeled internal standard, coumarin-d4. The time profiles of these isotopologues are vastly different. The authors rationalize that the species detected near m/z 147 with DIP-APCI-MS originated from the chemical modification of matrix components during the desorption/ionization process, likely due to the high temperatures applied directly to the sample.
Additional selectivity can be achieved without compromising the analysis time by introducting a complementary rapid ion separation/filter device such as IMS or differential mobility spectrometry (DMS) [30, 74, 75]. The biggest strength of DMS coupled with ADI-MS lies in the removal of background interferences in targeted analyses. The differential mobility system can be readily implemented in ADI-MS methods because it separates ions on the basis of a complementary mechanism. Clearly, however, it also adds complexity to the MS instrument and is associated with higher costs. Galhena et al. [33] demonstrated a hybrid DESI-DMS-MS platform on a commercial mass spectrometer. By allowing only ions of a specific mobility to pass through the DMS device, chemical noise was suppressed and analyte ions could be filtered from interfering ions of a similar mass.
The performance of DESI-DMS-MS was demonstrated for the separation and detection of the isobaric compounds 5-HMF and melamine. When the DMS device was off, the two species were marginally differentiated due to the relatively high resolving power of the TOF mass analyzer (cf. inset in Fig. 3). However, when the DMS cell was turned on and the compensation voltage swept during analysis, the isobaric species were baseline resolved and could be identified (cf. Fig. 3).

Application of DESI-DM-MS to the analysis of chemical standards on PTFE surfaces. The analysis of a binary mixture of melamine (100 μM) and 5-HMF (100 μM) in DM-on mode (SV = 900 V, CV = −15 to 5 V) is shown. The inset shows the marginally resolved TOF MS spectra of melamine and 5-HMF. Reprinted with permission from [33]
Another strategy to increase the selectivity and, by extension, the sensitivity of ADI-MS analyses is to alter the chemistry of the desorption/ionization process in what is often referred to as ‘reactive ambient mass spectrometry.’ Cotte-Rodríguez et al. [67] used additives in spray solvents of DESI to form characteristic adduct ions of explosives, which enhanced selectivity and improved LODs by up to an order of magnitude. In another case, direct and rapid detection of cholesterol in dried serum samples and animal tissue sections was drastically improved by the addition of betaine aldehyde to the DESI spray solvent [76]. The experiment combined desorption by DESI with in-situ chemical derivatization, as betaine aldehyde selectively and rapidly reacts with the hydroxyl group of cholesterol to form a hemiacetal salt. The study also demonstrated the quantitative analysis of free cholesterol in serum using reactive DESI with cholesterol-d7 as an internal standard [76]. An LOD of 1 ng was achieved for cholesterol and related compounds with this reactive DESI system.
In another example of reactive ADI-MS, the selective reaction of 2-phenyl-4,5,5,5-tetramethylimidazoline-1-oxyl-3-oxide with NO· was used for the quantification of exhaled nitric oxide (eNO) in human breath with extractive electrospray ionization–mass spectrometry (EESI-MS) [77]. The EESI-MS response of the 1-oxyl-2-phenyl-4,4,5,5-tetramethylimidazoline (PTI) product was used to calculate the eNO concentration. Quantification of eNO at the sub-ppb level (~0.02 ppbv) with a relative standard deviation of 11.6% was achieved.
A common misconception about the increased selectivity offered by instrumental approaches (e.g., MS/MS, HR-MS, and IMS) is related to its ability to address matrix effects, in particular ion suppression. Though these approaches often aid in filtering or separating ions produced from the sample matrix, ion suppression and other matrix effects still exist as they occur during the desorption/ionization step. As such, it is important not to confuse ion suppression with chemical-background interference, because ion suppression is a phenomenon that occurs in the ionization source and adversely affects the formation of the ions of interest. As the above instrumental approaches for improving selectivity are carried out after the desorption/ionization process, they cannot compensate for any ion suppression or other matrix-effect issues. Therefore, matrix effects need to be investigated and actively monitored, while appropriate quality-control procedures should also be implemented.
Even with the instrumental advantages discussed above, interferences and matrix effects remain the biggest issues limiting quantitative ADI-MS. The desorption and ionization processes can also give rise to interferences via the chemical modification of species in a sample. For example, the creation of 5-hydroxymethylfurfural (5-HMF, m/z 127.03897) from carbohydrates during the desorption/ionization process has complicated the detection of melamine (m/z 127.07267) in milk powder due to isobaric interferences [32, 78]. Fortunately, in that case, tandem MS and HRMS can be used to resolve those species.
Investigation and reduction of matrix effects
Without prior separation of analytes, matrix effects can severely degrade quantitative capabilities and even inhibit the identification of analytes in ADI-MS analyses. As mentioned by Cooks et al. [35], matrix effects ultimately limit the quantitative accuracy of MS methods. Although the matrix effects in plasma-based ADI sources are typically considered to be less severe than those in spray-based methods, they are still quite significant [62]. Shelley et al. [62] compared ionization matrix effects among three plasma-based ADI sources: FAPA, DART, and LTP. They found that all three methods suffer from ionization matrix effects, with FAPA being least susceptible to the ion-suppression process for species that only undergo proton-transfer ionization. In LTP and FAPA, a different type of matrix effect can occur, namely an inhibition of the formation of protonated reagent species due to the presence of nonpolar compounds [62].
Only a limited number of publications have thus far discussed matrix-effect theory and methods of mitigating this problem in ADI-MS. Song et al. [79] proposed a transient microenvironment mechanism (TMEM) to address matrix effects for DART. According to the mechanism, a transient microenvironment (TME) shields analytes from direct ionization when the DART gas stream impinges on the sample. Matrix molecules are ionized first and analytes are ionized later by gas-phase ion/molecule reactions with matrix ions. As little as 10 nL of liquid or 10 μg of solid material is reported to be sufficient to create a transient microenvironment. Furthermore, this TME can dictate the ionization pathways of analytes below a certain analyte-to-matrix ratio, depending on the DART temperature and the boiling points of the analyte and matrix.
Chen et al. [80] used a separate neutral gas stream with EESI-MS to sample the surfaces of solid biological objects such as human skin, frozen meat, and plant tissue without sample pretreatment. This approach produced a neutral aerosol that was subsequently analyzed online. Desorption of volatile and semivolatile analytes with the neutral gas stream effectively separated the sampling process from the ionization process in both time and space, which resulted in less ion suppression.
Harris et al. reported a spatial dependence of the sensitivity and ion suppression in a DART analysis of nerve-agent simulants [37]. Sampling locations with a high degree of analyte response (‘ion yield hot spots’) did not always correspond with the highest-temperature regions within the ionization space (cf. Fig. 4). Interestingly, they found that the volatility of analytes seemed to play a smaller role in ion suppression than differences in proton affinity.

Sensitivity-in-space maps at different DART gas temperatures and concentrations of DMMP. Maps a (50 μM), b (100 μM), and c (500 μM) were tested at 200 °C, maps d (50 μM), e (100 μM), and f (500 μM) were tested at 300 °C, and maps g (50 μM), h (100 μM), and i (500 μM) were tested at 400 °C. All averaged (n = 5) intensities were normalized to the highest intensity recorded for a given concentration. Reprinted with permission from [37]
No sample preparation was one of the initial selling points of ADI-MS. Today it is increasingly reported that tailored sample pretreatment seems to be more advantageous than no sample preparation at all. For example, when samples are directly analyzed from their native environment, improved sensitivity and selectivity can be achieved by the rapid preconcentration of analytes within the sample prior to analysis. Several preconcentration methods coupled with ADI-MS analyses have been reported [28, 66, 81, 82]. For example, quantitative analysis of mycotoxins was performed using matrix-matched standards or commercially available 13C-labeled internal standards [28]. The authors observed serious ion suppression by coextracted matrix compounds. To reduce these matrix effects, dispersive solid-phase extraction (SPE) with primary-secondary amine (PSA) sorbents and MgSO4 was employed to clean up the sample. A significant improvement in analyte signal response (deoxynivalenol, m/z 331.0943) as a function of the amount of PSA sorbent added (0–100 mg/mL of extract) is illustrated in Fig. 5. Ultimately, more severe matrix effects and poorer sensitivity (LOD of 648 μg/kg) was noted with DART-HRMS analyses as compared to LC-MS methods (LOD = 60 μg/kg), the standard approach for mycotoxin detection. Furthermore, slightly worse repeatability of the measurements with DART-HRMS (RSD between 7.9 and 12.0%) as compared to those with UPLC-TOFMS (RSD less than 5.6%) was achieved when examining certified reference material (CRM) extracts.

The impact of dispersive SPE clean-up employing PSA and MgSO4 on deoxynivalenol (m/z 331.0943 ± 4 ppm) signal intensity in a wheat extract (spike 500 μg/kg). The given sorbent amounts were used for 4 ml of acetonitrile extract containing the equivalent of 800 mg of the matrix; the solvent standard concentration was 100 ng/mL. Reprinted with permission from [28]
In another study, liquid-phase microextraction (LPME) in combination with DESI-MS was used for the identification and quantification of basic drugs in human urine [82]. A significant reduction in matrix effects was observed with the use of LPME compared to direct analysis with DESI-MS due to the selective extraction capabilities of three-phase LPME. The LPME extracts were deposited on porous Teflon, allowed to dry, and analyzed with DESI-MS. The limit-of-quantification for diphenhydramine was 140 ng/mL when an internal standard (diphenhydramine-d5) was used.
Preconcentration of UV-filter compounds in environmental water samples with stirbar sorptive extraction (SBSE) was performed, followed by DART-MS analysis [83]. After preconcentration with SBSE, LODs of better than 40 ng/L were achieved for several organic UV-filter standard solutions, but RSDs were as large as 30%. Micro Extraction by packed sorbent (MEPS) was used by Jagerdeo et al. [81] for the fast extraction and preconcentration of drugs of abuse followed by rapid analysis with DART-MS. Quantification of cocaine in human urine with internal standardization (isotopically labeled cocaine) was reported with a LOD of 4.0 ng/mL. In another study, molecularly imprinted polymers (MIPs) used as analyte sequesters were employed with easy sonic-spray ionization (EASI)-MS for the analysis of drugs in urine [84]. This method was advantageous for complex sample analysis due to the ability to selectively trap target analytes with well-designed MIPs, which results in reduced matrix effects.
Very recently, solid-phase microextraction (SPME) followed by thermal desorption of the extracted analytes to introduce the sample to a dielectric-barrier discharge ionization (DBDI) source was reported [85]. The method separates the thermal-desorption step from the ionization step, which enhances reproducibility and minimizes ion suppression. This approach features an in-line geometry (SPME, DBDI, interface) that results in improved analyte-ion transmission; LODs as low as 0.3 pg/mL were achieved for cocaine and diazepam.
Based on the reports discussed above, it is clear that microextraction techniques can improve matrix effects in the analysis of complex mixtures. In principle, however, they require additional and sometimes time-consuming preparation steps in the analytical protocol, a fact that conflicts with the initial concept of fast and direct ADI analyses.
Internal standardization
The detrimental effects of matrix components on quantification can be either reduced as outlined above or they can be compensated for using internal standardization. The use of an internal standard in ambient mass spectrometry was shown in the first publication on DESI [5]. Clearly, the selection of an appropriate standardization approach depends on the sample phase, surface composition, and the target analytes themselves. In liquid-phase analysis, internal standards can be added to the sample solution before the dried droplets are probed with ADI sources. Similarly to conventional mass spectrometric protocols, standardization with deuterated standards is feasible, although the limited availability and high cost of deuterated standards might be a concern. For example, DART was coupled to a linear ion trap mass spectrometer and successfully used to detect and quantify glucose through the use of a deuterated glucose standard [38].
In Fig. 6, extracted ion chromatograms (ammonium adduct of glucose [M + NH4]+ at m/z 198) of the analyte are depicted. The peak height and the peak profile were found to vary among nine repetitions, similar to what is observed with most ADI sources, but the peak area was successfully used for quantification after the calculation of a peak area ratio (PAR) that considered the response of the deuterated form of glucose at known concentrations. A linear range from 10 to 3000 μM was found. More details on the analytical performance (LOD, LOQ, etc.) were not reported.

Reproducibility experiments. a Extracted ion chronogram (m/z 198) for one trial where nine 50 mM glucose standards spiked with 40 mM of deuterated glucose were analyzed by DART-LIT. b Calculated peak area ratios (PAR) for standard solutions. Trial 1 is shown in a and additional trials represents a separate batch of samples (n = 9). Reprinted with permission from [38]
For natural surfaces, however, the use of internal standards is not straightforward. Applying an internal standard to an amorphous solid sample in a consistent way can be quite difficult. For example, it was reported that neither the addition of an internal standard to the solvent spray in DESI nor doping an internal standard by electrospray deposition yielded appropriate quantitative results [11]. Nyadong et al. showed that the internal standard-to-analyte response in the DESI-MS analysis of tablets is influenced by the hardness of the tablet samples [47].
Recent applications
Rapid and high-throughput analyses have expanded the field of ADI-MS to diverse applications. One interesting application is the rapid identification of pesticides in human oral fluids in self-poisoning cases. Lee et al. described a point-of-care method based on laser desorption (LD)-ESI-MS for the targeted detection of pesticides (methamidophos, methomyl, paraquat, dimethoate, and chlorpyrifos) by MS/MS using triple-quadrupole MS [39]. Pesticide–oral fluid mixtures were applied on a cotton swab and then transferred into methanol. A metallic probe was used to sample the methanol solution for subsequent LD-ESI-MS/MS analysis. Total analysis time (sampling, transfer, desorption, ionization, detection) was within 1 min. The LODs of the pesticides in oral fluid obtained from four human subjects were between 1 and 10 ppb, with a relative standard deviation of 10.7%.
ADI-MS has found its way into the field of environmental sciences as well. For example, DESI-MS was used for the quantification of organic acids in aerosols, and lengthy sample preparation steps such as extraction, concentration, and preseparation could be completely eliminated [40]. Li et al. reported that LODs of approximately 1 pg/mm2 were achieved for selected organic acids (oxalic and oleic acids) in atmospheric aerosols within as little as 5–10 s of sampling time [40]. Recently, the FAPA source was used for the analysis of organic aerosols [41]. Changes in aerosol composition and concentration were detected on a timescale of seconds and in the ng/m3 range. The FAPA-MS results from a field campaign in a mixed forest region were in good agreement with offline measurements of collected aerosols.
In recent years, ADI-MS techniques have increasingly been used for imaging applications. An indirect approach has been presented by Hemalatha et al. [42]. They used DESI-MS for imprint imaging. One prerequisite for imprint imaging is retention of spatial resolution. The study demonstrated the use of electrospun nanofiber mats made of nylon-6 surfaces for rapid detection or imaging by DESI-MS. The applicability of this method was demonstrated for six different examples, including patterns formed by single drops of dissolved dyes, marker pen inks, and printing inks. Imprints of plant parts showed the suitability of nanofiber mats as a substrate for identifying and preserving diverse classes of compounds. While the majority of the study focused on qualitative aspects of this approach, future experiments will have to evaluate (among other aspects) the completeness of analyte transfer for subsequent quantification.
Alternatively, ADI-MS analysis is increasingly being utilized to obtain spatial and molecular information from biological samples with minimal or no sample pretreatment. Zou et al. demonstrated the use of a picosecond infrared laser (PIRL) for small-molecule imaging, where the laser cut through biological tissues without causing significant thermal damage to nearby tissue [86]. The PIRL can be used as a standalone surgical scalpel with the added bonus of minimal postoperative scar tissue formation. The combination of PIRL ablation with ESI (PIR-LAESI) further improved the MS signals. The sensitivity of the method was estimated by analyzing aqueous solutions, with LODs in the range of 100 nM obtained for reserpine and better than 5 nM for verapamil.
The assessment of LODs in imaging applications is challenging. As described above, LODs were also determined using aqueous solutions by Lee et al. [87]. They present a laser desorption/ionization droplet delivery mass spectrometry (LDIDD-MS) method which is capable of single-cell analysis. For LDIDD-MS, a focused, pulsed UV laser was used for the desorption and ionization of target molecules deposited on a surface. The combined effect of photoionization by the UV laser and ESI increased the ionization of analytes in an analyte-dependent manner. For example, the caffeine signal was five times higher than obtained with photoionization alone, and more than ten times more intense than attained with ESI alone. The combination of UV photoionization and ESI was also applied to the amino acid lysine, and a LOD as low as 2 amol was obtained.
A detailed study of the capabilities of infrared-MALDESI for the quantification of the antiretroviral drug emtricitabine in incubated human cervical tissue was presented [18]. Stable-isotope-labeled emtricitabine was used for quantification, whereas a different but structurally similar compound, lamivudine, was used as an internal standard to account for voxel-to-voxel variation. The incorporation of a structurally similar normalization compound allows for the normalization of analyte-ion abundances on a per voxel basis. The quantitative IR-MALDESI analysis proved to be reproducible with an emtricitabine concentration of 17.2 ± 1.8 μg/g tissue. This amount corresponds to the detection of 7 fmol/voxel in the imaging experiment.
Groseclose and Castellino presented a detailed study of quantitative MALDI imaging. Instead of spotting a range of concentrations onto the tissue sample, they used a matrix-matched standardization approach [88]. A mimetic tissue model was proposed, consisting of a set of tissue homogenates spiked with a range of different drug concentrations that were frozen into a polymer support mold. Sections of the same thickness from the tissue model and the dosed tissue were collected. These were placed adjacent to each other on the MALDI target, which enabled the matrix application step and MALDI imaging acquisition to be conducted under identical conditions for both sections. A calibration curve was generated from the model and correlated with the intensities detected from the dosed tissue section to quantify the amount of drug present. The results for lapatinib- and nevirapine-dosed tissues from nonclinical species were compared with those generated by LC-MS quantification, and close agreement was observed.
One of the most challenging prospective applications with respect to available analysis time and sample complexity shown in the literature is the intraoperative molecular diagnosis of human brain tumors [89]. Eberlin et al. [89] developed a method to rapidly classify brain tumors based on lipid information obtained by DESI-MS, which could be used to determine the boundaries between healthy and neoplastic tissue. Oligodendroglioma, astrocytoma, and meningioma tumors of different histological grades and tumor cell concentrations were analyzed. The results obtained from mass-spectral imaging were in agreement with the histopathology diagnosis with very few exceptions. This method demonstrates the potential of ADI-MS to guide brain tumor surgery by providing rapid diagnosis and tumor margin assessment in near real time.
The group of Eberlin also developed and optimized an analytical approach integrating DESI and LMJ-SSP with a chip-based FAIMS device for imaging biological tissue samples [90]. This method allows the partial separation of singly charged metabolites, singly and doubly charged glycerophospholipids and glycosphingolipids, and multiply charged protein analytes after desorption or extraction from biological samples, resulting in decreased chemical noise and increased detection of selected molecular species. Reducing interferences through FAIMS separation improved the S/N for the species of interest, aiding in spectral interpretation and improving ion image quality. For example, lipid identification was improved by a 50% increase in S/N for all detected cardiolipin species.
The presented ADI-MS applications for bioimaging have one major characteristic in common: quantitative information is quite difficult to obtain. Therefore, it could be useful to also apply an analytical technique that provides more straightforward quantitative information which would complement the spatial and molecular information provided by ADI-MS. For example, laser ablation inductively coupled plasma time-of-flight mass spectrometry (LA-ICP-TOFMS) is successfully used for quantitatively imaging metal or heteroelement-containing species, sometimes with subcellular resolution [91,92,93,94]. While the quantitative sampling afforded by LA and the species-independent response of ICP-MS offer several advantages, information about molecular identity is completely lost upon atomization in the ICP source. However, combining tried-and-true quantitative analytical tools, such as LA-ICP-MS, with ADI approaches or alternative ways of using established ionization sources (e.g., see LA-APCI-MS/ICP-MS [95]) could provide a means to generate reliable quantitative information in ADI imaging applications. Though such a specific combination has not yet been presented in the literature, these sorts of multimodal analytical (and imaging) methodologies will likely begin to play an important role in ADI-MS analyses, particularly for obtaining reliable quantitative information.
Future perspectives
Ambient desorption/ionization mass spectrometry has already demonstrated tremendous potential in different fields of the analytical sciences. However, more effort is required to achieve reliable and quantitative information using these approaches. It is anticipated that key improvements to the analytical performance of ADI-MS will continue. However, our fundamental understanding of desorption and ionization processes and matrix effects is still very limited. As such, a greater push to understand these fundamentals is required for the field to progress and for the development of successful quantitative ADI-MS methods.
We expect continued development, with novel concepts not only for semiquantitative screening but also for accurate quantification. However, only the truly simple-to-operate, effective, and rugged sources are expected to have a long-lasting impact in the field, at least from a commercial standpoint. With an improved understanding of the advantages and limitations of a given ADI source (and compared to other sources), it would then be possible to define the useful range of applications, and to further improve the method’s selectivity, sensitivity, and reproducibility. To move the field forward, researchers should only publish data that was obtained following the guidelines and quality standards of the analytical sciences. For example, it should always be stated in a manuscript how quantitative results were obtained and if matrix effects were observed; these points are clearly lacking in many publications featuring ADI-MS methods. Also, new methodologies should be tested on certified reference materials and validated with an established method.
Clearly, ADI-MS will not replace techniques such as LC-MS or GC-MS, but it will become an indispensable and complementary tool, especially for in situ chemical analysis. For example, portability is something that traditional benchtop mass spectrometers do not offer, and it is assumed that portable miniature mass spectrometers equipped with ADI sources will become commercially available in the future. These unique instruments would open up new avenues for in situ chemical analysis, with applications in the fields of biomedicine, environmental science, and pharmaceuticals.
References
Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246(4926):64–71.
Carroll D, Dzidic I, Stillwell R, Haegele K, Horning E. Atmospheric pressure ionization mass spectrometry. Corona discharge ion source for use in a liquid chromatograph-mass spectrometer-computer analytical system. Anal Chem. 1975;47(14):2369–73.
Robb DB, Covey TR, Bruins AP. Atmospheric pressure photoionization: an ionization method for liquid chromatography-mass spectrometry. Anal Chem. 2000;72(15):3653–9.
Hanold KA, Fischer SM, Cormia PH, Miller CE, Syage JA. Atmospheric pressure photoionization. 1. General properties for LC/MS. Anal Chem. 2004;76(10):2842–51.
Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306(5695):471–3.
Cody RB, Laramée JA, Durst HD. Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal Chem. 2005;77(8):2297–302.
Badu-Tawiah AK, Eberlin LS, Ouyang Z, Cooks RG. Chemical aspects of the extractive methods of ambient ionization mass spectrometry. Annu Rev Phys Chem. 2013;64:481–505.
Albert A, Shelley JT, Engelhard C. Plasma-based ambient desorption/ionization mass spectrometry: state-of-the-art in qualitative and quantitative analysis. Anal Bioanal Chem. 2014;406(25):6111–27.
Shelley JT, Hieftje GM. Ambient mass spectrometry: approaching the chemical analysis of things as they are. J Anal At Spectrom. 2011;26(11):2153–9.
Chen H, Gamez G, Zenobi R. What can we learn from ambient ionization techniques? J Am Soc Mass Spectrom. 2009;20(11):1947–63.
Takats Z, Wiseman JM, Cooks RG. Ambient mass spectrometry using desorption electrospray ionization (DESI): instrumentation, mechanisms and applications in forensics, chemistry, and biology. J Mass Spectrom. 2005;40(10):1261–75.
Nanita SC, Kaldon LG. Emerging flow injection mass spectrometry methods for high-throughput quantitative analysis. Anal Bioanal Chem. 2016;408(1):23–33.
Venter A, Cooks RG. Desorption electrospray ionization in a small pressure-tight enclosure. Anal Chem. 2007;79(16):6398–403.
Chipuk JE, Brodbelt JS. Transmission mode desorption electrospray ionization. J Am Soc Mass Spectrom. 2008;19(11):1612–20.
IonSense. Installation and operations manual for ID-Cube source and power supply. Saugus: IonSense; 2014.
Jastrzembski JA, Sacks GL. Solid phase mesh enhanced sorption from headspace (SPMESH) coupled to DART-MS for rapid quantification of trace-level volatiles. Anal Chem. 2016;88(17):8617–23.
Shelley JT, Ray SJ, Hieftje GM. Laser ablation coupled to a flowing atmospheric pressure afterglow for ambient mass spectral imaging. Anal Chem. 2008;80(21):8308–13.
Bokhart MT, Rosen E, Thompson C, Sykes C, Kashuba ADM, Muddiman DC. Quantitative mass spectrometry imaging of emtricitabine in cervical tissue model using infrared matrix-assisted laser desorption electrospray ionization. Anal Bioanal Chem. 2015;407(8):2073–84.
Wachs T, Henion J. Electrospray device for coupling microscale separations and other miniaturized devices with electrospray mass spectrometry. Anal Chem. 2001;73(3):632–8.
Van Berkel GJ, Sanchez AD, Quirke JME. Thin-layer chromatography and electrospray mass spectrometry coupled using a surface sampling probe. Anal Chem. 2002;74(24):6216–23.
Yu S, Crawford E, Tice J, Musselman B, Wu J-T. Bioanalysis without sample cleanup or chromatography: the evaluation and initial implementation of direct analysis in real time ionization mass spectrometry for the quantification of drugs in biological matrixes. Anal Chem. 2008;81(1):193–202.
Orejas J, Pfeuffer KP, Ray SJ, Pisonero J, Sanz-Medel A, Hieftje GM. Effect of internal and external conditions on ionization processes in the FAPA ambient desorption/ionization source. Anal Bioanal Chem. 2014;406(29):7511–21.
Shelley JT, Wiley JS, Hieftje GM. Ultrasensitive ambient mass spectrometric analysis with a pin-to-capillary flowing atmospheric-pressure afterglow source. Anal Chem. 2011;83(14):5741–8.
McLafferty FW. Tandem mass spectrometry. Science. 1981;214(4518):280–7.
Yang S, Ding J, Zheng J, Hu B, Li J, Chen H, et al. Detection of melamine in milk products by surface desorption atmospheric pressure chemical ionization mass spectrometry. Anal Chem. 2009;81(7):2426–36.
Badal SP, Michalak SD, Chan GC-Y, You Y, Shelley JT. Tunable ionization modes of a flowing atmospheric-pressure afterglow (FAPA) ambient ionization source. Anal Chem. 2016;88(7):3494–503.
Wright JP, Heywood MS, Thurston GK, Farnsworth PB. The effects of added hydrogen on a helium atmospheric-pressure plasma jet ambient desorption/ionization source. J Am Soc Mass Spectrom. 2013;24(3):335–40.
Vaclavik L, Zachariasova M, Hrbek V, Hajslova J. Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta. 2010;82(5):1950–7.
Bennett RV, Gamage CM, Galhena AS, Fernández FM. Contrast-enhanced differential mobility-desorption electrospray ionization-mass spectrometry imaging of biological tissues. Anal Chem. 2014;86(8):3756–63.
Weston DJ, Bateman R, Wilson ID, Wood TR, Creaser CS. Direct analysis of pharmaceutical drug formulations using ion mobility spectrometry/quadrupole-time-of-flight mass spectrometry combined with desorption electrospray ionization. Anal Chem. 2005;77(23):7572–80.
Polfer NC, Valle JJ, Moore DT, Oomens J, Eyler JR, Bendiak B. Differentiation of isomers by wavelength-tunable infrared multiple-photon dissociation-mass spectrometry: application to glucose-containing disaccharides. Anal Chem. 2006;78(3):670–9.
Dane AJ, Cody RB. Selective ionization of melamine in powdered milk by using argon direct analysis in real time (DART) mass spectrometry. Analyst. 2010;135(4):696–9.
Galhena AS, Harris GA, Kwasnik M, Fernández FM. Enhanced direct ambient analysis by differential mobility-filtered desorption electrospray ionization-mass spectrometry. Anal Chem. 2010;82(22):9159–63.
Ferreres F, Llorach R, Gil-Izquierdo A. Characterization of the interglycosidic linkage in di-, tri-, tetra-and pentaglycosylated flavonoids and differentiation of positional isomers by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2004;39(3):312–21.
Cooks RG, Ouyang Z, Takats Z, Wiseman JM. Ambient mass spectrometry. Science. 2006;311(5767):1566–70.
Cody RB. Observation of molecular ions and analysis of nonpolar compounds with the direct analysis in real time ion source. Anal Chem. 2008;81(3):1101–7.
Harris GA, Falcone CE, Fernández FM. Sensitivity “hot spots” in the direct analysis in real time mass spectrometry of nerve agent simulants. J Am Soc Mass Spectrom. 2012;23(1):153–61.
Saang’onyo DS, Smith DL. Optimization of direct analysis in real time (DART) linear ion trap parameters for the detection and quantitation of glucose. Rapid Commun Mass Spectrom. 2012;26(3):385–91.
Lee CW, Su H, Chen PY, Lin SJ, Shiea J, Shin SJ, et al. Rapid identification of pesticides in human oral fluid for emergency management by thermal desorption electrospray ionization/mass spectrometry. J Mass Spectrom. 2016;51(2):97–104.
Li M, Chen H, Yang X, Chen J, Li C. Direct quantification of organic acids in aerosols by desorption electrospray ionization mass spectrometry. Atmos Environ. 2009;43(17):2717–20.
Bruggemann M, Karu E, Stelzer T, Hoffmann T. Real-time analysis of ambient organic aerosols using aerosol flowing atmospheric-pressure afterglow mass spectrometry (AeroFAPA-MS). Environ Sci Technol. 2015;49(9):5571–8.
Hemalatha RG, Ganayee MA, Pradeep T. Electrospun nanofiber mats as “smart surfaces” for desorption electrospray ionization mass spectrometry (DESI MS)-based analysis and imprint imaging. Anal Chem. 2016;88(11):5710–7.
Manicke NE, Yang Q, Wang H, Oradu S, Ouyang Z, Cooks RG. Assessment of paper spray ionization for quantitation of pharmaceuticals in blood spots. Int J Mass Spectrom. 2011;300(2):123–9.
Manicke NE, Abu-Rabie P, Spooner N, Ouyang Z, Cooks RG. Quantitative analysis of therapeutic drugs in dried blood spot samples by paper spray mass spectrometry: an avenue to therapeutic drug monitoring. J Am Soc Mass Spectrom. 2011;22(9):1501–7.
Nemes P, Vertes A. Laser ablation electrospray ionization for atmospheric pressure, in vivo, and imaging mass spectrometry. Anal Chem. 2007;79(21):8098–106.
Nyadong L, Galhena AS, Fernández FM. Desorption electrospray/metastable-induced ionization: a flexible multimode ambient ion generation technique. Anal Chem. 2009;81(18):7788–94.
Nyadong L, Late S, Green MD, Banga A, Fernández FM. Direct quantitation of active ingredients in solid artesunate antimalarials by noncovalent complex forming reactive desorption electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2008;19(3):380–8.
Chan GC-Y, Shelley JT, Wiley JS, Engelhard C, Jackson AU, Cooks RG, et al. Elucidation of reaction mechanisms responsible for afterglow and reagent-ion formation in the low-temperature plasma probe ambient ionization source. Anal Chem. 2011;83(10):3675–86.
Pfeuffer KP, Shelley JT, Ray SJ, Hieftje GM. Visualization of mass transport and heat transfer in the FAPA ambient ionization source. J Anal At Spectrom. 2013;28(3):379–87.
Pfeuffer KP, Ray SJ, Hieftje GM. Measurement and visualization of mass transport for the flowing atmospheric pressure afterglow (FAPA) ambient mass-spectrometry source. J Am Soc Mass Spectrom. 2014;25(5):800–8.
Schilling GD, Shelley JT, Barnes JH, Sperline RP, Denton MB, Barinaga CJ, et al. Detection of positive and negative ions from a flowing atmospheric pressure afterglow using a Mattauch-Herzog mass spectrograph equipped with a Faraday-strip array detector. J Am Soc Mass Spectrom. 2010;21(1):97–103.
Harris GA, Fernández FM. Simulations and experimental investigation of atmospheric transport in an ambient metastable-induced chemical ionization source. Anal Chem. 2008;81(1):322–9.
Wang H, Liu JJ, Cooks RG, Ouyang Z. Paper spray for direct analysis of complex mixtures using mass spectrometry. Angew Chem Int Ed. 2010;49(5):877–80.
Wang H, Ren Y, McLuckey MN, Manicke NE, Park J, Zheng L, et al. Direct quantitative analysis of nicotine alkaloids from biofluid samples using paper spray mass spectrometry. Anal Chem. 2013;85(23):11540–4.
Yang Q, Manicke NE, Wang H, Petucci C, Cooks RG, Ouyang Z. Direct and quantitative analysis of underivatized acylcarnitines in serum and whole blood using paper spray mass spectrometry. Anal Bioanal Chem. 2012;404(5):1389–97.
Maher S, Jjunju FP, Damon DE, Gorton H, Maher YS, Syed SU, et al. Direct analysis and quantification of metaldehyde in water using reactive paper spray mass spectrometry. Sci Rep. 2016;6:35643.
Zhang Z, Xu W, Manicke NE, Cooks RG, Ouyang Z. Silica coated paper substrate for paper-spray analysis of therapeutic drugs in dried blood spots. Anal Chem. 2012;84(2):931–8.
Sampson JS, Hawkridge AM, Muddiman DC. Generation and detection of multiply-charged peptides and proteins by matrix-assisted laser desorption electrospray ionization (MALDESI) Fourier transform ion cyclotron resonance mass spectrometry. J Am Soc Mass Spectrom. 2006;17(12):1712–6.
Brewer TM, Verkouteren JR. Atmospheric identification of active ingredients in over-the-counter pharmaceuticals and drugs of abuse by atmospheric pressure glow discharge mass spectrometry (APGD-MS). Rapid Commun Mass Spectrom. 2011;25(17):2407–17.
Bierstedt A, Panne U, Rurack K, Riedel J. Characterization of two modes in a dielectric barrier discharge probe by optical emission spectroscopy and time-of-flight mass spectrometry. J Anal At Spectrom. 2015;30(12):2496–506.
Andrade FJ, Shelley JT, Wetzel WC, Webb MR, Gamez G, Ray SJ, et al. Atmospheric pressure chemical ionization source. 1. Ionization of compounds in the gas phase. Anal Chem. 2008;80(8):2646–53.
Shelley JT, Hieftje GM. Ionization matrix effects in plasma-based ambient mass spectrometry sources. J Anal At Spectrom. 2010;25(3):345–50.
Jjunju FPM, Badu-Tawiah AK, Li AY, Soparawalla S, Roqan IS, Cooks RG. Hydrocarbon analysis using desorption atmospheric pressure chemical ionization. Int J Mass Spectrom. 2013;345:80–8.
Haapala M, Pól J, Saarela V, Arvola V, Kotiaho T, Ketola RA, et al. Desorption atmospheric pressure photoionization. Anal Chem. 2007;79(20):7867–72.
Xian F, Hendrickson CL, Marshall AG. High resolution mass spectrometry. Anal Chem. 2012;84(2):708–19.
Albert A, Kramer A, Scheeren S, Engelhard C. Rapid and quantitative analysis of pesticides in fruits by QuEChERS pretreatment and low-temperature plasma desorption/ionization orbitrap mass spectrometry. Anal Methods. 2014;6(15):5463–71.
Cotte-Rodríguez I, Takáts Z, Talaty N, Chen H, Cooks RG. Desorption electrospray ionization of explosives on surfaces: sensitivity and selectivity enhancement by reactive desorption electrospray ionization. Anal Chem. 2005;77(21):6755–64.
Reinhold VN, Reinhold BB, Costello CE. Carbohydrate molecular weight profiling, sequence, linkage, and branching data: ES-MS and CID. Anal Chem. 1995;67(11):1772–84.
Jackson AU, Werner SR, Talaty N, Song Y, Campbell K, Cooks RG, et al. Targeted metabolomic analysis of Escherichia coli by desorption electrospray ionization and extractive electrospray ionization mass spectrometry. Anal Biochem. 2008;375(2):272–81.
Bentayeb K, Ackerman LK, Begley TH. Ambient ionization-accurate mass spectrometry (AMI-AMS) for the identification of nonvisible set-off in food-contact materials. J Agric Food Chem. 2012;60(8):1914–20.
Sero R, Nunez O, Bosch J, Grases J, Rodriguez P, Moyano E, et al. Desorption electrospray ionization-high resolution mass spectrometry for the screening of veterinary drugs in cross-contaminated feedstuffs. Anal Bioanal Chem. 2015;407(24):7369–78.
Williams JP, Scrivens JH. Rapid accurate mass desorption electrospray ionisation tandem mass spectrometry of pharmaceutical samples. Rapid Commun Mass Spectrom. 2005;19(24):3643–50.
Krieger S, Hayen H, Schmitz OJ. Quantification of coumarin in cinnamon and woodruff beverages using DIP-APCI-MS and LC-MS. Anal Bioanal Chem. 2013;405(25):8337–45.
Myung S, Wiseman JM, Valentine SJ, Takats Z, Cooks RG, Clemmer DE. Coupling desorption electrospray ionization with ion mobility/mass spectrometry for analysis of protein structure: evidence for desorption of folded and denatured states. J Phys Chem B. 2006;110(10):5045–51.
D’Agostino PA, Chenier CL. Desorption electrospray ionization mass spectrometric analysis of organophosphorus chemical warfare agents using ion mobility and tandem mass spectrometry. Rapid Commun Mass Spectrom. 2010;24(11):1617–24.
Wu C, Ifa DR, Manicke NE, Cooks RG. Rapid, direct analysis of cholesterol by charge labeling in reactive desorption electrospray ionization. Anal Chem. 2009;81(18):7618–24.
Pan S, Tian Y, Li M, Zhao J, Zhu L, Zhang W, et al. Quantitative detection of nitric oxide in exhaled human breath by extractive electrospray ionization mass spectrometry. Sci Rep. 2015;5:8725.
Vaclavik L, Rosmus J, Popping B, Hajslova J. Rapid determination of melamine and cyanuric acid in milk powder using direct analysis in real time-time-of-flight mass spectrometry. J Chromatogr A. 2010;1217(25):4204–11.
Song L, Gibson SC, Bhandari D, Cook KD, Bartmess JE. Ionization mechanism of positive-ion direct analysis in real time: a transient microenvironment concept. Anal Chem. 2009;81(24):10080–8.
Chen H, Yang S, Wortmann A, Zenobi R. Neutral desorption sampling of living objects for rapid analysis by extractive electrospray ionization mass spectrometry. Angew Chem Int Ed. 2007;46(40):7591–4.
Jagerdeo E, Abdel-Rehim M. Screening of cocaine and its metabolites in human urine samples by direct analysis in real-time source coupled to time-of-flight mass spectrometry after online preconcentration utilizing microextraction by packed sorbent. J Am Soc Mass Spectrom. 2009;20(5):891–9.
Thunig J, Flø L, Pedersen-Bjergaard S, Hansen SH, Janfelt C. Liquid-phase microextraction and desorption electrospray ionization mass spectrometry for identification and quantification of basic drugs in human urine. Rapid Commun Mass Spectrom. 2012;26(2):133–40.
Haunschmidt M, Klampfl CW, Buchberger W, Hertsens R. Determination of organic UV filters in water by stir bar sorptive extraction and direct analysis in real-time mass spectrometry. Anal Bioanal Chem. 2010;397(1):269–75.
Figueiredo EC, Sanvido GB, Arruda MAZ, Eberlin MN. Molecularly imprinted polymers as analyte sequesters and selective surfaces for easy ambient sonic-spray ionization. Analyst. 2010;135(4):726–30.
Mirabelli MF, Wolf J-C, Zenobi R. Direct coupling of solid-phase microextraction with mass spectrometry: sub-pg/g sensitivity achieved using a dielectric barrier discharge ionization source. Anal Chem. 2016;88(14):7252–8.
Zou J, Talbot F, Tata A, Ermini L, Franjic K, Ventura M, et al. Ambient mass spectrometry imaging with picosecond infrared laser ablation electrospray ionization (PIR-LAESI). Anal Chem. 2015;87(24):12071–9.
Lee JK, Jansson ET, Nam HG, Zare RN. High-resolution live-cell imaging and analysis by laser desorption/ionization droplet delivery mass spectrometry. Anal Chem. 2016;88(10):5453–61.
Groseclose MR, Castellino S. A mimetic tissue model for the quantification of drug distributions by MALDI imaging mass spectrometry. Anal Chem. 2013;85(21):10099–106.
Eberlin LS, Norton I, Orringer D, Dunn IF, Liu X, Ide JL, et al. Ambient mass spectrometry for the intraoperative molecular diagnosis of human brain tumors. Proc Natl Acad Sci U S A. 2013;110(5):1611–6.
Feider CL, Elizondo N, Eberlin LS. Ambient ionization and FAIMS mass spectrometry for enhanced imaging of multiply charged molecular ions in biological tissues. Anal Chem. 2016;88(23):11533–41.
Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11(4):417.
Gundlach-Graham A, Burger M, Allner S, Schwarz G, Wang HAO, Gyr L, et al. High-speed, high-resolution, multielemental laser ablation-inductively coupled plasma-time-of-flight mass spectrometry imaging: part I. Instrumentation and two-dimensional imaging of geological samples. Anal Chem. 2015;87(16):8250–8.
Burger M, Gundlach-Graham A, Allner S, Schwarz G, Wang HAO, Gyr L, et al. High-speed, high-resolution, multielemental LA-ICP-TOFMS imaging: part II. Critical evaluation of quantitative three-dimensional imaging of major, minor, and trace elements in geological samples. Anal Chem. 2015;87(16):8259–67.
Mueller L, Herrmann AJ, Techritz S, Panne U, Jakubowski N. Quantitative characterization of single cells by use of immunocytochemistry combined with multiplex LA-ICP-MS. Anal Bioanal Chem. 2017;409(14):3667–76.
Herdering C, Wehe CA, Reifschneider O, Raj I, Ciarimboli G, Diebold K, et al. Laser ablation based bioimaging with simultaneous elemental and molecular mass spectrometry: towards spatially resolved speciation analysis. Rapid Commun Mass Spectrom. 2013;27(23):2588–94.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Shelley, J.T., Badal, S.P., Engelhard, C. et al. Ambient desorption/ionization mass spectrometry: evolution from rapid qualitative screening to accurate quantification tool. Anal Bioanal Chem 410, 4061–4076 (2018). https://doi.org/10.1007/s00216-018-1023-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-018-1023-9