
Abstract
A novel ultrasound-air-assisted demulsified liquid–liquid microextraction by solidification of a floating organic droplet (UAAD-LLM-SFO) followed by HPLC-UV detection was developed for the analysis of three antifungal drugs in water and biological samples. In this method, 1-dodecanol was used as the extraction solvent. The emulsion was rapidly formed by pulling in and pushing out the mixture of sample solution and extraction solvent for 5 times repeatedly using a 10-mL glass syringe while sonication was performed. Therefore, an organic dispersive solvent required in common microextraction methods was not used in the proposed method. After dispersing, an aliquot of acetonitrile was introduced as a demulsifier solvent into the sample solution to separate two phases. Therefore, some additional steps, such as the centrifugation, ultrasonication, or agitation of the sample solution, are not needed. Parameters influencing the extraction recovery were investigated. The proposed method showed a good linearity for the three antifungal drugs studied with the correlation coefficients (R 2 > 0.9995). The limits of detection (LODs) and the limits of the quantification (LOQs) were between 0.01–0.03 μg L−1 and 0.03–0.08 μg L−1, respectively. The preconcentration factors (PFs) were in the range of 107–116, respectively. The precisions, as the relative standard deviations (RSDs) (n = 5), for inter-day and intra-day analysis were in the range of 2.1–4.5% and 6.5–8.5%, respectively. This method was successfully applied to determine the three antifungal drugs in tap water and biological samples. The recoveries of antifungal drugs in these samples were 92.4–98.5%.

Ultrasound-air-assisted demulsified liquid–liquid microextraction by solidification of a floating organic droplet for the analysis of three antifungal drugs prior HPLC-UV
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Antifungal drugs are widely used in different pharmaceutical dosage forms to prevent the conversion of lanosterol to ergosterol as the main sterol of the fungal cell [1, 2]. Among the agents used, the imidazole group drugs (azole antifungals) have a wide range of actions [3, 4]. These groups of antifungal compounds such as clotrimazole (CT), miconazole (MC), and ketoconazole (KC) contain aromatic five-membered imidazole ring structures with two nitrogen atoms [1]. Due to the extensive usage and high emissions of relatively small absorption via the skin [5], the active ingredients can appear as drug residues in the aquatic environment [6]. There is a matter of public concern about their environmental impact because of possible negative effects [7, 8]. In order to know the occurrence of azole substances, an analytical method is required. Various chromatographic methods have been reported to be used for the determination of CT, KC, and MC level including high-performance liquid chromatography (HPLC) [9–12], capillary zone electrophoresis chromatography [13], UV-visible spectrophotometry [14], spectrofluorimetry [15], and high-performance thin-layer chromatography (HPTLC) [16]. However, due to pharmaceutical matrices, some sample preparation methods are still required. Different methods based on microextraction have been developed to overcome the limitations of the sample preparations such as the use of large volumes of organic solvents and long extraction times. In 2006, a novel microextraction method termed as dispersive liquid–liquid microextraction (DLLME) was developed [17]. DLLME is based on the ternary solvent system including a sample solution, a water immiscible solvent (extraction solvent), and a water miscible solvent (dispersive solvent). In this method, analytes are extracted into the fine droplets of the extractant formed by the disperser solvent [17]. The main limitation of DLLME is the use of hazardous solvents with high density. In addition, the use of high-density solvents as extractants limits a wider applicability of DLLME due to more limited choices since there are more low-density [18]. A novel DLLME method based on the solidification of floating organic droplets (DLLME-SFO) has been introduced [19]. In this proposed method, a low-density and low-toxic solvent with a melting point around room temperature was used as extraction solvent. However, special devices are required to easily collect the floated extraction phase after centrifugation [20, 21]. In addition, most DLLME methods need a centrifugation step for the phase separation. This time-consuming procedure makes the microextraction techniques difficult to be automated. Moreover, it is difficult to handle large volume centrifugation [22]. To overcome the mentioned limitations, a new method called the low-density solvent-based de-emulsification (SD-DLLME) has been introduced [23, 24]. In this method, the extraction process is finished with the addition of another portion of dispersive solvent as demulsifier into the solution to break up the emulsion without centrifugation [25]. Thus, the extraction time is decreased by the fact that no centrifugation is used for the phase separation [22, 25]. However, in the methods based on DLLME, polarity of the aqueous phase may decrease due to the use of a dispersive solvent that leads to a decrease in extraction efficiency [21]. Air-assisted liquid–liquid microextraction (AALLME) has been developed as disperser solvent-free LPME methods [26]. In this method, the extraction solvent is dispersed into the sample solution with withdrawing the sample into a syringe and pushing it out into the tube. By this action, fine organic droplets are formed without using a disperser solvent. In the presented work, a novel UAAD-LLM-SFO method was for the first time applied for the determination of three antidrugs. In this method, the low-density solvent, 1-dodecanol, was dispersed with AALLME. The ultrasound irradiation enhanced the rapid formation of fine droplets of the extractant in the sample solution, and the contact surface between both immiscible liquids was significantly enlarged. After a few minutes extraction, an aliquot of demulsifier solvent was injected into the solution to break the emulsion. Thus, the phase separation was performed without centrifugation. Then, the test tube was transferred into a beaker containing ice and the organic solvent was solidified after 2 min. The upper layer was collected and analyzed with HPLC-UV. It is the first time that air assistance has been used in combination with demulsified LLME for the determination of three antifungal drugs.
Experimental
Chemicals
The three antifungal drugs (clotrimazole (CZ), ketoconazole (KZ), and miconazole (MZ)) were purchased from the Department of Pharmaceutics of Tehran University (Tehran, Iran). HPLC-grade acetonitrile, sodium acetate, sodium chloride, 1-undecanol, hexadecane, and 1-dodecanol were obtained from Merck (Darmstadt, Germany). The stock standard solutions (100 mg L−1) were prepared by dissolving an appropriate amount of each standard in methanol. Working standard solutions were prepared by diluting the stock solution with ultra-pure water to the appropriate concentrations.
Instrumentation
The separation and analysis of the three antifungal drugs was performed by Younglin YL9100 HPLC instrument from Younglin Company (Korea) consisting of a YL9110 pump, a YL9120 UV-Vis detector, a Rheodyne 7725i (PerkinElmer, USA) injector, along with a 20-μL sample loop. The Clarity program for LC was used to perform data processing. A capital HPLC column (Scotland, UK) ODS-H C18 (150 mm × 4.6 mm, id. 5 μm) was employed for all separations. The mobile phase was a mixture of sodium acetate buffer (0.01 M, pH = 4.0) and acetonitrile (10:90, v/v) running at 1 mL min−1 in the isocratic mode with the detector’s wavelength set at 212 nm.
A Universal 320R centrifuge equipped with a swing out rotor (6-place, 5000 rpm, Cat. No. 1628A) was obtained from Hettich (Kirchlengern, Germany). The measurements of pH values were made with a pH-meter (model 692, Herisau, Switzerland) with a glass combined electrode.
Sample preparation
The biological samples containing the urine samples and the plasma samples (obtained from the Iranian Blood Transfusion Organization (Tehran, Iran)) were kept in glass tubes at 4 °C in the fridge.
In order to precipitate the proteins of the plasma samples, to 1 mL of human plasma, 0.5 ml of zinc sulfate solution (0.7 mol L−1) and 0.1 mL of 1 mol L−1 sodium hydroxide solution was added. The mixed solution was centrifuged for 10 min at 2000 rpm. The supernatants were transferred into another vial and diluted to 10 mL extraction solvent.
A urine sample was collected from a healthy volunteer. Before the extraction process, the sample was diluted 1:1 with ultra-pure water and its pH was adjusted to the optimum value by drop wise addition of a NaOH solution (2 mol L−1).
Tap water samples were collected from drinking water (Tehran, Iran) and 10 mL of sample was analyzed as soon as possible after sampling according to the recommended procedure.
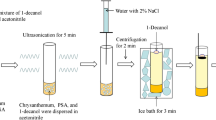
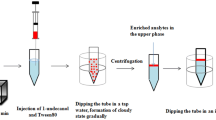
UAAD-LLM-SFO procedure
An aliquot of 10 mL of sample solution containing 100 μg L−1 of each antifungal drug was placed in a 15-mL screw cap glass tube with conical bottom. After adjusting the pH to 10, 100 μL of the extraction solvent (1-dodecanol) was rapidly injected and then the mixture pulled in and pushed out by a 10-mL glass syringe into a sample solution for 5 times while sonication was used for 30 s. Due to the dispersion of fine droplets of 1-dodecanol in the aqueous sample, analytes were extracted into the fine droplets in a few seconds. After a setting time, 200 μL of acetonitrile as demulsifier solvent was injected into the solution to break down the emulsion. The mixture cleared and separated into two phases. The test tube was transferred into a beaker containing ice and 1-dodecanol was solidified after 2 min (80 ± 5) μL. Afterwards, the solidified solvent was collected into a conical vial, and it melted immediately. Finally, 20 μL of the extraction solvent was directly injected to HPLC for analysis.
Results and discussion
In order to obtain a high extraction recovery and good precision of the UAAD-LLM-SFO procedure, the effect of different extraction parameters including the type and volume of extraction solvent, the type and volume of de-emulsifier solvent, and pH and extraction times was studied in terms of the extraction recovery of analytes.
The selection of extraction solvent
A crucial step is the choice of extraction solvent. It should have some characteristics such as high extraction affinity to the analytes, low solubility in the aqueous solution, low toxicity, low melting point close to room temperature, and lower density than water. Based on these facts, three organic solvents including 1-undecanol, 1-dodecanol, and hexadecane were investigated. A series of experiments were performed using different volumes of extraction solvents based on their solubility in aqueous solution and 200 μL acetonitrile as demulsification solvent. As it can be seen in Fig. 1, 1-dodecanol was found to give the highest extraction recoveries for all analytes.

Effect of type of extraction solvent on the extraction recovery of analytes. Conditions: sample volume, 10 mL; de-emulsifier solvent, acetonitrile 200 μL; pH 10; 100 μg L−1 clotrimazole, miconazole, and ketoconazole, n = 5
The effect of the extraction solvent volume on the extraction recoveries of three antifungal drugs was studied in the range of 50–150 μL. According to the results obtained in Fig. 2, the extraction recoveries of the drugs increased with the increase of extraction solvent volume from 50 to 100 μL and then decreased. This may be caused by the dilution effect when a larger volume of extraction solvent was used. Also, the volume of extraction solvent lower than 50 μL was difficult to collect in the extraction phase. Therefore, to achieve higher preconcentration factors and extraction recoveries, 100 μL of the extraction solvent was used as the optimal volume in subsequent experiments. The volume of the extraction phase was 80 ± 5 μL after extraction.

Effect of the extraction solvent volume on the extraction recoveries of analytes. Conditions: sample volume, 10 mL; de-emulsifier solvent, acetonitrile 200 μL; pH 10; 100 μg L−1 clotrimazole, miconazole, and ketoconazole, n = 5
Effect of simultaneous sonication and number of extraction times
It is necessary that the extraction solvent is dispersed as very fine droplets into the aqueous sample to achieve very high amounts of contact area and accelerate the extraction of analytes into the extraction solvent. In UAAD-LLM-SFO, the extraction solvent was dispersed into the sample solution without using a disperser solvent.
The use of ultrasound irradiation in the UAAD-LLM-SFO method can lead to the rapid formation of fine droplets of the extractant in the aqueous solution, and enhance the interfaces between 1-dodecanole and the aqueous sample, which causes a significant increase in the analytes mass transfer into the extractant. In the UAAD-LLM-SFO method, the extraction times were defined as the number of the operations that the mixture of extraction solvent and sample solution was pulled into a 10-mL glass syringe and then pushed out into the test tube. The effect of the number of extraction times on the extraction recoveries of analytes was examined in the range of 1–12 times, while the syringe and test tube had been immersed in an ultrasound bath. The results obtained showed that after 5 times in 30 s of the sonication (∼6 s for each time), the extraction recoveries remained constant.
Effect of the pH
The pH values of the sample can influence the ratio of ionic form to molecular form of the weak acid or weak alkali analytes. For weakly basic drugs such as the three antifungal drugs, the sample solution must be alkaline to convert the analytes in their neutral form and consequently reduce their solubility in the sample and enhance their transfer to the organic solvent. The effect of pH on the extraction recoveries for the three antifungal drugs was studied in the range of 3–12 times. As shown in Fig. 3, the analytes were ionized at pH values lower than the optimum value, so the extraction recoveries decreased. Therefore, pH values of the solutions were adjusted to 10 for subsequent experiments.

Effect of pH on the extraction recovery of analytes. Conditions: sample volume, 10 mL; de-emulsifier solvent, acetonitrile 200 μL; extraction solvent (1-dodecanol) volume, 100 μL; 100 μg L−1 clotrimazole, miconazole, and ketoconazole, n = 5
Investigation of type and volume of de-emulsifier solvent
The emulsion breaking process and the phase separation without centrifugation were performed with using a demulsifier. The solvent used as demulsifier should be able to participate into both extraction and aqueous phases, and have low surface tension and high surface activity [25]. Therefore, different solvents containing methanol, acetonitrile, ethanol, and acetone were studied. The results showed (Fig. 4) that acetonitrile has the highest extraction recoveries compared to the other solvents. Therefore, acetonitrile was adopted as the de-emulsifier solvent. Volumes of acetonitrile were varied between 100 and 1000 μL. According to the obtained results, 200 μL acetonitrile solvent was applied in the subsequent experiments.

Effect of the type of de-emulsifier solvent on the extraction recovery of analytes. Conditions: sample volume, 10 mL; extraction solvent volume (1-dodecanol), 100 μL; pH 10; 100 μg L−1 clotrimazole, miconazole, and ketoconazole, n = 5
Analytical performance
The analytical performance of the proposed method was investigated in the terms of limits of detection (LODs), limits of quantification (LOQs), linearity, the correlation of determination (R 2), repeatability, preconcentration factors (PFs), and extraction recoveries (ERs). The obtained results are summarized in Table 1. The calibration curves of the method were evaluated for the extraction of three antifungal drugs with a series of concentrations in the range of 0.03–500 μg L−1 for ketoconazole and clotrimazole and 0.08–500 μg L−1 for miconazole. Correlation coefficients were R 2 ≥ 0.9995 for the three drugs, thus confirming the linearity of the method. LODs and LOQs, calculated as three and ten times the standard deviation of the blank signal, were in the range of 0.01–0.03 μg L−1 and 0.03–0.08 μg L−1, respectively. Precision of the method (RSD%) was determined by spiking samples at a concentration level of 100 μg L−1 at the same day and at different days. Relative standard deviations (RSDs) were founded between 2.1 and 4.5% for intra-day (n = 6) and 6.5 and 8.5% for inter-days precision (n = 6), respectively.
PF was defined as the ratio of the concentration of analytes in the extraction solvent (c org) and the initial concentration of the analytes (c aq) in the sample by Eq. (1). C org was obtained from a calibration curve by direct injection of drugs standard solution in the extraction solvent.
ER was calculated by Eq. (2),
where V org and V aq are the volume of the organic extractant and the volume of the aqueous sample solution, respectively. Based on the mentioned equations, PFs and ERs for the three drugs were obtained in the range of 107–116% and 85.65–92.82%, respectively.
A comparison of the proposed method with other reported methods for preconcentration and determination of clotrimazole, ketoconazole, and miconazole was presented in Table 2. Its simplicity and high sensitivity with acceptable extraction recoveries made this method more efficient for determination of the analytes. The proposed method required a short period of time (<3 min) to achieve full extraction due to the large surface area between the fine droplets of the extraction solvent and the aqueous sample.
Analysis of real samples
The performance of the presented method was applied with various real samples such as tap water, blood, and urine samples under the optimum conditions (Table 3). The selected samples were examined to be free of antifungal drugs. Thus, plasma, urine, and water samples were spiked with the analytes at different concentration levels 10 and 50 μg L−1.
Relative recovery (RR) was calculated by Eq. (3)
where C found s the concentration of the analytes after spiking a known amount of standard to the real samples, C real is the concentration in the sample prior to spiking, and C added is a concentration of standard solution that was spiked in the real samples. The RR’s percent for the spiked real samples was in the range of 92.4 and 98.5% and the RSDs were between 2.3 and 6.1%. The results of three replicate analyses of each real sample showed that the proposed method was in good agreement with the spiking amounts. The error percentages (E%) as accuracy of the method for the analytes in the range of −1.5 to −7.2% in different real samples presented that the proposed method offers acceptable accuracy even in complicated matrices.
Chromatograms obtained of the standard mixed solution of three anti-fungal drugs non-spiked, 20 µg L−1 spiked tap water and 10 and 20 µg L−1 urine samples after extraction under optimal conditions were shown in Fig. 5. There were no interferences observed in the region of interest where the analytes were eluted.

HPLC-UV chromatograms of the standard mixed solution of three antifungal drugs (5 mg L−1) (1) ketoconazole, (2) clotrimazole, and (3) miconazole (A); drugs extracted from spiked 20 μg L−1 tap water (B); drugs extracted from 20 μg L−1 spiked urine samples (C), 10 μg L−1 spiked urine samples (D); and non-spiked tap water sample (E)
Conclusion
A novel ultrasound-air-assisted demulsified liquid–liquid microextraction by solidification of a floating organic droplet coupled to HPLC-UV was developed for the analysis of the three antifungal drugs in water and biological samples. Air-assisted dispersion was applied as an alternative approach to disperse the extraction solvent as the fine droplets into the aqueous sample without the need of organic dispersive solvent. Therefore, the use of larger amounts of the dispersive solvent compared to conventional DLLME was eliminated. The use of 1-dodecanol as the extraction solvent and acetonitrile as demulsifier in the proposed method is advantageous since these solvents are less toxic than the solvent used in DLLME. Ultrasound was used to accelerate the formation of the fine droplets. In addition, extraction time was reduced by the fact that no centrifugation was required for phase separation. This method also proposed a low detection limit, simple operation, good precision, and accuracy. Finally, a high extraction recovery and a high preconcentration factor were obtained in less than 3 min.
References
Moradi M, Yamini Y, Vatanara A, Saleh A, Hojati M, Seidi S. Monitoring of trace amounts of some anti-fungal drugs in biological fluids by hollow fiber based liquid phase microextraction followed by high performance liquid chromatography. Anal Methods. 2010;2:387–92.
Ebrahimpour B, Yamini Y, Esrafili A. Extraction of azole antifungal drugs from milk and biological fluids using a new hollow fiber liquid-phase microextraction and analysis by GC-FID. Chromatographia. 2011;74:281–9.
Beggs WH. Mycopathologia. Protonation of ketoconazole in relation to fungistatic activity. 1991; 116:3–4.
Zarn JA, Bruschweiler BJ, Schlatter JR. Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14 alpha-demethylase and aromatase. Environ Health Perspect. 2003;111:255–61.
Letzel M, Metzner G, Letzel T. Exposure assessment of the pharmaceutical diclofenac based on long-term measurements of the aquatic input. Environ Int. 2009;35:363–8.
Huanga Q, Yua Y, Tanga C, Peng X. Determination of commonly used azole antifungals in various waters and sewage sludge using ultra-high performance liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2010;1217:3481–8.
Sagristà E, Larsson E, Ezoddin M, Hidalgo M, Salvadó V, Jönsson JÅ. Determination of non-steroidal anti-inflammatory drugs in sewage sludge by direct hollow fiber supported liquid membrane extraction and liquid chromatography–mass spectrometry. J Chromatogr A. 2010;1217:6153–8.
Larsson N, Petersson E, Rylander M, Jönsson JÅ. Continuous flow hollow fiber liquid-phase microextraction and monitoring of NSAID pharmaceuticals in a sewage treatment plant effluent. Anal Methods. 2009;1:59–67.
Nguyet ANM, Tallieu L, Plaizier-Vercammen J, Massart DL, Heyden YV. Validation of an HPLC method on short columns to assay ketoconazole and formaldehyde in shampoo. J Pharm Biomed Anal. 2003;32:1–19.
de Bruijn P, Kehrer DFS, Verweij J, Sparreboom A. Liquid chromatographic determination of ketoconazole, a potent inhibitor of CYP3A4-mediated metabolism. J Chromatogr B Biomed Sci Appl. 2005;753:395–400.
Kedor-Hackmann ERM, Santoro MIRM, Singh AK, Peraro AC. First-derivative ultraviolet spectrophotometric and high performance liquid chromatographic determination of ketoconazole in pharmaceutical emulsions. Rev Bras Cienc Farm. 2006;42:91–8.
Mousa BA, El-Kousy NM, El-Bagary RI, Mohamed NG. Stability indicating methods for the determination of some anti-fungal agents using densitometric and RP-HPLC methods. Chem Pharm Bull. 2008;56:143–9.
Crego AL, Marina ML, Lavandera JL. Optimization of the separation of a group of antifungals by capillary zone electrophoresis. J Chromatogr A. 2001;917:337–45.
El Shabouri SR, Emara KM, Khashaba PY, Mohamed AM. Charge-transfer complexation for spectrophotometric assay of certain imidazole antifungal drugs. Anal Lett. 1998;31:1367–85.
Khashaba PY, El-Shabouri SR, Emara KM, Mohamad AM. Analysis of some antifungal drugs by spectrophotometric and spectrofluorimetric methods in different pharmaceutical dosage forms. J Pharm Biomed Anal. 2000;22:363–76.
Parmar P, Mehta A. Development and validation of HPTLC method for the estimation of clotrimazole in bulk drug and tablet formulation. Indian J Pharm Sci. 2009;71:451–4.
Rezaee M, Assadi Y, Hosseini MRM. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A. 2006;1116:1–9.
Guo L, Lee HK. Low-density solvent-based solvent demulsification dispersive liquid–liquid microextraction for the fast determination of trace levels of sixteen priority polycyclic aromatic hydrocarbons in environmental water samples. J Chromatogr A. 2011;1218:5040–6.
Leong MI, Huang SD. Dispersive liquid–liquid microextraction method based on solidification of floating organic drop combined with gas chromatography with electron-capture or mass spectrometry detection. J Chromatogr A. 2008;1211:8–12.
Saleh A, Yamini Y, Faraji M, Rezaee M, Ghambarian M. Ultrasound-assisted emulsification microextraction method based on applying low density organic solvents followed by gas chromatography analysis for the determination of polycyclic aromatic hydrocarbons in water samples. J Chromatogr A. 2009;1216:6673–9.
Farajzadeh MA, Seyedi SE, Shalamzari MS, Bamorowat M. Dispersive liquid-liquid microextraction using extraction solvent lighter than water. J Sep Sci. 2009;32:3191–200.
Majidi B, Shemirani F. Solvent-based de-emulsification dispersive liquid–liquid microextraction of palladium in environmental samples and determination by electrothermal atomic absorption spectrometry. Talanta. 2012;93:245.
Chen H, Chen R, Li S. Low-density extraction solvent-based solvent terminated dispersive liquid–liquid microextraction combined with gas chromatography-tandem mass spectrometry for the determination of carbamate pesticides in water samples. J Chromatogr A. 2010;1217:1244–8.
Zacharis CK, Tzanavaras PD, Roubos K, Dhima K. Solvent-based de-emulsification dispersive liquid–liquid microextraction combined with gas chromatography–mass spectrometry for determination of trace organochlorine pesticides in environmental water samples. J Chromatogr A. 2010;1217:5896–900.
Seebunrueng K, Santaladchaiyakit Y, Srijaranai S. Vortex-assisted low density solvent based demulsified dispersive liquid–liquid microextraction and high-performance liquid chromatography for the determination of organophosphorus pesticides in water samples. Chemosphere. 2013;103:51–8.
Farajzadeh MA, Mogaddam MRA. Air-assisted liquid–liquid microextraction method as a novel microextraction technique; Application in extraction and preconcentration of phthalate esters in aqueous sample followed by gas chromatography–flame ionization detection. Anal Chim Acta. 2012;728:31–8.
Adlnasab L, Ebrahimzadeh H, Yamini Y, Mirzajani F. Optimization of a novel method based on solidification of floating organic droplet by high-performance liquid chromatography for evaluation of antifungal drugs in biological samples. Talanta. 2010;83:370–8.
Xia Y, Zhi X, Wang X, Chen M, Cheng J. Ultrasound-enhanced surfactant-assisted dispersive liquid-liquid microextraction and high-performance liquid chromatography for determination of ketoconazole and econazole nitrate in human blood. Anal Bioanal Chem. 2012;402:1241–7.
Adlnasab L, Ebrahimzadeh H. A novel salt-controlled homogenous ionic liquid phase microextraction based on the salting out effect and optimization of the procedure using the experimental design methodology. Anal Methods. 2013;5:5165–71.
Gordien JB, Pigneux A, Vigouroux S, Tabrizi R, Accoceberry I, Bernadou JM, et al. Simultaneous determination of five systemic azoles in plasma by high-performance liquid chromatography with ultraviolet detection. J Pharm Biomed Anal. 2009;50:932–8.
Khan Beigi A, Imani M, Payehghadr M, Hosseini H. SPE-HPLC method for determination of ketoconazole and clotrimazole residues in cow’s milk. Braz Chem Soc. 2011;22:1679–85.
Acknowledgments
Support of this investigation by the research council of Payame Noor University is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have are no conflicts of interest.
Informed consent
Written informed consent was obtained from the healthy volunteer who donated urine samples.
Ethical approval
Ethical approval for the study was obtained from the Ethics Committee of the Iranian Blood Transfusion Organization before collection and analysis of human blood samples.
Rights and permissions
About this article

Cite this article
Ezoddin, M., Shojaie, M., Abdi, K. et al. Ultrasound-air-assisted demulsified liquid–liquid microextraction by solidification of a floating organic droplet for determination of three antifungal drugs in water and biological samples. Anal Bioanal Chem 409, 2119–2126 (2017). https://doi.org/10.1007/s00216-016-0158-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-0158-9