
Abstract
A screening method for analyzing environmental waters contaminated with UV filters using direct analysis in real-time mass spectrometry (DART-MS) was developed. To demonstrate the suitability of DART-MS a test set of seven organic UV filters, namely benzophenone-3 (BP-3), ethylhexyl dimethyl p-aminobenzoate (OD-PABA), 4-t-butyl-4′-methoxydibenzoylmethane (BM-DBM), homomethyl salicylate (HMS), 2-(ethylhexyl) salicylate (EHS), octocrylene (OC), and 4-methylbenzylidene camphor (4-MBC), was defined. In the first step, standard solutions of the analytes prepared in methanol were investigated in order to determine optimum parameters for the DART-MS. Because of the very low concentrations of UV filters expected in environmental water samples, a pre-concentration step using stir bar sorptive extraction was performed. DART-MS allows the direct, simple and rapid semi-quantitative analysis of the analytes enriched on the surface of the polydimethylsiloxane-coated stir bars. The optimized method provided calibration curves with correlation coefficients R > 0.959, repeatability from 5% (for 4-MBC) to 30% (for BM-DBM) relative standard deviation and limits of detection lower than 40 ng L−1 for all analytes. Finally, real lake water samples from locations with typical leisure activities were analyzed. Results obtained with the developed DART-MS method were cross-checked by confirmatory analysis using thermodesorption gas chromatography mass spectrometry (TD-GC-MS). Thereby, it could be demonstrated that both analytical methods provide comparable concentrations for the UV filters in the lake water samples.

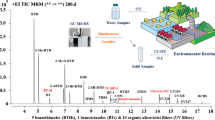
DART-MS measurement of UV filters enriched by stir bar sorptive extraction.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The rising awareness of the risk of direct exposure to sunlight has led to increased use of personal care products such as sunscreens containing UV filters. Sunscreens provide protection from UV radiation by producing a thin layer on the skin in which UV light is absorbed by organic compounds or scattered and reflected by inorganic micro particles. With the increasing use of sunscreens, their environmental impact has become an important issue. The sunscreen ingredients, which are applied to the skin, may enter the surface water directly (when released from the skin during water activities) or indirectly via wastewater treatment plants (when released during showering or washed from textiles).
Because of very low concentrations of UV filters in environmental water samples, pre-concentration has to be performed to allow a reliable analysis. Liquid–liquid extraction [1, 2], micellar-mediated extraction [3], membrane-assisted liquid–liquid extraction [4, 5], solid-phase extraction [6–9], solid-phase microextraction [10–13], and stir bar sorptive extraction (SBSE) [14, 15] are suitable techniques for pre-concentrating organic UV filters and analyzing them by GC-MS [16–18] or GC-MS/MS [12]. Besides, GC-MS also liquid chromatography coupled with UV detection [3, 8, 19–21] or with mass spectrometry (either with electrospray ionization [22] or with atmospheric pressure photo ionization [23]) is a suitable technique for analyzing this class of compounds.
Separation by HPLC or GC often takes more than 30 min depending on the complexity of the sample. A much more straightforward and less time-consuming technique is direct analysis in real-time mass spectrometry (DART-MS) [24]. The ionization is based on the interactions of long-lived electronic or vibronic excited-state atoms or molecules with the analyte and the atmospheric gases. It is suitable for the detection of gases, liquids, and solids. DART provides immediate information, a critical requirement for screening or high throughput. It has been successfully used for different classes of analytes including chemical agents, pharmaceuticals, explosives, and toxic industrial chemicals. A unique application is the direct detection of chemicals on surfaces without any sample preparation. Therefore, it seems interesting to investigate its applicability to the detection of analytes pre-concentrated by sorptive extraction techniques, using the polymeric or solid sorbent directly for the MS analysis.
In this paper, the suitability of DART-MS is demonstrated for UV filters in environmental water samples after pre-concentration on polydimethylsiloxane-coated stir bars and direct ionization, identification and semi-quantification without any further sample pre-treatment.
The reliability of the developed DART-MS method was confirmed by additional thermodesorption (TD) GC-MS experiments.
Experimental
Standard solutions and samples
The names and structures of the analytes included in this work are summarized in Table 1. All UV filters and the internal standard benzyl cinnamate were obtained from Merck (Darmstadt, Germany). Methanol, acetonitrile, dichloromethane, and acetone (all of chromatographic analysis grade) were purchased from JT Baker (Deventer, The Netherlands).
For stock solutions, 50 mg of each of UV filter were dissolved in 50 mL of methanol. A mixed standard solution of UV filters with a concentration of 1 µg mL−1 each was prepared in methanol. This solution was again diluted to the final working concentrations 12.5 µg L−1, 25.0 µg L−1, 50.0 µg L−1, 75.0 µg L−1, 125.0 µg L−1, 175.0 µg L−1, and 250.0 µg L−1 by using methanol as solvent, whereby an appropriate volume of a stock solution of the internal standard benzyl cinnamate (BC) was added to obtain a concentration of 100.0 µg L−1.
For preparation of spiked water samples, 1 mL of each working standard was added to 250 mL water, thereby obtaining samples with a final concentration of 50, 100, 200, 300, 500, 700, or 1,000 ng L−1 of each UV filter and 400 ng L−1 of BC, respectively. All standard solutions were prepared by using glassware only, which was washed with acetone, methanol, and deionized water before use.
Lake water from locations with typical leisure activities was collected in brown bottles, which were pre-cleaned with acetone and methanol. Directly after sampling 0.5% acetonitrile was added to stabilize the samples. The water samples were stored at 4 °C in a refrigerator. Before adding the internal standard and performing the SBSE, the water samples were filtrated through a 0.45 µm membrane filter. Finally, 1 mL internal standard (concentration of 100 µg L−1) was added to the lake water sample.
Instrumentation
DART-MS conditions
Measurements were performed with a DART ion source from IonSense, Inc. (Saugus, MA, USA) coupled to a JMS T100 (AccuTOF-LC-plus) time-of-flight mass spectrometer (JEOL Ltd, Tokyo, Japan). The following voltages were applied to the ion source: orifice 1, 10 V; orifice 2, 5 V; ring electrode, 10 V. Orifice 1 was operated at a temperature of 120 °C and the ion guide potential was set to 600 V. For operation of the DART source, helium gas (4.6) was employed, whereas nitrogen gas was used in the standby mode.
Tuning of the DART was performed by positioning the DART orifice and the MS orifice in a way that the highest signal intensity for water-clusters (m/z 37.0290) was obtained. Mass calibration was carried out by using polyethylene glycol 600 (PEG) from Baker (Deventer, The Netherlands). For this purpose, a Dip-IT glass rod (IonSense), was dipped into a solution of 50 µl of PEG dissolved in 10 mL methanol/dichloromethane (50:50; v/v) and placed into the DART source. Additionally, internal mass drift compensation was carried out, employing bis(ethylhexyl phthalate) (m/z 391.2848) which generally occurs in a common background spectrum in the positive ion mode, to get better mass accuracy. Data acquisition was performed in a range from 200 to 400 m/z for all analytes. An in-lab modified holder, providing a much more rigid guidance of the glass rod compared to the design by IonSense that allowed too much play (resulting in fluctuation of signal intensities), was used to obtain reproducible positioning. The Twister stir bars (see below) were inserted manually into the DART source by using tweezers.
TD-GC-MS conditions
The instrumentation consisted of a 6890N gas chromatograph coupled to a 5973 inert XL MSD mass spectrometer from Agilent (Waldbronn, Germany). The thermodesorption device used as inlet system during this work was a Gerstel TDS 2 system (Gerstel, Mühlheim, Germany) equipped with a Gerstel TDS A autosampler and a Gerstel Cooled Injection System (CIS) 4.
The program for thermodesorption started with an initial temperature of 30 °C which was increased to 275 °C at a rate of 60 °C s−1 and held for 10 min. The desorbed compounds were cryofocused at −100 °C. After the desorption, the temperature of the CIS increased from −100 °C to 280 °C (held for 10 min) at 12 °C s−1. The injection was performed in the solvent vent mode. Separation was carried out on a HP-5ms fused silica column (30 m × 0.25 mm i.d.; 0.25 µm film thickness, Agilent) in constant flow. The oven program started at 70 °C (held for 2 min), and the temperature was increased in the first step to 160 °C at 20 °C min−1 and in the second step to 280 °C (held for 10 min) at 8 °C min−1. Helium (4.6) was used as carrier gas with a column flow rate of 1 mL min−1. The mass spectrometer was first operated in the scan mode and subsequently the following ions (in each case, the numbers in italics indicate the m/z used for quantitative analysis and the second m/z ratio was used as qualitative criterion by calculating the ratio of intensities of the two ions) were monitored in the selected ion monitoring mode: 120, 138 for EHS; 120, 261 for HMS; 151, 227 for BP-3; 211, 254 for 4-MBC; 131, 238 for BC; 165, 277 for OD-PABA; 249, 360 for OC.
Stir bar sorptive extraction
For the extraction, Twister stir bars from Gerstel of 2 cm length coated with a 0.5-mm layer of polydimethylsiloxane (PDMS) were employed for all measurements. Before use, the coated stir bars were placed into a cleaning solution containing methanol and dichloromethane (50:50, v/v) for 1 h. Afterwards, the stir bars were put into thermodesorption glass tubes and were conditioned for 1 h at 300 °C under nitrogen flow in a Gerstel tube conditioner TC 2. After the cleaning procedure the Twister stir bars can be directly immersed into the water sample. After a defined stirring time (1, 4, 6, or 20 h; 1,050 rpm), the stir bar was removed from the water sample, dried with lint-free cloth, and directly used for DART-MS or for thermodesorption gas chromatography mass spectrometry (TD-GC-MS) measurement.
Results and discussion
Analysis of standard solutions
In a first step, a test set of seven commonly employed UV filters was selected and standard solutions for each compound were prepared. These solutions were employed for the optimization of DART parameters. A Dip-IT glass rod was dipped into the sample solution and subsequently positioned in the helium gas stream of the DART for acquisition of mass spectra. The following parameters were investigated: temperature of the gas heater on the ceramic tube, 50–450 °C (in 50 °C-steps); the helium gas flow; the needle voltage, 2,000–4,000 V (in 500 V-steps); the discharge electrode and grid electrode voltages, 50–300 V (in 50 V-steps). The heater temperature and the gas flow rate are generally considered as crucial parameters with respect to the desolvation and the vaporization of the analyte molecules. Due to the fact that high temperatures may lead to pyrolysis of the sample, the signal intensity decreases upon exceeding a certain temperature. Furthermore, too high gas flow rates result in a decrease of the signal intensity simply because of physically blowing away the liquid sample from the glass rod, so the analyte molecules collide with the sprayer shield instead of entering the orifice of the MS. Best results were obtained with the following settings: needle voltage 3,500 V, discharge electrode and grid electrode voltages 100 and 50 V, respectively. The optimum heater temperature varied from 150 to 250 °C for the different analytes, so a compromise temperature of 200 °C was chosen. Under these conditions, all analytes were detected as [M+H]+. A list of the UV filters together with measured exact masses in the positive ion mode is provided in Table 2.
After optimization of the DART-MS parameters, the possibility of direct analysis of the UV filters enriched on Twisters stir bars was checked. Therefore, SBSE of spiked deionized water (with UV filter concentrations of 700 ng L−1 each and an internal standard concentration of 400 ng L−1) was performed. All tested solutes could be ionized directly from the stir bar and therefore determined with DART-MS (a spectrum is depicted in Fig. 1). It might be of interest that, maybe due to the short contact time of the heated He gas stream and the stir bars, no PDMS degradation products were observed in the MS spectrum. In the next step, spiked lake water samples were investigated. Despite the more complex matrix, no interferences were observed in the DART mass spectrum.

DART-MS spectrum of deionized water spiked with a mixture containing seven UV filters (each 700 ng L−1) and the internal standard (400 ng L−1); conditions are given in the text of the experimental part; peaks: 1 BP-3, 2 OD-PABA, 3 BM-DBM, 4 HMS, 5 EHS, 6 OC, 7 4-MBC, 8 BC
An important parameter in the whole procedure is the dependence of extraction recovery upon time. Therefore, the effect of stirring time on both methods (DART-MS and TD-GC-MS) was investigated. Using TD-GC-MS, the signal intensity increased upon increasing the stirring time until equilibrium between the two phases (water sample and PDMS) was reached, which was achieved by stirring the sample for 20 h. For DART-MS, the situation is different. Increasing the stirring time from 1 to 6 h resulted in increasing signal intensities, but longer extraction time led to a decrease of signal intensity. This effect can be explained by diffusion phenomena of the analytes within the stir bar. In the early stage of the extraction process, the analytes are enriched near the surface of the PDMS coating. Later, the analyte molecules diffuse into the inner of the PDMS coating. Thereby, the total amount of analyte in the stir bar may still increase although the amount near the surface may actually be decreased. DART-MS can only detect those species near the surface so that contrary to TD-GC-MS long extraction times do not provide any further improvements.
To increase the extraction efficiency, the addition of sodium chloride (10%) was tested. It is worth noting that no improvements were observed with respect to the signal intensity for all tested analytes. Additionally, different pH values (pH 2, pH 7, pH 10) were investigated with optimum results with respect to average signal intensities obtained at pH 7. Recoveries after 4 h stirring time (for DART-MS) were between 18% (BP-3) and 67% (EHS) and between 50% (BP-3) and 100% (OC) after 20 h.
Method performance
The unique combination of SBSE and DART-MS is a useful method for screening of pollution in water by UV filters. Even more, semi-quantitative results can be obtained by this technique. With respect to reproducibility and quantification, the position of the Twister in the DART source is one of the most crucial points. Due to the fact that no autosampler was available, the positioning of the Twister in the ion source was not fully reproducible. Therefore, only a semi-quantitative analysis was possible, but it still made fast preliminary information about the approximate concentration of a certain analyte available.
For the investigation of the DART-MS method performance, the optimized parameters (250 mL sample stirred for 4 h at 1,050 rpm) for extraction together with the optimized DART-MS parameter mentioned above were used. For calibration the standard solutions with concentrations of 50, 100, 200, 300, 700, or 1000 ng L−1 UV filter, and 400 ng L−1 internal standard were employed. The calibration curves were determined by plotting analyte signal intensities, relative to the signal obtained for the internal standard (to minimize the influence of the stir bar quality), versus concentration. Calibration curves with correlation coefficients of R > 0.959 were achieved which seems to be sufficient for semi-quantitative analysis. The limits of detection for DART-MS measurements (from extracted mass traces by taking the signal equal or better 3× noise) were estimated from a spiked deionized water sample (with a concentration of 50 ng L−1). BP-3, EHS, OD-PABA, and 4-MBC were detectable above 20 ng L−1, whereas HMS, BM-DBM, and OC only could be detected above 40 ng L−1. Linearity was checked by plotting signal intensities/concentration versus concentration and evaluating the resulting graphs. For DART-MS, the linear range was between 50 (4-MBC, OD-PABA, OC, BM-DBM) or 100, and 1000 ng L−1. The repeatability was measured for a water sample spiked with 500 ng L−1 and ranged from 5% (for 4-MBC) to 30% (for BM-DBM) relative standard deviation.
TD-GC-MS measurements (based on a method developed recently by our group [25]) were performed to confirm the results of DART-MS. For calibration of TD-GC-MS, the same standard solutions as for the DART-MS calibration were employed. As mentioned above, maximum signal intensity was achieved after extraction for 20 h. If sensitivity can be sacrificed to some extent, a shorter extraction time may be possible. Nevertheless, repeatability of measurements after 4 h was poorer than for samples after extraction for 20 h. Correlation coefficients for calibration were between 0.979 and 0.999 for the different analytes after 4 h extraction, and between 0.988 and 0.999 after 20 h extraction (peak areas of analytes were divided by the peak area of the internal standard). The limits of detection ranged from 0.28 (for OC) to 4.3 ng L−1 (for 4-MBC) and the repeatability from 4% (for BP-3) to 11% (for OC) at a level of 50 ng L−1(n = 10) The only exception herein was BM-DBM showing a much higher detection limit in comparison to the other test compounds. Therefore, no calibration was performed for this analyte.
Analysis of lake water samples
Finally, lake water was analyzed from locations where contamination of the water by UV filters could be expected. A typical DART spectrum of an unspiked water sample measured with DART-MS is depicted in Fig. 2. As can be seen from this figure, two analytes (BP-3 and OC) were detected in the lake water. The same sample was also analyzed with TD-GC-MS, whereby five substances were found. A chromatogram is shown in Fig. 3. Three of the five analytes, namely EHS, 4-MBC, and OD-PABA could not be detected by DART-MS, because of too low concentrations (below 10 ng L−1). The concentration of BP-3 (40 ng L−1) and OC (1,400 ng L−1) measured with DART-MS are in the same order of magnitude compared to the results obtain with TD-GC-MS (32 ng L−1 BP-3 and 1,710 ng L−1 OC). OC is the UV filter which was found to be present in the highest concentration of all detected substances. The determined levels are rather similar to those reported in the literature for environmental water samples [4].

DART-MS spectrum of a real lake water sample taken during the summer season. For conditions, see Fig. 1; peaks: 1 BP-3, 6 OC, 8 BC

TD-GC-MS chromatogram of the lake water sample shown in Fig. 2. Conditions are given in the text of the experimental part; peaks: 1 BP-3, 4 HMS, 5 EHS, 6 OC, 7 4-MBC, 8 BC
Conclusion
In the present paper, it could be demonstrated that DART-MS is suited for direct measurements of UV filter traces enriched on PDMS-coated stir bars. Thereby, an attractive complement to chromatographic methods is provided that allows fast screening of environmental water samples contaminated by UV filter. DART-MS cannot fully replace the more time-consuming TD-GC-MS method since only semi-quantitative results are obtained and detection limits are poorer by one to two orders of magnitude depending on the analyte. Nevertheless, DART-MS after stir bar sorptive extraction is appropriate for the detection of UV filters in the mid nanogram per liter range which corresponds to levels typically found in surface water affected by swimming activities. Preliminary screening by DART-MS can help to substantially reduce the number of samples that have to be analyzed by TD-GC-MS.
References
Jeon HK, Chung Y, Ryu JC (2006) J Chromatogr A 1131:192–202
Kupper T, Plagellat C, Braendli RC, de Alencastro LF, Grandjean D, Tarradellas J (2006) Water Res 40:2603–2612
Giokas DL, Sakkas VA, Albanis TA, Lambropoulou DA (2005) J Chromatgr A 1077:19–27
Rodil R, Schrader S, Moeder M (2009) J Chromatogr A 1216:4887–4894
Moeder M, Lange F (2007) LC-GC Europe 20:97–103
Balmer ME, Buser HR, Müller MD, Poiger T (2005) Environ Sci Technol 39:953–962
Poiger T, Buser HR, Balmer ME, Bergqvist PA, Müller MD (2004) Chemosphere 55:951–963
Giokas DL, Sakkas VA, Albanis TA (2004) J Chromatogr A 1026:289–293
Cuderman P, Heath E (2007) Anal Bioanal Chem 387:1343–1350
Lambropoulou DA, Giokas DL, Sakkas VA, Albanis TA, Karayannis MI (2002) J Chromatogr A 967:243–253
Sakkas VA, Giokas DL, Lambropoulou DA, Albanis TA (2003) J Chromatogr A 1016:211–222
Negreira N, Rodríguez I, Ramil M, Rubí E, Cela R (2009) Anal Chim Acta 638:36–44
Felix T, Hall BJ, Brodbelt JS (1998) Anal Chim Acta 371:195–203
Kawaguchi M, Ito R, Endo N, Sakui N, Okanouchi N, Saito K, Sato N, Shiozaki T, Nakazawa H (2006) Anal Chim Acta 557:272–277
Rodil R, Moeder M (2008) J Chromatogr A 1179:81–86
Giokas DL, Salvador A, Chisvert A (2007) Trends Anal Chem 26:360–374
Peck AM (2006) Anal Bioanal Chem 386:907–939
Díaz-Cruz MS, Llorca M, Barceló D (2008) Trends Anal Chem 27:873–887
Chisvert A, Pascual-Martí M-C, Salvador A (2001) Fresenius J Anal Chem 369:638–641
Salvador A, Chisvert A (2005) Anal Chim Acta 537:1–14
Salvador A, Chisvert A (2005) Anal Chim Acta 537:15–24
Rodil R, Quintana JB, Lopez-Mahia P, Muniategui-Lorenzo S, Prada-Rodriguez D (2008) Anal Chem 80:1307–1315
Rodil R, Schrader S, Moeder M (2009) Rapid Commun Mass Spectrom 23:580–588
Cody RB, Laramée JA, Durst HD (2005) Anal Chem 77:2297–2302
Zaborsky P (2006) Doctoral Thesis, Johannes-Kepler-University Linz (Austria)
Acknowledgment
The authors gratefully acknowledge JEOL (Europe) BV, Zaventem, Belgium for providing the DART-AccuTOF.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haunschmidt, M., Klampfl, C.W., Buchberger, W. et al. Determination of organic UV filters in water by stir bar sorptive extraction and direct analysis in real-time mass spectrometry. Anal Bioanal Chem 397, 269–275 (2010). https://doi.org/10.1007/s00216-009-3438-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3438-9