
Abstract
Stable isotope dilution assays (SIDAs) are becoming ever commoner in mycotoxin analysis, and the number of synthesized or commercially available isotopically labelled compounds has greatly increased in the 7 years since our last review dealing with this topic. Thus, this review is conceived as an update for new applications or improvements of SIDAs for compounds discussed earlier, but the main focus is on newly introduced labelled substances and the development of SIDAs for, for example, fusarin C, moniliformin or the enniatins. Mycotoxin research has concentrated on the emerging group of Alternaria toxins in recent years, and a series of SIDAs have been developed, including ones for tenuazonic acid, alternariol, altertoxins and tentoxin that are discussed in detail in this review. Information about synthetic routes, isotopic purity and mass-spectrometric characterization of labelled compounds is given, as well as about the development and validation of SIDAs and their application to foods, feeds or biological samples. As the number of commercially available labelled standards is increasing continuously, a general tendency for the use of analytical methods based on liquid chromatography coupled with mass spectrometry capable of identifying a series of mycotoxins simultaneously (“multimethods”) and using one or more labelled internal standards can be observed. An overview of these applications is given, thus demonstrating that SIDAs are increasingly being used in routine analysis.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction: state-of-the-art of stable isotope dilution assays
Mycotoxin contamination of food and feedstuffs is regulated in many parts of the world, and particularly low maximum limits are stipulated in the European Union. Owing to severe legal consequences when maximum limits are exceeded, accurate and reliable analytical methods for these contaminants are essential. Moreover, as the maximum limits for many mycotoxins are at sub-parts per billion levels, e.g. 0.05 μg/kg for aflatoxin M1, accuracy at these trace levels poses an additional challenge. In 2008 we described in our previous review [1] the particular benefits that stable isotope dilution assays (SIDAs) offer, especially for liquid chromatography (LC)–mass spectrometry (MS) methods, and here we present an update. The previous review introduced the principle of SIDAs in mycotoxin analysis along with the prerequisites for and limitations of the use of stable isotopically labelled internal standards and possible calibration procedures. Moreover, the state of SIDA applications at that time for the analysis of trichothecenes, zearalenone (ZEN), fumonisins, patulin, and ochratoxin A (OTA) was described. In the subsequent years, this method has seen tremendous expansion and appreciation. The principles of access to stable-isotope-labelled analogues as internal standards, the prerequisites and the limitations have remained the same, and thus we describe here new stable-isotope-labelled analogues and SIDAs that have been developed since then.
Update on applications of stable isotope dilution assays to mycotoxins
Fusarium toxins
Zearalenone
ZEN is a mycoestrogen produced by Fusarium species. The synthesis of deuterated ZEN and its use (together with commercially available labelled standards of zearalenols and zearalanols) in a SIDA of environmental samples was reported in 2007 [2], and was included in our previous review [1]. The method used to synthesize labelled ZEN was base-catalyzed protium–deuterium exchange. However, as protons at C-3, C-5, C-5′ and C-7′ of the ZEN molecule are prone to protium–deuterium exchange, a mixture of different labelled compounds was obtained, with [2H5]ZEN being the predominant isotopologue (38.3 %). Aware of the discovery that the protons at C-3 and C-5 are always almost completely exchanged against deuterium [3], a modified synthetic route was developed, in which the carbonyl moiety at C-6′ is protected as a 1,3-dioxolane prior to base-catalyzed protium–deuterium exchange [4]. Hence, with the partial protium–deuterium exchange at C-5′ and C-7′ being avoided, doubly labelled [2H2]ZEN was obtained with high isotopic purity (94.4 %), the rest being singly deuterated (5.4 %) or unlabelled (0.1 %) (Fig. 1). However, the presence of the unlabelled compound in the labelled standard and spectral overlap of the doubly labelled standard with the naturally occurring 13C2-labelled isotopologue (2.9 %) required quadratic regression to obtain a linear calibration curve between analyte-to-standard ratios of 1:10 to 10:1. The recovery rates for ZEN were between 104 ± 3 % and 101 ± 2 % for spiking levels ranging from 5 to 300 μg/kg. The precision, limit of detection (LOD) and limit of quantitation (LOQ) were not determined. The SIDA developed was applied to the analysis of several food commodities from the German retail market with a ZEN content not higher than 45 μg/kg in a sample of taco shells.

It is generally assumed that Fusarium species biosynthesize ZEN with the side chain double bond only trans configured; however, the change to cis configuration has been reported after artificial UV irradiation [5] or irradiation with natural sunlight [3]. Data on the estrogenic relevance and occurrence in food and feed of this isomer are limited. However, after synthesis by UV irradiation, purification and characterization of pure cis-ZEN as an analytical standard [6, 7], it was reasonable to prepare an isotopically labelled standard in the same way. After UV irradiation of commercially available [U-13C18]ZEN, isomeric [U-13C18]-cis-ZEN was obtained with a yield of about 50 % [8]. The SIDA developed showed low LODs (0.35 μg/kg for trans-ZEN; 0.28 μg/kg for cis-ZEN) and LOQs (1.17 μg/kg for trans-ZEN; 0.93 μg/kg for cis-ZEN) and excellent recovery (104 ± 4 % for trans-ZEN; 104 ± 2 % for cis-ZEN). However, a survey of 15 edible oils from the German retail market showed low contamination with trans-ZEN (two of 15 oils), with only one sample exceeding the LOD for cis-ZEN, but not the LOQ [8].
Moniliformin
Semisquaric acid or, systematically, 1-hydroxycyclobut-1-ene-3,4-dione, was originally isolated from Fusarium moniliforme in 1973 [9], from which its common name “moniliformin” (MON) was formed, and soon after was structurally characterized [10]. However, many other Fusarium species have been found to be capable of producing MON, e.g. F. avenaceum, F. tritinctum, and F. proliferatum [11], and contamination of cereal samples with MON (as the sodium or potassium salt) has been reported worldwide [12]. Although the principal mode of action is still under discussion, studies with ducklings, mice and rats revealed MON to be an acute cardiotoxin with LD50 that are comparable with those of the most toxic Fusarium mycotoxins T-2 toxin and HT-2 toxin [13–15]. Recent studies have shown rapid urinary excretion of administered MON in rats and no signs of accumulation, but subchronic toxic effects could not be excluded [16]. Synthetic routes to MON have been reported [17, 18], in the latter case based on a [2 + 2] cycloaddition that was also used in the synthesis of [13C2]MON [19]. In the [2 + 2] cycloaddition 2,3-dihydro-1,4-dioxine was reacted with dichloro-[13C2]ketene, which was generated in situ by reductive dehalogenation of trichloro-[13C2]acetyl chloride with activated zinc. After hydrolysis and purification, [13C2]MON was isolated with a yield of 15 % and an isotopic purity of 99 % (Fig. 2).

Synthesis of [13C2]moniliformin (black square: 13C) [19]
With use of this labelled standard, a SIDA was developed that was based on the measurement of the base peaks of the [M − H]– ions of MON and [13C2]MON with high-resolution MS (HRMS) after LC [20]. It is known that the use of triple-quadrupole mass spectrometers is critical for the analysis of MON as only one significant mass transition is formed and selectivity is therefore derogated [21, 22]. The use of HRMS has been shown to ensure selectivity of the MON analysis by the exact mass measurement [23]. Besides the use of [13C2]MON as an internal standard, selectivity was further improved by the monitoring of the ratio between MON and its naturally occurring 13C1-labelled isotopologue [20]. Unfortunately, little information was given about the linear range of the calibration, and the SIDA developed was principally limited by addition of the labelled standard just before the mass-spectrometric measurement. For cereal matrices, a mean recovery of 75.3 % was calculated, but was not used to correct the results for the samples. The LOD (0.7 μg/kg) and LOQ (2.5 μg/kg) of the SIDA determined by the study authors showed sensitivity similar to that of recent conventional methods using LC–MS/MS [24]. The precision of the SIDA (6.5 %) also differs slightly from that of published conventional methods [22, 24, 25], although deeper assessment is difficult as method validation was not systematically done or reported in most studies.
Fusarin C
Fusarin C (Fig. 3) is a mycotoxin produced by several Fusarium species, including the commonly occurring F. avenaceum, F. culmorum, F. graminearum and F. sporotrichoides, and many others [26, 27]. It was isolated from corn infected with F. moniliforme in the early 1980s, and was soon found to be mutagenic [28, 29]. Data on contamination of food and feed with this toxin are limited, but corn seems to be especially affected [30, 31]. High-performance LC (HPLC) with UV detection was used for the analysis of this toxin until the recent development of an HPLC method with tandem mass-spectrometric detection [32]. Sample preparation was based on dispersive solid-phase extraction (quick, easy, cheap, effective, rugged and safe, QuEChERS), and the LOD was 2 μg/kg, the LOQ was 7 μg/kg and the recovery was 80 %. Fusarin C was detected in corn-based food and feed, with maximum values up to 28 μg/kg in a popcorn sample. However, in that study quantitation had to be done by matrix-matched calibration, which is time-consuming and expensive. The same group of authors subsequently developed a SIDA for fusarin C [33]. Using carboxyfusarin C, a biochemical precursor of fusarin C that was enriched in cultures of a knockout strain of F. fujikuroi, they synthesized fusarin C by methylation with deuterated diazomethane. Although the authors took every precaution against deuterium–protium back-exchange, the isotopic purity was low, as the synthesis yielded only 62 % [2H3]fusarin C. However, as the synthesized standard contained the unlabelled compound at a level as low as 0.3 %, it could be used for development of a SIDA based on the earlier LC–MS/MS method. With use of the SIDA, the LOD and the LOQ decreased (1 and 4 μg/kg, respectively), and the recovery was almost complete (99 %). Unfortunately, no systematic precision studies were performed in both cases. However, on the basis of the data given for, for example, whole corn, an improvement of the precision from 55 % (11 ± 6 μg/kg, matrix calibration [32]) to 3 % (11.5 ± 0.3 μg/kg, SIDA [33]) can be calculated.

Structure of fusarin C (R is CH3) and its labelled isotopologue (R is C[2H]3) [33]
Depsipeptides
Some Fusarium species, most commonly F. tricinctum and F. avenaceum, are able to produce a series of cyclic depsipeptides [34–36], of which the enniatins (ENNs) and beauvericin (BEA) are the most frequently analysed ones. These compounds have recently been evaluated by the European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain [37]. According to this evaluation, grain and grain-based products are often contaminated with ENNs and BEA, but the lack of relevant toxicity data prevented a risk assessment. However, as these compounds show distinct biological effects, e.g. acting as ionophores, antimicrobials or enzyme inhibitors, the EFSA Panel on Contaminants in the Food Chain considered short-term and long-term exposure as being a possible concern for human health. The preferred analytical method for the analysis of ENNs and BEA in food, feed and agricultural goods has been LC–MS [38–41]. Depending on the matrix, both ion suppression [41] and signal enhancement [38] have been described, thus underlining the need for an isotopically labelled standard for compensation. Although it has been shown that chemical synthesis of ENNs and BEA is possible in principle (e.g. reviewed in [42]), the introduction of labels into the molecules was easily achieved biosynthetically by Hu and Rychlik [43] by growing Fusarium strains on Czapek–Dox liquid minimal medium with Na15NO3 as the only nitrogen source. Thus, [15 N3]BEA was isolated from a culture of F. fujikuoi, and [15 N3]ENN A, [15 N3]ENN A1, [15 N3]ENN B and [15 N3]ENN B1 were isolated from a culture of F. sambucinum. The SIDA developed had LODs (1.9–4.4 μg/kg) and LOQs (5.8–13.1 μg/kg) as usually achievable with modern LC–MS/MS instrumentation. However, both the recovery (96–110 %) and the intraday precision (1.35–7.21 %) of the SIDA were much better than for conventional LC–MS/MS methods (e.g. recovery 64–121 % and precision 7–18 % according to [39]).
Alternaria toxins
The saprophytic moulds of the genus Alternaria are ubiquitous plant pathogens invading several agricultural crops, as well as food and feed derived therefrom. Mould growth is accompanied by the production of more than 70 toxins, of which the most relevant ones can be classified into the two major groups of benzopyrones and perylenequinones, along with the phytotoxin tentoxin (TEN) and the amino acid derivative tenuazonic acid (TA) [44].
Benzopyrones
Of all the Alternaria toxins, the benzopyrone group is the best examined, and was the first to be analysed. This group encompasses the two major toxins alternariol (AOH) and alternariol monomethyl ether (AME) besides altenuene and altenuisol. Both AOH and AME have been reported to inhibit topoisomerase I and topoisomerase IIa [45] and to be mutagenic in cell culture [46].
As the first labelled isotopologues, deuterated AOH and AME were synthesized by palladium-catalyzed protium–deuterium exchange from the unlabelled toxins (Fig. 4) [47]. The aromatic protons are most easily exchanged against deuterium, leading to the preferred formation of the 2H4-labelled isotopologue of AOL (43 % isotopic purity) and AME (59 % isotopic purity). However, as the three benzylic protons are also affected by the exchange reaction, a mixture of isotopologues with mass increments of up to M + 7 was obtained. But as the internal standards contained no unlabelled material, this isotopic fractionation resulted only in the slope of the response curve deviating from the ideal value of 1, whereas the linearity was not affected.

Structure of the 2H4-labelled isotopologues of alternariol (R is H) and alternariol methyl ether (R is CH3) [47]
On the basis of the use of [2H4]AOH and [2H4]AME as internal standards, SIDAs were developed and applied to the determination of AOH and AME in several food commodities using LC–MS/MS [47]. Method validation for fruit juices revealed complete recovery (100.5 ± 3.4 % for AOH and 107.3 ± 1.6 % for AME), low LODs (0.03 μg/kg for AOH and 0.01 μg/kg for AME) and LOQs (0.09 μg/kg for AOH and 0.03 μg/kg for AME) and excellent precision (4.0 % for AOH and 2.3 % for AME). The values for both benzopyrones in fruit juices were low in general (less than 1 μg/kg for AOH and less than 0.3 μg/kg for AME), but higher values of the toxins were found in wine and vegetable juices, amounting to 7.82 μg/kg and 0.79 μg/kg for AOH and AME, respectively.
In another study by the same group of authors, cereal, fruit and vegetable products were analysed for contamination with both benzopyrones with the SIDA developed [48]. Both toxins were practically not detectable in cereals and cereal products from the German retail market. Higher values were found in cereals intended for animal nutrition (13–250 μg/kg for AOH and 3–100 μg/kg for AME). However, AOH and AME were also detected in about 50 % of all analysed vegetable products at rather high levels (2.6–25 μg/kg for AOH and 0.1–5 μg/kg for AME). Tomato products were especially affected. The highest contents of AOH (25 μg/kg) and AME (5 μg/kg) were found in triply concentrated tomato paste.
Most recently, uniformly 13C-labelled AOH and AME have been synthesized by a microbiological procedure using a modified Czapek–Dox medium with low concentrations of [13C6]glucose and [13C2]acetate as the only carbon sources and ammonium sulfate as the only source of nitrogen [49]. Thus, [13C14]AOH and [13C15]AME were isolated in high isotopic purity, albeit in low yield (5 μg absolute for each of them) and applied to develop SIDAs for different food commodities. As by-products [13C15]altenuene and [13C14]altenuisol were also isolated, but have not been used as internal standards in SIDAs so far.
The alternative labelling of AOH and AME enabled Liu and Rychlik [49] to clarify the fragmentation of both benzopyrones in LC–MS/MS. For AOH, the fragments of the 2H4-labelled isotopologue and the 13C14-labelled isotopologue led to the conclusion that the quantifier fragment at m/z 147 of AOH must have lost five carbon atoms and one aromatic hydrogen (Fig. 5). For AME, the assignment was much more complicated. From the deuterated isotopologue alone no structural deduction of the fragments was possible [47]. However, the main fragment ions m/z 255 and 256 of AME were accordingly observed with the 13C15-labelled isotopologue (m/z 269 and 270), whereas the 2H4-labelled isotopologue showed three signals (m/z 258, 259 and 260). Therefore, the loss of the O-methyl radical and a hydrogen radical is reasonable. The latter must originate mainly from the hydroxyl group, and, according to the appearance of a small signal at m/z 258 in the [2H4]AME spectrum, a minor amount may originate from the aromatic ring (Fig. 6). With use of both uniformly 13C-labelled benzopyrenes, a SIDA was developed and validated for cereal matrices. LODs and LOQs of 0.36 μg/kg and 1.1 μg/kg, respectively, for AOH and 0.09 μg/kg and 0.27 μg/kg, respectively, for AME were determined. The values for recovery (98–103 % for AOH and 109–112 % for AME) and precision (4.1 % for AOH and 5.8 % for AME) once more proved the advantages of the SIDA method. A survey on a series of food commodities from the German market confirmed the rather low contamination with these two toxins. However, paprika powder (AOH, 17–46 μg/kg; AME, 14–26 μg/kg) and sorghum-based animal feed (AOH, 347–757 μg/kg; AME, 109–215 μg/kg), especially, contained both AOH and AME in amounts that may be critical for human and animal health.

Proposed fragmentation of labelled and unlabelled alternariol to the main quantifier ions in liquid chromatography (LC)–tandem mass spectrometry (MS/MS) [49]. The respective m/z values of the completely 13C-labelled isotopologue are given in parentheses, and the respective m/z values of the isotopologue with 2H labelling at four positions in the aromatic rings are given in square brackets

Proposed fragmentation of labelled and unlabelled alternariol monomethyl ether to the main quantifier ions in LC–MS/MS [49]. The respective m/z values of the completely 13C-labelled isotopologue are given in parentheses, and the respective m/z values of the isotopologue with 2H labelling at four positions in the aromatic rings are given in square brackets
Tenuazonic acid
The tetramic acid derivative l-TA [(5S,8S)-3-acetyl-5-sec-butyl-pyrrolidine-2,4-dione] is one of the major mycotoxins produced by a series of different Alternaria spp. [44], and has attracted increasing attention in recent years. It is regarded as the most acutely toxic metabolite produced by Alternaria [50], and is known to occur in high concentrations in commodities infected with this mould [44]. Until now, two different methods leading to isotopologues of TA have been described.
[13C6,15 N]Tenuazonic acid
The first stable-isotope-labelled TA was synthesized from [13C6,15 N]methyl isoleucinate by Dieckmann intramolecular cyclization after acetoacetylation with diketene, yielding sevenfold-labelled [13C6,15 N]TA (Fig. 7, pathway I) [51]. The labelled standard obtained had excellent isotopic purity; however, the low yield (about 30 %) of the synthesis was acceptable only because the labelled amino acid is commercially available at a relatively moderate price. Additionally, the conditions used for the ring closure led to epimerization, the labelled standard therefore being a mixture of the 5S,8S and 5R,8S diastereomers. However, this reaction is reported to occur with the analyte itself in aqueous solution, but the few analytical methods that are able to separate the diastereomers are not routinely performed [52]. Thus, the labelled standard can be used for general purposes, but if, for example, immunochemical devices become available for sample preparation, this cannot be taken for granted in every case. With use of this stable-isotope-labelled standard, a SIDA was developed and validated. The method was based on derivatization of TA with 2,4-dinitrophenylhydrazine during extraction, solid-phase clean-up using C18 material and LC before mass-spectrometric detection. The use of the internal standard during the extraction and derivatization procedure greatly improved the recovery, which ranged between 104 ± 2.5 % and 108 ± 1.6 % for tomato products. Without an internal standard the recovery was 79 ± 11 % for different cereal matrices and 90 ± 22 % for beer [53, 54]. Different tomato products (n = 16) from the German market were analysed for their content of TA with the SIDA developed. The values were between 15 and 195 μg/kg (tomato ketchup, n = 9), 363 and 909 μg/kg (tomato paste, n = 2) and 8 and 247 μg/kg (pureed tomatoes and comparable products, n = 5).

Synthetic pathways leading to stable-isotope-labelled tenuazonic acid (black square: 13C): I acetoacylation of [13C6,15 N]methyl isoleucinate with in situ generated diketene followed by Dieckmann intramolecular condensation [47]; II condensation of isoleucine with Meldrum’s acid, followed by thermal cyclization, decarboxylation and introduction of the label by C-3 acylation with labelled acetyl chloride [63]. BOC tert-butyloxycarbonyl
In a further application of the [13C6,15 N]TA isotopologue, the median contents in different foods were found to be 1.8 μg/kg (fruit juices), 16 μg/kg (cereals) and 500 μg/kg (spices). The amount of positive samples was 86 % (fruit juices), 92 % (cereals) and 87 % (spices) [55], which highlighted the ubiquitous occurrence of TA.
In another study, infant foods and beverages were analysed by SIDA [56]. The median content of TA in infant tea infusions (n = 12) was 2 μg/L, but values up to 20 μg/L were found in fennel tea infusions. In puree infant food in jars (n = 12), the median content of TA was 7 μg/kg, but higher values were detected in products containing tomato (25 μg/kg), banana and cherry (80 μg/kg) and sorghum (20 μg/kg). Infant cereals based on wheat and/or oats, rice, spelt and barley (n = 4) did not contain TA at values higher than 30 μg/kg, but if sorghum was the major ingredient (n = 12) the mean content of TA was 550 μg/kg and the maximum level was 1200 μg/kg. This finding raised concerns about the safety of millet infant cereals, and the risk was assessed on the basis of the threshold of toxicological concern (TTC) approach [57] by the European Food Safety Authority with a TTC of 1500 ng TA per kilogram of body weight per day [58]. The TTC is likely to be exceeded by infants consuming these cereals and, therefore, the Bavarian Health and Food Safety Authority stipulated a warning limit for these kinds of products of 500 μg/kg, which led to withdrawal of the highly contaminated products from the Bavarian market.
[13C6,15 N]TA was also used as an internal standard for the quantitation of TA analogues derived from valine (ValTA), leucine (LeuTA), alanine and phenylalanine that were synthesized from the respective amino acids [59]. Two analytical methods based on HPLC and mass-spectrometric detection were developed, one with derivatization with 2,4-dinitrophenylhydrazine and one without. Besides TA, the analogues LeuTA (about 4 % of TA content) and ValTA (about 10 % of TA content) were found in highly contaminated sorghum infant cereals and sorghum grains. Other analogues were not detected.
Further studies using SIDA were directed towards unravelling the toxic potential of TA. In toxicokinetic studies in humans, the content of TA in human urine was determined by a SIDA extensively revalidated for urine matrix [60]. It was very difficult to obtain blank urine samples as TA is constantly consumed because of its ubiquitous occurrence. For a blank urine sample, a diet devoid of spices, cereals, vegetables and fruits was necessary. The method was then applied to two studies dealing with urinary excretion of TA: In the first study, TA was quantified in the 24-h urine of six volunteers from Germany (three female, three male) having their usual diet in a concentration range of 1.3–17.3 μg/L or 2.3–10.3 ng/mg creatinine, respectively. In the second study, two volunteers (one female, one male) ingested 30 μg TA by consumption of naturally contaminated whole meal sorghum infant cereals and tomato juice, respectively. The urinary excretion of the ingested TA was 54–81 % after 6 h, depending on the matrix and the volunteer. After 24 h, 87–93 % of the ingested amount of TA had been excreted, but the fate of the remaining about 10 % was not revealed.
In another toxicokinetic study of TA (Fraeyman S, Devreese M, Broekaert N, De Mil T, Antonissen G, De Baere S, De Backer P, Rychlik M, Croubels S. In vivo hazard characterization of the Alternaria mycotoxin tenuazonic acid in broiler chickens and pigs: comparative toxicokinetics and absolute oral bioavailability, submitted for publication) in the plasma of pigs and broiler chickens, each animal received a single dose of 0.05 mg TA per kilogram body weight orally and intravenously in a two-way crossover design. The toxicokinetic profile of TA measured by the SIDA using [13C6,15 N]TA as an internal standard markedly differed between pigs and broiler chickens. TA was completely absorbed in pigs as well broiler chickens, with an absolute oral bioavailability of 147 and 124 %, respectively. However, absorption was slower in broiler chickens, with a mean time to maximal plasma concentration of 2.60 h. TA was more rapidly eliminated in pigs than in broiler chickens, since the mean elimination half-life after intravenous administration was 0.51 h and 2.03 h for pigs and broiler chickens, respectively. This slower elimination is mainly due to the significantly lower total body clearance of TA in broiler chickens, i.e. 448.4 mL/h/kg and 59.3 mL/h/kg after intravenous administration for pigs and broiler chickens, respectively. Possible hypotheses are zero-order biotransformation kinetics of TA and/or inhibition of biotransformation enzymes in broiler chickens. Furthermore, the higher area under the plasma concentration–time curve, a measurement of body exposure, in broiler chickens could imply differences in species susceptibility to the harmful effects of TA, although no clinical signs of intoxication were observed in either species after single-bolus administration.
A study from Belgium used [13C6,15 N]TA to analyse a series of derived tomato products with a SIDA [61]. The samples had previously been screened for the mycotoxins AOH, AME, ochratoxin A (OTA), fumonisin B1, fumonisin B2 and fumonisin B3 by LC–HRMS. All samples that contained AOH or AME were also contaminated with TA (0.7–4.8 mg/kg). A dietary exposure assessment was performed for TA with Belgian consumption data, and the mean value obtained (4230 ng/kg body weight/day) was higher than the TTC of 1500 ng/kg bod weight/day set by the EFSA [58].
A UPLC–positive/negative electrospray ionization MS/MS method for the simultaneous determination of some free [AOH, AME, altenuene, TA, TEN, altertoxin (ATX) I] and conjugated (sulfates and glucosides of AOH and AME) Alternaria toxins in cereals and cereal products (rice, oat flakes and barley) has recently been described [62], applying isotopically labelled internal standards ([2H4]AME and [13C6,15 N]TA) for SIDAs of AME and TA. [13C6,15 N]TA was also used as an internal standard for all other surveyed toxins. Extensive validation of the method was done very thoroughly for all compounds with use of different matrices and spiking levels. Good values for recovery (more than 95 %) and precision (less than 10 %) were obtained, in general, but even better validation data were obtained for TA and AME that were analysed with a SIDA. Finally, 24 commercially available cereal-based foodstuffs were analysed for all ten toxins. Besides some trace levels of AOH and TEN in rice samples, TA was the toxin most often detected. A total of 71 % (22/31) of the rice samples, and 31 % (5/16) of the oat flake samples were contaminated with concentrations in ranges 1.9–113 μg/kg and 2.1–39 μg/kg, respectively.
[13C2]Tenuazonic acid
Another synthetic route towards labelled TA was reported recently [63]. [13C2]TA was produced in a three-step procedure starting from unlabelled tert-butyloxycarbonyl-protected isoleucine followed by condensation with Meldrum’s acid, thermal cyclization, decarboxylation and finally introduction of the labelled carbons via 3-C acetylation with [13C2]acetyl chloride (Fig. 7 pathway II). Apart from the good yield (67 %), the particular benefit of this procedure was the introduction of the label in the last step via a rather cheap reagent. However, epimerization at C-5 also occurred during the last step of this approach, so no stereochemically pure labelled standard for TA has been prepared yet. With use of the 13C2-labelled isotopologue, a SIDA was developed for tomato and pepper products based on sample preparation using the QuEChERS procedure. No derivatization was applied, and therefore the method was less sensitive (LOD 0.86 μg/kg; LOQ 2.89 μg/kg). Recovery was determined for different matrices and ranged between 91.0 ± 1.2 % (pepper paste) and 102 ± 4.4 % (tomato products). No data on the precision of the method and the linear range of the calibration were given. The method was applied to 26 tomato samples and four bell pepper samples from the German market, and TA was found in each sample, with levels ranging from 3 to 2330 μg/kg.
Perylenequinones
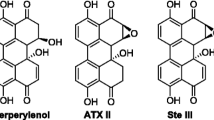
Derivatives of perylenquinone such as ATX I, ATX II, ATX III, alterperylenol (APOL; synonym “alteichin”), and stemphyltoxin (STX) III (Fig. 8) are minor metabolites of Alternaria spp., but are regarded as very critical because of their mutagenic properties [64, 65]. Because of the lack of available reference compounds, in particular for ATXs, analytical studies are rare. Therefore, biosynthesis of ATXs and other perylenes (Fig. 8) by Alternaria was studied in a modified Czapek–Dox medium with a low level of glucose as the carbon source and ammonium sulfate as the sole nitrogen source [49]. Under these conditions the perylenequinones were the most abundant metabolites produced by the A. alternata strain used, and the required compounds were isolated from the culture broth, purified by preparative HPLC and used as analytical standards in the following investigations. Labelled ATXs were synthesized in the same way with [13C6]glucose. However, the 13C-labelled isotopologues obtained were not completely labelled as unlabelled citric acid in the trace element solution contributed with one 12C2 unit to the biosynthetic pathway and caused the production of 13C18-labelled isotopologues, which amounted to about 40 % of the 13C20-labelled isotopologues for all compounds. Unfortunately, the mass increment of 18 Da caused an unexpected mass spectral overlap between the internal standards and the unlabelled ATXs as the hydroxyl group of C-12a (Fig. 9) in the bay corner is easily eliminated during ionization, thus leading to a loss of water equivalent to 18 Da in the ion source. Thus, the fragments shown in Fig. 9, structure (a) were not suitable for quantitation, and the fragments shown in Fig. 9, structure (c) were chosen instead. SIDAs were developed for ATX I, ATX II and APOL, and validation resulted in good values for precision (3.8–9.9 %) and recovery (96–105 %). With use of SIDAs, 35 food samples from the German market were analysed, and ATX I and APOL were detected in organic whole grains and paprika powder with maximum values of 4.7 μg/kg (ATX I) and 5.2 μg/kg (APOL). ATX II, ATX III and STX III were not detectable. In addition, if the samples were contaminated with ATX, they were likely to be co-contaminated with other Alternaria toxins such as AOH, AME and TEN, but not necessarily vice versa. Much higher levels of perylenequinones were found in feed samples based on sorghum. The maximum concentrations of ATX I and APOL were 43 and 58 μg/kg, respectively, and in some cases ATX II was also detected (maximum 1.7 μg/kg).

Structures of perylenequinones produced by Alternaria species. ATX altertoxin, APOL alterperylenol, STTX stemphyltoxin

Spectral interferences between altertoxin I, [13C18]altertoxin I and [13C20]altertoxin I in mass spectrometry [49]. The respective m/z values of the 13C-labelled isotopologue (first value [13C18], second value [13C20]) are given in parentheses. Arrows below indicate the respective spectral overlap
Tentoxin
The cyclic tetrapeptide TEN is produced as a secondary metabolite by some Alternaria species along with dihydrotentoxin (DHT) and isotentoxin (isoTEN) (Fig. 10) [66, 67]. Their structures differ at the unsaturated bond of the N-methyldehydrophenylalanine moiety, which is hydrogenated into a single bond in DHT and E configured in isoTEN. All three compounds are considered as phytotoxins, with TEN being the most potent one, inhibiting photophosphorylation and inducing chlorosis [68]. However, no toxicological data are available for mammals, and the data on the occurrence of this toxin in food and feed are limited as well. Triply deuterated TEN was prepared via total synthesis from simple building blocks (Fig. 11), and [2H3]DHT and [2H3]isoTEN were synthesized starting from [2H3]TEN by hydrogenation or isomerization, respectively [69]. The label was introduced with [2H3]iodomethane in the last step before cyclization. Although the yield of the whole synthetic procedure was only 0.24 %, it can be calculated to be about 10 % for the steps after introduction of the label, which is reasonable and emphasizes the need for thorough planning of the synthetic routes leading to labelled compounds. The method was applied to 103 food samples, including bread, cereals, crisps, juice, nuts, oil, sauce, seeds, and spices. Of these, 85 % were contaminated with TEN and 55 % were contaminated with DHT, whereas isoTEN was not quantifiable. The maximum concentrations of TEN and DHT were 52.4 and 36.3 μg/kg, respectively, and both were detected in paprika powder.

Structures of cyclic tetrapeptides produced by Alternaria species. DHT dihydrotentoxin, isoTEN isotentoxin, TEN tentoxin

The last two steps (introduction of the label and ring closure) of the synthesis of [2H3]tentoxin [69]. The respective origin of the building blocks leading to the intermediate products is given in the dotted areas. BA benzaldehyde, D 2H, DMF dimethylformamide, Gly glycine, HBTU O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate, Leu leucine, MeAla N-methylalanine, tB tert-butyl, TFA trifluoroacetic acid
Ochratoxin A
Since the first report on the synthesis of a labelled OTA analogue, namely [2H5]OTA [70], two further isotopologues have been made commercially available or have been reported in the literature. The first one, [13C20]OTA, is commercially available, and has been used since its generation in several studies either on contamination by multiple mycotoxins presented in Table 1 or specifically on OTA, e.g. in coffee [71], or on toxicokinetics in rats [72]. Whereas [13C20]OTA has been prepared microbiologically and can be expected to be enantiomerically pure in its 3R,14S configuration, in chemical syntheses the stereochemistry of the OTA prepared has to be considered. Owing to its two stereo centres at C-3 and C-14, four diastereomers are possible. The first isotopologue reported, [2H5]OTA [70], has not been characterized with regard to its stereochemistry. However, because of its preparation by base-catalyzed coupling of ochratoxin alpha (OTα) with L-[2H5]phenylalanine methyl ester, one can assume there is partial racemization of the phenylalanine moiety, thus giving the respective pair of diastereomers. This isomerization has also been observed to occur thermally during roasting of coffee [73] and, for accurate analysis, the respective (14R)-[2H5]OTA was prepared by thermal isomerization of enantiomerically pure (14S)-[2H5]OTA. This stereo differentiation will allow the contribution of heated foods containing OTA to the overall exposure to be evaluated when blood analyses are used to assess the “internal exposure” of humans to the mycotoxin. For OTA, stable isotopologues are increasingly used also in clinical analysis [74]. In the latter study—for the first time in mycotoxin analysis—the quantitation of exact amounts of standards by quantitative NMR was introduced, thus providing better accuracy also for SIDAs. During analysis of OTA, stereochemistry is crucial as often immunoaffinity clean-up is applied, and the latter may be enantiospecific. This effect has been observed during the use of the 3S diastereomer as a possible internal standard for OTA in a diastereomeric dilution assay as an alternative to a SIDA. For this study, both racemic unlabelled and deuterated OTα were prepared chemically [75] and coupled to l-phenylalanine. As a result, the respective diastereomeric dilution assay was found to be significantly less accurate than the SIDA used in parallel [76]. In a complementary approach, Gabriele et al. [77] synthesized racemic OTα in a three-step procedure, and coupled racemic OTα with protected L-[2H5]phenylalanine to give a mixture of 3S and 3R diastereomers of OTA, which were separated by preparative thin-layer chromatography. In contrast, stereoisomerically pure OTα was prepared in another study to be used as an educt for labelled OTA after its coupling with labelled phenylalanine. Two new routes for the synthesis of enantiomerically pure (3R)-OTα have been presented [78]. The key step of both routes is the directed ortho metallation of unprotected and suitably functionalized aromatic carboxylic acids, followed by an alkylation–cyclization reaction with (R)-propylene oxide (Fig. 12).

Synthetic routes leading to enantiomerically pure (3R)-ochratoxin alpha to be used as educt to produce stable isotopologues of ochratoxin A [70]. LTMP lithium tetramethylpiperidine, THF tetrahydrofuran
Commercially available labelled standards for mycotoxins
The emerging availability of commercial labelled standards has led to increased use in mycotoxin analysis. Of the 30 published LC–MS methods covering the years 2008–2013 that have been reviewed recently [79], ten of them use at least one commercially labelled standard for correction of the analytical results [80–89]. More recent methods follow this trend and perform SIDAs for all or at least some analysed mycotoxins (Table 1). At present, labelled standards are commercially available for all mycotoxins, the maximum contents of which are internationally regulated in foods and feeds, and are provided as uniformly 13C-labelled ([U-13C]) isotopologues, in general. The aflatoxins may serve as an example, as for them the labelled internal standards [U-13C]aflatoxin B1, [U-13C]aflatoxin B2, [U-13C]aflatoxin G1, [U-13C]aflatoxin G2 and [U-13C]aflatoxin M1 can be purchased from well-known suppliers of chemicals. Further examples are [U-13C]patulin, [U-13C]OTA, some trichothecenes ([U-13C]T-2 toxin and [U-13C]HT-2 toxin) and fumonisins B1, B2 and B3, which can all be purchased as uniformly 13C-labelled isotopologues as well. However, some other labelled mycotoxins that are not legally controlled routinely are also commercially available at the moment, e.g. [U-13C]nivalenol and [U-13C]kojic acid and an increasing number of others that may serve the specific purpose of the researcher, but whose long-term availability is in doubt owing to lack of general demand. Although the range of labelled compounds does not seem to point to a certain “most needed” lacking compound, most likely researchers will not find their desired compound commercially available and will have to develop individual strategies towards its synthesis.
Concerning the reliability of the results, a closer look reveals that most of the recent methods tend to add the labelled standard to the sample matrix directly at the beginning of the sample preparation (Table 1), which is the best method to counterbalance analyte losses during the analytical procedure. However, in some methods, the labelled standard is added just before the LC–MS measurement, which compensates for suppression or enhancement effects in the ion source of the mass spectrometer, but does not account for analyte losses during sample preparation and, therefore, is less reliable.
Discussion of perspectives
Since our first review on SIDAs in mycotoxin analysis, the stock of available stable isotopologues has expanded tremendously. All mycotoxins with maximum limits in foods are commercially available labelled with stable isotopes. When reviewing the synthetic strategies towards labelled mycotoxins, we can distinguish biosynthesis by fungi versus chemical synthesis. As a guideline, the more complex the molecule is, the more efficient a biotechnologically approach must be. For example, isotopologues for the small molecule TA up to now have only been synthesized chemically. In contrast, biosynthesis offers the possibility to obtain fully labelled isotopologues, labelled either with 13C (e.g. ATXs) or with 15 N (e.g. ENNs). Most of the commercially available labelled mycotoxins are produced biotechnologically as fully 13C-labelled isotopologues, and are applied more often than the chemically synthesized ones.
Generally, it can be recognized that SIDAs have gained the status of a reference method because of several benefits. The excellent attributes of SIDAs in terms of recovery and precision are essential if low maximum limit trace amounts have to be accurately quantitated. Moreover, the versatile and sensitive LC–MS/MS instrumentation has become standard laboratory equipment, facilitating the low LODs and LOQs required in supervision of maximum limits, but its susceptibility to matrix interference does not leave many alternatives other than the use of stable-isotope-labelled isotopologues as internal standards. Finally, more and more analysts are realizing that the price of the labelled standard is negligible, if calculated for the respective sample and compared with personnel labour costs and general costs for consumables (solvents, columns for clean-up, etc.) and for acquiring and running LC–MS/MS equipment.
However, the prices of stable-isotope-labelled standards are still too high for the breakthrough of multiple SIDAs. This is due to mycotoxins occurring in very different contents in food matrices, and, therefore, a multi-SIDA would require high standard additions for the most abundant mycotoxins, at least when added in the initial step of a SIDA in its pure philosophy. The addition of the labelled standard immediately before the LC–MS/MS measurement is therefore sometimes performed (Table 1), but analyte losses during sample preparation are not compensated for any more in this case, requiring determination of and correction for recovery. Moreover, maximum limits require highly varying sensitivity and linear ranges of the LC–MS/MS method (up to four orders of magnitude), which is not easy to achieve with standard instruments and calibrations. Thus, specifically combined ones rather than multiple SIDAs will still be used in the medium term.
Further challenges arise from the “emerging mycotoxins” such as Alternaria toxins, for which the commercial availability of stable isotopologues is still limited. However, research groups are increasingly willing to cooperate and exchange labelled isotopologues noncommercially.
Moreover, the issue of “modified mycotoxins” is emerging; these were formerly known as “masked mykotoxins”, but the latter term is now accepted to be strained [90]. New strategies for identification of modified mycotoxins are required, with a promising approach being nontargeted metabolomics, as exemplified for deoxynivalenol [91]. Risk assessment of modified mycotoxins is limited because of the lack of analytical methods, which in turn restricts studies on toxicology and exposure surveys. The first opinions of risk assessment authorities on modified mycotoxins dealt with the best studied compounds, i.e. with deoxynivalenol and its acetates [92] and, more recently, with ZEN and its metabolites [93].
References
Rychlik M, Asam S (2008) Stable isotope dilution assays in mycotoxin analysis. Anal Bioanal Chem 390(2):617–628
Hartmann N, Erbs M, Wettstein FE, Schwarzenbach RP, Bucheli TD (2007) Quantification of estrogenic mycotoxins at the ng/L level in aqueous environmental samples using deuterated internal standards. J Chromatogr, A 1138(1–2):132–140
Miles CO, Erasmuson AF, Wilkins AL, Towers NR, Smith BL, Garthwaite I, Scahill BG, Hansen RP (1996) Ovine metabolism of zearalenone to α-zearalanol (zeranol). J Agric Food Chem 44(10):3244–3250
Cramer B, Bretz M, Humpf H (2007) Stable isotope dilution analysis of the Fusarium mycotoxin zearalenone. J Agric Food Chem 55(21):8353–8358
Peters CA (1972) Photochemistry of zearalenone and its derivatives. J Med Chem 15(8):867–868
Köppen R, Riedel J, Emmerling F, Koch M (2012) (3S,11Z )-14,16-Dihydroxy-3-methyl-3,4,5,6,9,10-hexahydro-1 H-2-benzoxacyclotetradecine-1,7(8 H)-dione (cis-zearalenone): a redetermination. Acta Crystallogr E Struct Rep Online 68(3):o832
Köppen R, Riedel J, Proske M, Drzymala S, Rasenko T, Durmaz V, Weber M, Koch M (2012) Photochemical trans-/cis-isomerization and quantitation of zearalenone in edible oils. J Agric Food Chem 60(47):11733–11740
Drzymala S, Riedel J, Köppen R, Garbe L, Koch M (2014) Preparation of 13C-labelled cis-zearalenone and its application as internal standard in stable isotope dilution analysis. World Mycotoxin J 7(1):45–52
Cole RJ, Kirksey JW, Cutler HG, Doupnik BL, Peckham JC (1973) Toxin from Fusarium moniliforme: effects on plants and animals. Science 179(4080):1324–1326
Springer JP, Clardy J, Cole RJ, Kirksey JW, Hill RK, Carlson RM, Isidor JL (1974) Structure and synthesis of moniliformin, a novel cyclobutane microbial toxin. J Am Chem Soc 96(7):2267–2268
Schütt F, Nirenberg HI, Demi G (1998) Moniliformin production in the genus Fusarium. Mycotoxin Res 14(1):35–40
Sharman M, Gilbert J, Chelkowski J (1991) A survey of the occurrence of the mycotoxin moniliformin in cereal samples from sources worldwide. Food Addit Contamin 8(4):459–466
Kriek N, Marasas W, Steyn P, van Rensburg S, Steyn M (1977) Toxicity of a moniliformin-producing strain of Fusarium moniliforme var. subglutinans isolated from maize. Food Cosmet Toxicol 15(6):579–587
Burmeister HR, Ciegler A, Vesonder RF (1979) Moniliformin, a metabolite of Fusarium moniliforme NRRL 6322: purification and toxicity. Appl Environ Microbiol 37(1):11–13
Jonsson M, Jestoi M, Nathanail AV, Kokkonen U, Anttila M, Koivisto P, Karhunen P, Peltonen K (2013) Application of OECD guideline 423 in assessing the acute oral toxicity of moniliformin. Food Chem Toxicol 53:27–32
Jonsson M, Atosuo J, Jestoi M, Nathanail AV, Kokkonen U, Anttila M, Koivisto P, Lilius E, Peltonen K (2015) Repeated dose 28-day oral toxicity study of moniliformin in rats. Toxicol Lett 233(1):38–44
Bellus D, Fischer H, Greuter H, Martin P (1978) Synthesen von Moniliformin, einem Mycotoxin mit Cyclobutendion-Struktur. Helv Chim Acta 61(5):1784–1813
Fétizon M, Hanna I (1990) 2,3-Dihydro-1,4-dioxin in organic chemistry. Part X. 1 A new synthesis of 3-hydroxy-3-cyclobutene-1,2-dione (semisquaric acid). Synthesis 1990:583–584
Lohrey L, Murata T, Uemura D, Humpf H (2011) Synthesis of isotopically labeled Fusarium mycotoxin 13C2-moniliformin [1-hydroxycyclobut-1-ene-3,4-dione]. Synlett 2011(15):2242–2244
Von Bargen KW, Lohrey L, Cramer B, Humpf H (2012) Analysis of the Fusarium mycotoxin moniliformin in cereal samples using 13C2-moniliformin and high-resolution mass spectrometry. J Agric Food Chem 60(14):3586–3591
Jestoi M, Rokka M, Rizzo A, Peltonen KM (2003) Moniliformin in finnish grains: analysis with LC-MS/MS. Asp Appl Biol 68:211–216
Jin P, Han Z, Cai Z, Wu Y, Ren Y (2010) Simultaneous determination of 10 mycotoxins in grain by ultra-high-performance liquid chromatography–tandem mass spectrometry using 13C15-deoxynivalenol as internal standard. Food Addit Contamin Part A 27(12):1701–1713
Sørensen JL, Nielsen KF, Thrane U (2007) Analysis of moniliformin in maize plants using hydrophilic interaction chromatography. J Agric Food Chem 55(24):9764–9768
Scarpino V, Blandino M, Negre M, Reyneri A, Vanara F (2013) Moniliformin analysis in maize samples from north-west Italy using multifunctional clean-up columns and the LC-MS/MS detection method. Food Addit Contamin Part A 30(5):876–884
Sewram V, Nieuwoudt TW, Marasas WFO, Shephard GS, Ritieni A (1999) Determination of the mycotoxin moniliformin in cultures of Fusarium subglutinans and in naturally contaminated maize by high-performance liquid chromatography–atmospheric pressure chemical ionization mass spectrometry. J Chromatogr A 848(1–2):185–191
Thrane U (1988) Screening for fusarin C production by European isolates of Fusarium species. Mycotoxin Res 4(1):2–10
Farber JM, Sanders GW (1986) Production of fusarin C by Fusarium spp. J Agric Food Chem 34(6):963–966
Wiebe LA, Bjeldanes LF (1981) Fusarin C, a mutagen from Fusarium moniliforme grown on corn. J Food Sci 46(5):1424–1426
Gelderblom WCA, Thiel PG, van der Merwe KJ, Marasas WFO, Spies HSC (1983) A mutagen produced by Fusarium moniliforme. Toxicon 21(4):467–473
Gelderblom WCA, Thiel PG, Marasas WFO, van der Merwe KJ (1984) Natural occurrence of fusarin C, a mutagen produced by Fusarium moniliforme, in corn. J Agric Food Chem 32(5):1064–1067
Thiel PG, Gelderblom WCA, Marasas WFO, Nelson PE, Wilson TM (1986) Natural occurrence of moniliformin and fusarin C in corn screenings known to be hepatocarcinogenic in rats. J Agric Food Chem 34(5):773–775
Kleigrewe K, Söhnel A, Humpf H (2011) A new high-performance liquid chromatography–tandem mass spectrometry method based on dispersive solid phase extraction for the determination of the mycotoxin fusarin C in corn ears and processed corn samples. J Agric Food Chem 59(19):10470–10476
Kleigrewe K, Niehaus E, Wiemann P, Tudzynski B, Humpf H (2012) New approach via gene knockout and single-step chemical reaction for the synthesis of isotopically labeled fusarin C as an internal standard for the analysis of this Fusarium mycotoxin in food and feed samples. J Agric Food Chem 60(34):8350–8355
Logrieco A, Rizzo A, Ferracane R, Ritieni A (2002) Occurrence of beauvericin and enniatins in wheat affected by Fusarium avenaceum head blight. Appl Environ Microbiol 68(1):82–85
Morrison E, Kosiak B, Ritieni A, Aastveit AH, Uhlig S, Bernhoft A (2002) Mycotoxin production by Fusarium avenaceum strains isolated from Norwegian grain and the cytotoxicity of rice culture extracts to porcine kidney epithelial cells. J Agric Food Chem 50(10):3070–3075
Thrane U, Adler A, Clasen P, Galvano F, Langseth W, Lew H, Logrieco A, Nielsen KF, Ritieni A (2004) Diversity in metabolite production by Fusarium langsethiae, Fusarium poae, and Fusarium sporotrichioides. Int J Food Microbiol 95(3):257–266
Benford D, Ceccatelli S, Cottrill B, Dinovi M, Dogliotti E, Edler L, Farmer P, Furst P, Hoogenboom L, Knutsen HK, Lundebye A, Metzler M, Nebbia CS, O'Keeffe M, Rietjens I, Schrenk D, Silano V, van Loveren H, Vleminckx C, Wester P (2014) Scientific opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J 12(8):3802
Sørensen JL, Nielsen KF, Rasmussen PH, Thrane U (2008) Development of a LC-MS/MS method for the analysis of enniatins and beauvericin in whole fresh and ensiled maize. J Agric Food Chem 56(21):10439–10443
Uhlig S, Ivanova L (2004) Determination of beauvericin and four other enniatins in grain by liquid chromatography–mass spectrometry. J Chromatogr, A 1050(2):173–178
Sewram V, Nieuwoudt TW, Marasas WFO, Shephard GS, Ritieni A (1999) Determination of the Fusarium mycotoxins, fusaproliferin and beauvericin by high-performance liquid chromatography–electrospray ionization mass spectrometry. J Chromatogr, A 858(2):175–185
Jestoi M, Rokka M, Järvenpää E, Peltonen K (2009) Determination of Fusarium mycotoxins beauvericin and enniatins (A, A1, B, B1) in eggs of laying hens using liquid chromatography–tandem mass spectrometry (LC–MS/MS). Food Chem 115(3):1120–1127
Bräse S, Encinas A, Keck J, Nising CF (2009) Chemistry and biology of mycotoxins and related fungal metabolites. Chem Rev 109(9):3903–3990
Hu L, Rychlik M (2012) Biosynthesis of 15N3-labeled enniatins and beauvericin and their application to stable isotope dilution assays. J Agric Food Chem 60(29):7129–7136
Bottalico A, Logrieco A (1998) Toxigenic Alternaria species of economic importance. In: Kaushal K, Bhatnagar D (eds) Mycotoxins in agriculture and food safety. Dekker, New York, pp 65–108
Fehr M, Pahlke G, Fritz J, Christensen MO, Boege F, Altemöller M, Podlech J, Marko D (2009) Alternariol acts as a topoisomerase poison, preferentially affecting the IIα isoform. Mol Nutr Food Res 53(4):441–451
Brugger E, Wagner J, Schumacher DM, Koch K, Podlech J, Metzler M, Lehmann L (2006) Mutagenicity of the mycotoxin alternariol in cultured mammalian cells. Toxicol Lett 164(3):221–230
Asam S, Konitzer K, Schieberle P, Rychlik M (2009) Stable isotope dilution assays of alternariol and alternariol monomethyl ether in beverages. J Agric Food Chem 57(12):5152–5160
Asam S, Konitzer K, Rychlik M (2011) Precise determination of the Alternaria mycotoxins alternariol and alternariol monomethyl ether in cereal, fruit and vegetable products using stable isotope dilution assays. Mycotoxin Res 27(1):23–28
Liu Y, Rychlik M (2015) Biosynthesis of seven carbon-13 labeled Alternaria toxins including altertoxins, alternariol, and alternariol methyl ether, and their application to a multiple stable isotope dilution assay. Anal Bioanal Chem 407(5):1357–1369
Pero RW, Posner H, Blois M, Harvan D, Spalding JW (1973) Toxicity of metabolites produced by the Alternaria. Environ Health Perspect 4:87–94
Asam S, Liu Y, Konitzer K, Rychlik M (2011) Development of a stable isotope dilution assay for tenuazonic acid. J Agric Food Chem 59(7):2980–2987
Siegel D, Merkel S, Bremser W, Koch M, Nehls I (2010) Degradation kinetics of the Alternaria mycotoxin tenuazonic acid in aqueous solutions. Anal Bioanal Chem 397(2):453–462
Siegel D, Rasenko T, Koch M, Nehls I (2009) Determination of the Alternaria mycotoxin tenuazonic acid in cereals by high-performance liquid chromatography-electrospray ionization ion-trap multistage mass spectrometry after derivatization with 2,4-dinitrophenylhydrazine. J Chromatogr, A 1216(21):4582–4588
Siegel D, Merkel S, Koch M, Nehls I (2010) Quantification of the Alternaria mycotoxin tenuazonic acid in beer. Food Chem 120(3):902–906
Asam S, Lichtenegger M, Liu Y, Rychlik M (2012) Content of the Alternaria mycotoxin tenuazonic acid in food commodities determined by a stable isotope dilution assay. Mycotoxin Res 28(1):9–15
Asam S, Rychlik M (2013) Potential health hazards due to the occurrence of the mycotoxin tenuazonic acid in infant food. Eur Food Res Technol 236(3):491–497
Kroes R, Renwick AG, Cheeseman M, Kleiner J, Mangelsdorf I, Piersma A, Schilter B, Schlatter J, van Schothorst F, Vos JG, Wurtzen G (2004) Structure-based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Food Chem Toxicol 42(1):65–83
Alexander J, Benford D, Boobis A, Ceccatelli S, Cottrill B, Cravedi J, Di Domenico A, Doerge D, Dogliotti E, Edler L, Farmer P, Filipic M, Fink-Gremmels J, Furst P, Guerin T, Knutsen HK, Machala M, Mutti A, Schlatter J, Rose M, van Leeuwen R, Metzler M, Rauscher-Gabernig E, van Peteghem C, Civera AV, Cioacata G, Curtui V, Eskola M, Heppner C (2011) Scientific opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J 9(10):2407
Asam S, Lichtenegger M, Muzik K, Liu Y, Frank O, Hofmann T, Rychlik M (2013) Development of analytical methods for the determination of tenuazonic acid analogues in food commodities. J Chromatogr, A 1289:27–36
Asam S, Habler K, Rychlik M (2013) Determination of tenuazonic acid in human urine by means of a stable isotope dilution assay. Anal Bioanal Chem 405(12):4149–4158
Van de Perre E, Deschuyffeleer N, Jacxsens L, Vekeman F, Van der Hauwaert W, Asam S, Rychlik M, Devlieghere F, De Meulenaer B (2014) Screening of moulds and mycotoxins in tomatoes, bell peppers, onions, soft red fruits and derived tomato products. Food Control 37:165–170
Walravens J, Mikula H, Rychlik M, Asam S, Ediage EN, Di Mavungu JD, Van Landschoot A, Vanhaecke L, De Saeger S (2014) Development and validation of an ultra-high-performance liquid chromatography tandem mass spectrometric method for the simultaneous determination of free and conjugated Alternaria toxins in cereal-based foodstuffs. J Chromatogr, A 1372:91–101
Lohrey L, Marschik S, Cramer B, Humpf H (2013) Large-scale synthesis of isotopically labeled 13C2-tenuazonic acid and development of a rapid HPLC-MS/MS method for the analysis of tenuazonic acid in tomato and pepper products. J Agric Food Chem 61(1):114–120
Stack ME, Prival MJ (1986) Mutagenicity of the Alternaria metabolites altertoxins I, II, and III. Appl Environ Microbiol 52(4):718–722
Fleck SC, Burkhardt B, Pfeiffer E, Metzler M (2012) Alternaria toxins: altertoxin II is a much stronger mutagen and DNA strand breaking mycotoxin than alternariol and its methyl ether in cultured mammalian cells. Toxicol Lett 214(1):27–32
Templeton GE, Grable CI, Fulton ND, Bollenbacher K (1967) Factors affecting the amount and pattern of chlorosis caused by a metabolite of Alternaria tenuis. Phytopathology 57(5):516–518
Kono Y, Gardner JM, Takeuchi S (1986) Nonselective phytotoxins simultaneously produced with host-selective ACTG-toxins by a pathotype of Alternaria citri causing brown spot disease of mandarins. Agric Biol Chem 50(9):2401–2403
Arntzen CJ (1972) Inhibition of photophosphorylation by tentoxin, a cyclic tetrapeptide. Biochim Biophys Acta 283(3):539–542
Liu Y, Rychlik M (2013) Development of a stable isotope dilution LC–MS/MS method for the Alternaria toxins tentoxin, dihydrotentoxin, and isotentoxin. J Agric Food Chem 61(12):2970–2978
Lindenmeier M, Schieberle P, Rychlik M (2004) Quantitation of ochratoxin A in foods by a stable isotope dilution assay using high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr, A 1023:57–66
Noba S, Uyama A, Mochizuki N (2009) Determination of ochratoxin A in ready-to-drink coffee by immunoaffinity cleanup and liquid chromatography-tandem mass spectrometry. J Agric Food Chem 57(14):6036–6040
Han Z, Zhao Z, Shi J, Liao Y, Zhao Z, Zhang D, Wu Y, De Saeger S, Wu A (2013) Combinatorial approach of LC–MS/MS and LC–TOF-MS for uncovering in vivo kinetics and biotransformation of ochratoxin A in rat. J Chromatogr, B 925:46–53
Cramer B, Königs M, Humpf HU (2013) Identification and in vitro cytotoxicity of Ochratoxin A degradation products formed during coffee roasting. J Agric Food Chem 56(14):5673–5681
Korn M, Frank O, Hofmann T, Rychlik M (2011) Development of stable isotope dilution assays for ochratoxin A in blood samples. Anal Biochem 419:88–94
Bouisseau A, Roland A, Reillon F, Schneider R, Cavelier F (2013) First synthesis of a stable isotope of Ochratoxin A metabolite for a reliable detoxification monitoring. Org Lett 15(15):3888–3890
Roland A, Bros P, Bouisseau A, Cavelier F, Schneider R (2014) Analysis of ochratoxin A in grapes, musts and wines by LC–MS/MS: First comparison of stable isotope dilution assay and diastereomeric dilution assay methods. Anal Chim Acta 818(1):39–45
Gabriele B, Attya M, Fazio A, Di Donna L, Plastina P, Sindona G (2009) A new and expedient total synthesis of ochratoxin A and d5-ochratoxin A. Synthesis 2009(11):1815–1820
Lenz CA, Rychlik M (2013) Efficient synthesis of (R)-ochratoxin alpha, the key precursor to the mycotoxin ochratoxin A. Tetrahedron Lett 54(8):883–886
Pereira VL, Fernandes JO, Cunha SC (2014) Mycotoxins in cereals and related foodstuffs: a review on occurrence and recent methods of analysis. Trends Food Sci Technol 36(2):96–136
Herebian D, Zühlke S, Lamshöft M, Spiteller M (2009) Multi-mycotoxin analysis in complex biological matrices using LC-ESI/MS: experimental study using triple stage quadrupole and LTQ-Orbitrap. J Sep Sci 32(7):939–948
Gottschalk C, Barthel J, Engelhardt G, Bauer J, Meyer K (2009) Simultaneous determination of type A, B and D trichothecenes and their occurrence in cereals and cereal products. Food Addit Contamin Part A 26(9):1273–1289
Vaclavik L, Zachariasova M, Hrbek V, Hajslova J (2010) Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta 82(5):1950–1957
Zachariasova M, Lacina O, Malachova A, Kostelanska M, Poustka J, Godula M, Hajslova J (2010) Novel approaches in analysis of Fusarium mycotoxins in cereals employing ultra performance liquid chromatography coupled with high resolution mass spectrometry. Anal Chim Acta 662(1):51–61
Tam J, Pantazopoulos P, Scott P, Moisey J, Dabeka R, Richard I (2011) Application of isotope dilution mass spectrometry: determination of ochratoxin A in the Canadian Total Diet Study. Food Addit Contamin Part A 28(6):754–761
Pettersson H, Brown C, Hauk J, Hoth S, Meyer J, Wessels D (2011) Survey of T-2 and HT-2 toxins by LC–MS/MS in oats and oat products from European oat mills in 2005–2009. Food Addit Contamin Part B 4(2):110–115
Juan C, Ritieni A, Mañes J (2012) Determination of trichothecenes and zearalenones in grain cereal, flour and bread by liquid chromatography tandem mass spectrometry. Food Chem 134(4):2389–2397
Varga E, Glauner T, Koeppen R, Mayer K, Sulyok M, Schuhmacher R, Krska R, Berthiller F (2012) Stable isotope dilution assay for the accurate determination of mycotoxins in maize by UHPLC-MS/MS. Anal Bioanal Chem 402(9):2675–2686
Barthel J, Gottschalk C, Rapp M, Berger M, Bauer J, Meyer K (2012) Occurrence of type A, B and D trichothecenes in barley and barley products from the Bavarian market. Mycotoxin Res 28(2):97–106
Köppen R, Bremser W, Rasenko T, Koch M (2013) Development and certification of a reference material for Fusarium mycotoxins in wheat flour. Anal Bioanal Chem 405(14):4755–4763
Rychlik M, Humpf H, Marko D, Dänicke S, Mally A, Berthiller F, Klaffke H, Lorenz N (2014) Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res 30(4):197–205
Kluger B, Bueschl C, Lemmens M, Berthiller F, Häubl G, Jaunecker G, Adam G, Krska R, Schuhmacher R (2013) Stable isotopic labelling-assisted untargeted metabolic profiling reveals novel conjugates of the mycotoxin deoxynivalenol in wheat. Anal Bioanal Chem 405(15):5031–5036
Joint FAO/WHO Expert Committee on Food Additives (2011) Safety evaluation of certain contaminants in food / prepared by the seventy-second meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). WHO food additives series: 63
European Food Safety Authority (2014) Evaluation of the increase of risk for public health related to a possible temporary derogation from the maximum level of deoxynivalenol, zearalenone and fumonisins for maize and maize products. EFSA J 12(5):3699–3730
Liao C, Wong JW, Zhang K, Yang P, Wittenberg JB, Trucksess MW, Hayward DG, Lee NS, Chang JS (2014) Multi-mycotoxin analysis of finished grain and nut products using ultrahigh-performance liquid chromatography and positive electrospray ionization–quadrupole orbital ion trap high-resolution mass spectrometry. J Agric Food Chem. doi:10.1021/jf505049a
Mata AT, Ferreira JP, Oliveira BR, Batoreu MC, Barreto Crespo MT, Pereira VJ, Bronze MR (2015) Bottled water: analysis of mycotoxins by LC-MS/MS. Food Chem 176:455–464
Kirincic S, Skrjanc B, Kos N, Kozolc B, Pirnat N, Tavcar-Kalcher G (2015) Mycotoxins in cereals and cereal products in Slovenia - official control of foods in the years 2008-2012. Food Control 50:157–165
Brezina U, Rempe I, Kersten S, Valenta H, Humpf H, Daenicke S (2014) Diagnosis of intoxications of piglets fed with Fusarium toxin-contaminated maize by the analysis of mycotoxin residues in serum, liquor and urine with LC-MS/MS. Arch Anim Nutr 68(6):425–447
Zhang K, Wong JW, Jia Z, Vaclavikova M, Trucksess MW, Begley TH (2014) Screening multimycotoxins in food-grade gums by stable isotope dilution and liquid chromatography/tandem mass spectrometry. J AOAC Int 97(3):889–895
Zhang K, Wong JW, Krynitsky AJ, Trucksess MW (2014) Determining mycotoxins in baby foods and animal feeds using stable isotope dilution and liquid chromatography tandem mass spectrometry. J Agric Food Chem 62(36):8935–8943
Zhao Z, Rao Q, Song S, Liu N, Han Z, Hou J, Wu A (2014) Simultaneous determination of major type B trichothecenes and deoxynivalenol-3-glucoside in animal feed and raw materials using improved DSPE combined with LC-MS/MS. J. Chromatogr B Anal Technol Biomed Life Sci 963:75–82
Pietruszka K, Sell B, Burek O, Wisniewska-Dmytrow H (2013) Simultaneous determination of multi - component mycotoxin in feeds by liquid chromatography - tandem mass spectrometry. Bull Vet Inst Pulawy 57(4):567–572
Han Z, Feng Z, Shi W, Zhao Z, Wu Y, Wu A (2014) A quick, easy, cheap, effective, rugged, and safe sample pretreatment and liquid chromatography with tandem mass spectrometry method for the simultaneous quantification of 33 mycotoxins in Lentinula edodes. J Sep Sci 37(15):1957–1966
Desmarchelier A, Tessiot S, Bessaire T, Racault L, Fiorese E, Urbani A, Chan W, Cheng P, Mottier P (2014) Combining the quick, easy, cheap, effective, rugged and safe approach and clean-up by immunoaffinity column for the analysis of 15 mycotoxins by isotope dilution liquid chromatography tandem mass spectrometry. J Chromatogr, A 1337:75–84
Blajet-Kosicka A, Twaruzek M, Kosicki R, Sibiorowska E, Grajewski J (2014) Co-occurrence and evaluation of mycotoxins in organic and conventional rye grain and products. Food Control 38:61–66
Abia WA, Warth B, Sulyok M, Krska R, Tchana A, Njobeh PB, Turner PC, Kouanfack C, Eyongetah M, Dutton M, Moundipa PF (2013) Bio-monitoring of mycotoxin exposure in Cameroon using a urinary multi-biomarker approach. Food Chem Toxicol 62:927–934
Gratz SW, Duncan G, Richardson AJ (2013) The human fecal microbiota metabolizes deoxynivalenol and deoxynivalenol-3-glucoside and may be responsible for urinary deepoxy-deoxynivalenol. Appl Environ Microbiol 79(6):1821–1825
Osselaere A, Devreese M, Goossens J, Vandenbroucke V, De Baere S, De Backer P, Croubels S (2013) Toxicokinetic study and absolute oral bioavailability of deoxynivalenol, T-2 toxin and zearalenone in broiler chickens. Food Chem Toxicol 51:350–355
Al-Taher F, Banaszewski K, Jackson L, Zweigenbaum J, Ryu D, Cappozzo J (2013) Rapid method for the determination of multiple mycotoxins in wines and beers by LC-MS/MS using a stable isotope dilution assay. J Agric Food Chem 61(10):2378–2384
Zhang K, Wong JW, Hayward DG, Vaclavikova M, Liao C, Trucksess MW (2013) Determination of mycotoxins in milk-based products and infant formula using stable isotope dilution assay and liquid chromatography tandem mass spectrometry. J Agric Food Chem 61(26):6265–6273
De Baere S, Goossens J, Osselaere A, Devreese M, Vandenbroucke V, De Backer P, Croubels S (2011) Quantitative determination of T-2 toxin, HT-2 toxin, deoxynivalenol and deepoxy-deoxynivalenol in animal body fluids using LC-MS/MS detection. J Chromatogr, B: Anal Technol Biomed Life Sci 879(24):2403–2415
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Asam, S., Rychlik, M. Recent developments in stable isotope dilution assays in mycotoxin analysis with special regard to Alternaria toxins. Anal Bioanal Chem 407, 7563–7577 (2015). https://doi.org/10.1007/s00216-015-8904-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8904-y