
Abstract
This review summarizes the advances in environmental analysis by liquid chromatography–high-resolution mass spectrometry (LC–HRMS) during the last decade and discusses different aspects of their application. LC–HRMS has become a powerful tool for simultaneous quantitative and qualitative analysis of organic pollutants, enabling their quantitation and the search for metabolites and transformation products or the detection of unknown compounds. LC–HRMS provides more information than low-resolution (LR) MS for each sample because it can accurately determine the mass of the molecular ion and its fragment ions if it can be used for MS–MS. Another advantage is that the data can be processed using either target analysis, suspect screening, retrospective analysis, or non-target screening. With the growing popularity and acceptance of HRMS analysis, current guidelines for compound confirmation need to be revised for quantitative and qualitative purposes. Furthermore, new commercial software and user-built libraries are required to mine data in an efficient and comprehensive way. The scope of this critical review is not to provide a comprehensive overview of the many studies performed with LC–HRMS in the field of environmental analysis, but to reveal its advantages and limitations using different workflows.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The challenges that can be encountered when analyzing polar organic pollutants in environmental samples are diverse. One of the challenges is that there are a plethora of organic contaminants of environmental concern with different physicochemical properties. The number of organic pollutants reported in the literature is increasing annually, with new pollutants, the so-called “contaminants of emerging concern”, which are frequently present at low concentrations in the environment, making the analysis of environmental samples evermore challenging. This is mainly caused by the necessity of including large numbers of contaminants in a single analysis because this reduces analysis times. Liquid chromatography coupled to mass spectrometry (LC–MS) is a sophisticated hyphenation of analytical techniques which enables the determination of organic pollutants in complex environmental matrices because of its selectivity and sensitivity. Since the 1990s, when LC coupled to triple-quadrupole mass spectrometry (QqQ-MS) started to be marketed, operating in multiple-reaction-monitoring (MRM) mode, this technique has been the first choice for quantitative analysis because of its high sensitivity and selectivity against a complex matrix background. However, high-resolution mass spectrometry (HRMS) has in recent years become more accessible to research laboratories with the development of Orbitrap-MS-based instruments and the improvements to time-of-flight (TOF) MS systems. Several publications describing the new generation of HRMS systems [1] and revealing differences between low-resolution (LR) MS and high-resolution capabilities [2,3] testify to the growing importance of HRMS. It should be kept in mind that HRMS analyzers can also be coupled with gas chromatography, but it has been revealed that LC enables analysis of a broader range of contaminants with very different polarities and chemical properties. The use of HRMS enables rapid, selective, and robust analysis in both qualitative and quantitative applications. The ease of setup and the possibility of storing a high volume of full-scan and MS–MS data of high mass accuracy enables retrospective analysis without the need to re-run samples. In addition, the data can be processed using target analysis, suspect screening, and non-target screening (Fig. 1). The reliability of these approaches relies on the main features of the mass analyzer, including mass accuracy, precision, and resolving power; the combination of these characteristics is crucial for correctly measuring ion masses in the presence of interfering matrix components occurring in complex environmental samples. In addition, the creation of compound spectra libraries in conjunction with software packages makes the data analysis and interpretation easier. This opens up the possibility of different specialized types of analysis including screening of suspects, target and non-target analysis, and structural elucidation of novel metabolites and transformation products (TPs).

From target to non-target analysis
Several studies have been performed in recent years using LC–HRMS for environmental analysis of different classes of pollutants, and some reviews have been published which discuss these works [4–7]. For these reasons, in this paper we focus on the most recent published articles on environmental analysis using LC–HRMS for qualitative and quantitative purposes. Particular attention has been dedicated to recent aspects related to the use of HRMS for quantitative analysis in comparison to low-resolution analyzers, and to the latest trends in detection and structural identification of contaminants and their related metabolites and TPs, a field where HRMS is surely the standard technique.
Quantitative analysis by LC–HRMS
The sensitive detection and reliable quantitation of a large number of contaminants at low concentrations in environmental studies has been always one of the most important challenges. The use of LC–LRMS in MRM mode requires as a first step the definition of the target analytes to be monitored, and then standards have to be acquired to optimize compound-specific MS conditions including fragment-ion masses, ion-source voltages, and collision energies. In addition the LC gradient has to be adjusted such that all compounds can be accommodated in a large number of retention-time windows. Breaking up the chromatographic time into several experiments is essential to minimize the number of MRM transitions, because this has a direct effect on the sensitivity. Although with modern QqQ-MS analyzers the inter-channel dead time is reduced to a few milliseconds, the increasing number of compounds and therefore of MRM transitions always comes with a loss of sensitivity. When optimizing MRM conditions, several data points per peak always have to be critically considered. Instead of detecting compounds by one or more compound-specific transitions, no pre-selection of precursor ions is necessary when working in scan mode with HRMS because a high selectivity can be achieved in full-scan mode, and thus an unlimited number of analytes can be detected. This means that a theoretically unlimited numbers of analytes can be searched for without compromising sensitivity, because the acquisitions have been made as “all ions all the time”. This is useful for both target and so-called post-target analysis where the presence of additional contaminants can be also evaluated by retrospective analysis of the data. Then, if there are positive findings, further quantification can be performed, reducing the cost and time of analysis.
Unlike QqQ-LRMS, where selectivity is achieved by selecting one or more suitable MRM transitions, HRMS including (Q)TOFMS and Orbitrap MS usually relies only on the detection of the ions (isotope cluster and isotope spacing can be used as additional criteria for confirmation). Therefore, it is essential that the instrument is operated at a high resolving power such that potentially interfering peaks are resolved. If this is not achieved, mass accuracy is compromised, and when using a mass extraction window of a few ppm (or mDa) the interfering compound is included in this window, resulting in increased signal of the targeted ion and eventually an overestimation of its concentration. This has been emphasized in recent publications that compare selectivity provided by HRMS instruments and QqQ-LRMS [3,8]. In these works the objective of the authors was to identify the HRMS resolution necessary to achieve the same selectivity as the LRMS detector. Using the HRMS Orbitrap Exactive, they concluded that resolution above 50,000 (full width at half maximum, FWHM, at m/z 200) afforded selectivity higher than that provided by current unit-resolving QqQ-MS instruments in MRM mode. A relatively novel MS technique is the ABSciex instrument Triple TOFMS, which is designed to offer MRMHR mode for quantitative analysis which measures the accurate mass of the product ions of a selected precursor ion. Furthermore, the Triple TOFMS in MRMHR aids compound identification with highest confidence via comparison of theoretical and measured mass accuracy and isotopic pattern. Equal or even slightly better quantitative and confirmative performance can be observed for the HRMS instruments [3]. In conclusion, if the resolution of the instrument is not sufficient, false-positive findings can occur even if the experimentally measured exact mass seems to match the ion mass of the compound of interest. The risk of inaccurate quantification is particularly high when two chromatographically coeluting compounds have almost identical m/z values, because at insufficient resolving power their intensities combine to form a single peak which eventually leads to overestimation of the analyte concentration. By contrast, underestimation of the concentration may occur when a very narrow mass window is used to extract the analyte mass from such a mixed mass peak. Although the presence of an analyte and unresolved interference might still be detected in a mass spectrum recorded in profile mode (visual inspection reveals a shoulder peak), centroided data can completely shift out of the expected mass-accuracy window. Ferrer and Thurman [9] revealed the possibility of distinguishing analytes from interferences and analytes with very close m/z value (i.e. analytes with the same nominal mass) with HRMS. They analyzed lamotrigine and the metabolite of bupropion, hydroxy-bupropion, with the same nominal mass in wastewater extracts. Both compounds eluted at very similar retention times and the exact masses of the protonated molecules differed by 0.0948 mass units. The authors calculated that the resolving power needed to separate the two compounds was 6000, and performed a reliable quantification with a QTOFMS instrument. Assuming the resolving power is sufficiently high to achieve the necessary selectivity, the variables to be evaluated for method validation are very similar to those for LRMS. These include linearity, repeatability, reproducibility, recovery, matrix effects, and limits of detection (LOD) and quantification.
One of the most commonly used methods to estimate the LOD in LRMS is based on the signal-to-noise (S/N) ratios [10]. By contrast, in HRMS instruments the background noise in the chromatogram is often very low, or even zero for Orbitrap MS instruments in which by default a baseline noise cut-off is applied by the instrument control software to reduce data-file size, making the S/N approach sometimes impracticable. For this reason, alternative methods to calculate LODs have been proposed. One of them defines the LOD as the concentration that gives a peak intensity of at least 1.0 × 104 counts [11]. In another approach [12], instead of LOD, the authors propose to use the limit of identification, which is calculated as the minimum concentration that provides a fragment ion with a mass error <5 ppm [13]. The S/N ratio criterion, however, is still applied in HRMS [14].
As regards the comparison of the linear range, in which (Q)TOFMS and Orbitrap MS provide accurate quantitative measurement over a wide range of analyte concentrations, the Orbitrap has been proved to offer a linear range of at least four orders of magnitude. Modern TOF instruments achieve comparable performance to Orbitrap with respect to linear range, but this is not currently achievable on common hybrid QTOFMS systems. A notable exception is the “Triple TOF” series, which competes directly with triple-quadrupole MS in the field of quantitative analysis. This QTOF-based MS operates at an acquisition frequency of as high as 100 Hz, and promises a linear range of greater than five orders of magnitude and low limits of quantitation equivalent to high-performance triple quadrupoles.
To guarantee reliable confirmation, rules or guidelines are necessary not only for LRMS but also for approaches based on HRMS. In the latter case, the most commonly applied rules for confirming the presence of a target compound in the sample are:
-
1.
detection with a mass error of less than 5 ppm;
-
2.
a matching retention time with respect to the authentic standard retention time within 2.5 % and
-
3.
an isotopic pattern in line with the elemental composition.
Wille et al. [12] quantified pesticides and pharmaceuticals in environmental samples using only the exact mass of the molecular ions deriving from the full-scan acquisition that fulfilled the aforementioned restrictions. Unlike LRMS, for which well-defined criteria have been established for confirming analyte presence, the corresponding criteria for HRMS are still under discussion. According to the EU guideline [15], the presence of a banned drug is confirmed when a specific number of identification points has been achieved. Working with QqQ-MS, one molecular ion gives 1.0 identification point and each of the two transitions 1.5 identification points. In addition, the measured retention time of the suspected peak has to match that of the standard. Finally, the area ratio between the two monitored MRM traces must be identical for the sample and for the standard. As for HRMS, the fact that resolving power may vary largely between instruments and/or techniques makes the definition of general criteria difficult. The EU guidelines state that a monitored ion measured at a resolution of 10,000 yields two identification points. However, this resolution refers to sector MS instruments, for which resolution is defined at 10 % of the peak height. Modern HRMS instruments, including TOF and Orbitrap, use as reference value the peak width at 50 % height. Hence, 10,000 at 10 % corresponds approximately to 20,000 at 50 % (FWHM). Another point to be taken into account is the m/z-value-dependent resolution; i.e., in TOF analyzers the resolution is fairly constant over a broad m/z range, whereas in the Orbitrap analyzer resolution drops exponentially with increasing m/z [16–19]. These aspects are discussed in detail in Ref. [20].
The reliable confirmation of an analyte measured by HRMS has been much improved with the availability of hybrid instruments including QTOFMS, Orbitrap MS, and Triple TOFMS. As well as all the characteristics of full-scan acquisition, the selection of precursor ions and subsequent generation of product-ion spectra are highly valuable because they provide compound-specific fragmentation patterns [21]. In fact, there are different ways of generating fragment ions in an unbiased manner. One way is the MSE mode available on some QTOFMS instruments, consisting of alternating full-spectrum acquisitions at low and high collision energies. The analogous experiment on Orbitrap MS is the so-called all-ion fragmentation (AIF), in which a full MS scan is combined with a nonselective precursor-ion fragmentation performed in the high-energy-collisional-dissociation (HCD) cell. AIF using HCD in Orbitrap MS and collision-induced dissociation (CID) on QqQ-MS often share common main fragments; however, CID and HCD fragmentations in the hybrid Velos Pro-hybrid Ion Trap-Orbitrap MS may be complementary [21,22]. As with QqQ-MS, the extent of fragmentation is compound-specific, and therefore a compromise in the collision-energy setting has to be made to provide as much confirmatory information as possible [23]. Although the generation of fragment ions without precursor-ion selection requires a more careful interpretation when assigning fragment ions to precursor ions, it has proved to be very helpful in confirming compound identity.
As far as the standard approach to precursor-ion selection is concerned, this is undoubtedly the most reliable method and the confirmation of analyte identity is straightforward. Depending on the MS technique the product-ion spectra can contain accurate mass data, as generated by default by QTOFMS and some instruments from the Orbitrap family, or can be low-resolution spectra, e.g. when fragment ions are generated in the ion trap but, instead of being sent to the Orbitrap analyzer, are detected at the ion-trap detector. In contrast with the aforementioned unbiased ion fragmentation (MSE and AIF), the acquisition of true product-ion spectra always requires a criterion to define the selection of the ion to be isolated for subsequent fragmentation. This can be based on a predefined precursor-ion list or simply based on ion abundance. During this so-called information-dependent acquisition (IDA) or data-dependent acquisition (DDA), the instrument automatically switches after a full-scan-mode acquisition to a product-ion scan mode as the second scan event in the scan cycles.
Literature on this aspect is increasing, and works on the analysis of pharmaceuticals or pesticides in the environment using hybrid instruments have recently been published [11,24]. The high-mass-resolution capabilities of the LTQ Orbitrap MS including an HCD cell were exploited for the determination of trace contaminants, enabling straightforward discrimination between analytes and matrix, and the dependent-scan functions of the Orbitrap MS using LIT MS and an HCD cell were evaluated and compared for the confirmation of analytes at trace concentrations. The authors concluded that data-dependent scanning using LIT MS is more suitable for trace environmental analysis than a data-dependent scan using HCD because of the slower scan times of the latter [24]. The LODs of the HRMS approach were in the low ng L−1 range (0.0007–0.0088 μg L−1), revealing a sensitivity comparable to that of the data produced using the QqQ-MS instruments which have been the standard methods for quantitative analysis [24].
It can therefore be concluded that the most important advantage of using hybrid mass spectrometers, rather than QqQ-MS instruments or single-stage HRMS techniques, is to combine different kinds of study in a single method. This is particularly useful in environmental applications where the tasks of target analysis and of structural elucidation of transformation products and determination of their occurrence are equally important. In the recent work by Negreria et al. [25] the applicability of this combined approach was revealed for the elucidation of biotransformation products and subsequent quantitative target analysis.
It should be kept in mind, however, that quantitative studies always require reference standards of the targeted analytes, contaminants to be determined, and their metabolites or transformation products. In many cases their availability is a severe problem: they are either not commercially available, or prohibitively high prices make the purchase unrealistic. A possible work-around of this challenge is in-silico studies which attempt to predict the ionization efficiency in the electrospray-ionization source [26,27]. This interesting approach, however, requires more research to better understand the relevant factors defining ESI efficiency [28].
Qualitative analysis by LC–HRMS
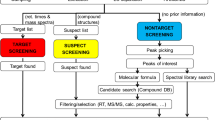
HRMS systems have been primarily used for qualitative environmental analysis. LC–HRMS has mostly been used as a powerful tool for the identification of TPs or metabolites of different contaminants generated in laboratory-scale studies under controlled conditions [29,30]. This approach consists of a manual comparison of control samples and treated samples or detection of the TPs by creating a list of possible TPs from the literature or with in-silico methods (Fig. 2). Alignment and comparison of the total ion chromatograms of treated and control samples from lab-scale experiments enable detection of new peaks originating from the transformation of the test compounds [31,32]. Mass-accuracy errors of up to 5 ppm are usually accepted regardless of the mass analyzers used. These accurate mass measurements and complementary data, including the isotope cluster of the molecular ion, the comparison of the fragmentation patterns of the related parent compounds, and retention times, enable elucidation of the compound structure (Fig. 2). Eventually the identified compounds are included in the procedures for environmental monitoring [33,34]. A study using four MS techniques (LIT MS, QqQ-MS, LTQ Orbitrap, and TOFMS) for the identification of human metabolites of amitriptyline and verapamil was performed by Rousu et al. [35]. They compared their suitability for metabolite screening, considering the abilities to detect metabolites and to provide structural information for their identification, and addressed additional aspects of the workload of each instrument including time consumption and data processing. For the detection of metabolites different approaches were used: scan mode for HRMS instruments, a combination of neutral-loss and product-ion spectra for QqQ-MS, and information-dependent acquisition (IDA) with MRM for LIT MS. Data from each instrument was processed separately. Using the TOFMS approach, 28 and 69 metabolites were confirmed for amitriptyline and verapamil, respectively. However, with the other three instruments less than 50 % of these metabolites were detected. Although LTQ Orbitrap had better mass accuracy, TOFMS had more sensitivity and faster data acquisition than LTQ Orbitrap MS because of the inherently low data-acquisition rates in full-scan-type analysis for the latter. The QTRAP and triple-quadrupole MS had poor sensitivity and time-consuming sample analysis [35]. Therefore, the high sensitivity in full-scan mode has made LC–HRMS the most suitable tool for the development of screening methods, enabling the detection of a large number of emerging contaminants without prior selection of compounds [36]. Thus, LC–HRMS-based screening methods are unquestionably an important advance in the determination of environmental pollutants, with hundreds to thousands of organic pollutants and their TPs or metabolites under scrutiny. In general, what is now expected from HRMS instruments is to provide as much as information as possible from a single run of each sample without the need for reanalysis. In 2010 Krauss et al. [5] classified qualitative analysis of environmental samples into two categories: suspect screening and non-target screening (Fig. 3). Both methods are usually used in a complementary way, enabling the detection of suspect pollutants and the identification of unknowns [37]. Hernandez et al. proposed the additional term post-target for when the search for pollutants is performed after HRMS acquisition without prior information.

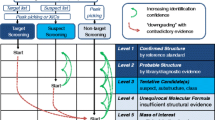
General workflow for detection of relevant TPs in environmental samples with identification confidence levels in accordance with Schymanski et al. 2014 [38]

General workflow for qualitative HRMS approaches
For suspect analysis, elemental composition and structure are used to create databases of exact ion masses for the expected protonated or deprotonated molecules or their common adducts (Fig. 3). These libraries are created with suspect emerging pollutants and, sometimes, completed with their metabolites or TPs, either manually or by use of transformation-prediction software. After accurate mass extraction of expected ions, specific information on each compound is used to confirm plausible identities. Isotopic pattern, MS–MS spectra, fragmentation pathways, retention times, or mass defects can be found in the literature or predicted by appropriate software. The probability of false-positive findings decreases when more compound-specific properties are available. Schymanski et al. [38] proposed a system of five levels of confidence in the identification. In the end, reference standards must be used to achieve the top level of confirmation of the compound identity.
For example, Wode et al. [39] recently developed a method for the screening of 2188 compounds with Orbitrap MS. The TraceFinder software provided isotopic-pattern matching of the accurate mass measurement. Suspects with an isotopic-pattern score below 80 % were regarded as false-positive. Finally, 55 compounds were identified by analytical standards. The authors could not take advantage of spectral libraries for the verification of other suspects because there is no possibility of obtaining an MS2 spectrum with a single-stage Orbitrap. In fact, the use of single-stage mass spectrometers in AIF mode, where all precursor ions are fragmented without the possibility of isolating precursor ions, limits the study to all fragment ions derived from the fragmentation in the HCD cell.
With respect to the aforementioned limitation, hybrid HRMS systems are gaining popularity because of their ability to accurately measure product-ion masses. High-quality MS–MS spectra are produced by the two different approaches mentioned previously for quantitative confirmation: non-selective fragmentation and data-dependent MS–MS acquisitions. The major disadvantage of the first approach is the difficulty of assigning fragment ions to the molecular ion, because there is no connection between them; sometimes this task can be delegated to deconvolution software. In contrast, MS–MS acquisitions can provide information for a small set of compounds which then can be searched for in spectra libraries. Inclusion lists are often applied to the screening methods to perform pre-target suspect analysis. Coupling an Orbitrap with an LIT (hybrid LIT series) enables the combination of two complementary product-ion scans in HRMS, HCD, and CID, obtaining more structural data for the identification of TPs and metabolites. Thus, with this approach there is a gain in product-ion information that cannot be obtained on typical ion-trap instruments. The two product-ion scans complement each other by generating different spectra. For instance, in a recent paper the application of dual collision cells in LTQ Orbitrap MS, HCD, and CID in a data-dependent scan enhanced the structural elucidation of human metabolites of selected pharmaceuticals [40]. Such an acquisition procedure can be an advantage over the approaches that can be performed with QqQ-MS and (Q)TOFMS instruments.
Regardless of whether AIF or data-dependent MS–MS switching modes are run, the use of specific software to streamline the data-mining process, and ultimately the confirmation, is regarded as essential.
As an example of new approaches with Q Exactive Orbitrap HRMS for TP detection and identification, Zonja et al. [41] used the standard approach based on lab-scale experiments combined with suspect screening of real surface-water samples. In this study, a sunlight simulator was used to degrade relevant pollutants of emerging concern, six iodinated contrast media (ICM), in surface-water samples. SIEVE software was used for the chromatographic-peak alignment and enabled the detection of 108 photoproducts in the photodegraded samples. Instead of performing the structural elucidation of and search for all TPs in environmental samples, a database was created with experimentally determined accurate masses, retention times, and MS–MS data of the 108 TPs for further suspect screening of real surface-water extracts. This approach enabled the prioritization of eleven environmentally relevant TPs, on the basis of their detection frequency in real samples, for subsequent structure elucidation. Finally, standards of the TPs were obtained by semipreparative LC, and quantitative analysis of the parent compounds and their prioritized TPs was performed in real surface-water extracts.
In contrast with the lab-scale methods, Kern et al. [42] used a different approach to determine the occurrence of TPs directly in natural waters by HRMS using LTQ Orbitrap. To this end, the authors created a list containing known aquatic contaminants, and predicted their TPs by means of the University of Minnesota Pathway Prediction System (UM-PPS) and literature information. Suspect screening was performed using an accurate database of 1794 compounds. When the target list contains a large number of compounds, comparison with blanks and estimated retention times and isotope patterns is essential to filter for positive results. This approach enabled the identification of 19 TPs, later confirmed by reference standards. In a subsequent study, Kern et al. [43] combined detection of the predicted TPs, degradation in batch reactors, and the calculated mass balances of wastewater treatment plants (WWTPs) to quantify transformation rates.
Wang et al. [44] developed a suspect-screening method to identify phase II metabolites of pharmaceuticals in reclaimed water with Q Exactive Orbitrap MS. In this study, the MetWorks software was used to find expected phase II metabolites of the most commonly detected pharmaceuticals. The identification of metabolites was only performed when the parent drug was detectable. As a criterion for positive findings, they expected an earlier retention time of a metabolite except in the case of acetylation. HRMS analysis was performed in DDA mode, meaning peaks from full-scan chromatograms with intensities lower than 1 × 105 counts were not considered. This study reported the detection and identification of sulfamethoxazole glucuronide and acetylsulfamethoxazole, two phase II metabolites of sulfamethoxazole.
Figure 2 summarizes the approaches used for detection and identification of TPs in environmental samples. Recently, Bletsou et al. [45] reviewed the state-of-the-art of LC–HRMS for the identification of TPs in the aquatic environment. A prioritization step is essential to optimize our efforts in the elucidation of more relevant TPs, and clearly software development is crucial for the prediction of transformations and for detection of candidates and their elucidation. As an example, Jeon et al. [46] used UM-PPS and Meteor Environmental Pathway Prediction System (Lhasa Limited, UK) for biotransformation prediction, MetWorks and SIEVE for candidate detection, Xcalibur for data analysis, Mass Frontier (HighChem, Ltd., Bratislava, Slovakia) and MetFrag for the prediction of MS–MS fragmentation, and even MOLGEN for molecular-structure generation. In this study 23 of the 360 predicted metabolites were detected and identified. Out of the 19 oxidation products identified, 12 metabolites were predicted by Meteor and seven by UM-PPS. Consequently, the combination of different software programs and the development of new ones are required.
In particular, software designed for the management of mass-spectral data provides another possibility for the detection of phase II metabolites or other compounds that provide common fragment ions through fragment-ion search. From these ions, related compounds are detected and tentatively identified [47]. Several authors refer to this approach as a semi-non-target approach because one searches for known fragment ions of unknown pollutants. In this way, Hernandez et al. [6] differentiated between unbiased non-target analysis and biased non-target analysis. In general, the objective of non-target screening is to analyze environmental samples without any information on the compounds present in the sample, and to detect and identify relevant compounds. Thus, the prioritization of compounds that will be selected for identification becomes an important step of non-target analysis [48]. In unbiased non-target analysis, peaks for the identification could be selected on the basis of intensity, detection frequency, and toxicity (for bioassay-directed analysis) and/or in a comparison of samples and blanks. In biased non-target analysis, the method screens for compounds with concrete properties including specific mass defects and distinctive isotope pattern or fragment ions.
The general workflow for non-target studies consists of automatic peak detection, prioritization, determination of elemental composition, structural elucidation, and library spectrum matching and ranking of the candidates (Fig. 3). Automated peak detection and spectra deconvolution algorithms are required, and the success is limited to the prioritized compounds. The lack of comprehensive (LC–)MS libraries hampers the identification process [48]. In short, non-target screening can be summarized in two steps: the reduction of peaks included in the identification procedure and the reduction and ranking of candidates [37]. Whereas in suspect screening compound identities are known, in non-target analysis calculated molecular formulas must be evaluated. Consequently, the application of non-target screening requires a large investment in data analysis. Some software packages, including MOLGEN and the Seven Golden Rules (7GR) afford an automated analysis [49]. Currently, the identification of unknown pollutants remains a tedious and time-consuming task. Researchers usually have to rely on a combination of complementary software packages, user-built libraries, and literature information to propose plausible compound identities.
In addition, as mentioned above, the use of publicly available LC–HRMS databases, including MassBank, ChemSpider, NIST, HMDB, METLIN, and MetFrag, for the identification of “known unknown” compounds is helpful but still limited.
Usually, qualitative studies have performed non-target, suspect, and target analysis together [50,51] in so-called “all in one” analysis. For instance, the studies by Schymansky et al. [52] revealed the capability of LC–HRMS to perform analysis of wastewaters by taking advantage of these different approaches. First, an accurate mass list was compiled with enviMass and processed by the R package “non-target” (Eawag, Switzerland). This software selects the most intense peak of each unknown compound and creates groups with all related peaks into one component to associate isotope and adduct peaks with the compound. After blank and noise subtraction, an intensity-based prioritization was performed. In (−)ESI MS, four of the 30 most intense peaks were determined by their target screening method; also, 15 of these peaks contained S, enabling the identification of seven compounds by searching a list of 394 compounds which contain sulfur. Finally, non-target workflows were used for the elucidation of the remaining unknown compounds. As a result, of the 26 most intense non-target peaks, seven were tentatively identified and one identified and confirmed by use of a reference standard.
As a promising approach for the screening of unknown toxicant pollutants, LC–HRMS identification could be prioritized through effect-directed analysis (EDA). Weiss et al. [53] used SIEVE to discriminate the peaks between active and non-active fractions and followed a non-target workflow to identify the most toxic unknowns. This approach enabled the identification of eight androgen-disrupting compounds.
As well as false-positive findings, which are reduced by different strategies, false-negative findings are also plausible in qualitative analysis without reference standards. Several compounds might not be ionized or are not extracted by the sample-preparation method. To this end, sample procedures including direct-injection or large-volume-injection LC–HRMS of water samples can be an alternative to avoid selective pre-concentration [54,55]. To increase the range of polarity and volatility, recent studies combined HRMS with both LC and gas chromatography [56].
Conclusions and future advances
In summary, LC–HRMS is a powerful tool for quantitative and qualitative analysis, enabling the quantitation of target compounds and the search for and identification of suspected or unknown pollutants. A recent trend in LC–HRMS-based environmental studies is to perform “all-in-one” analysis, i.e. simultaneous target analysis (quantitative), suspect screening, and non-target determination (qualitative). Comparing the performance for quantitative analysis, LC–HRMS has sensitivity comparable to that of LC–QqQ-MS, whereas the selectivity—achieved on QqQ-MS by choosing suitable MRM transitions—is largely dependent on the resolving power of the instrument. Only sufficiently high resolving power can guarantee that co-eluting matrix components can be distinguished from compounds of interest. The high sensitivity in conjunction with the wealth of unbiased information gathered means that LC–HRMS is increasingly competing with LC–QqQ-MS in environmental research laboratories. With the growing popularity and acceptance, one of the aspects that needs to be addressed is the criteria used in compound confirmation.
There are some differences between different HRMS instruments for quantitative analysis. (Q)TOFMS instruments were initially used for qualitative analysis. However, the latest instruments on the market have improved quantitative capabilities, providing a good approach for quantification combined with the power of exact mass. However, the Orbitrap instruments try to achieve higher sensitivity with higher mass accuracy. A disadvantage of the Orbitrap mass analyzer is that the mass resolution of Orbitrap is inversely related to scan speed, meaning high-resolving-power settings imply low acquisition frequencies. By contrast, TOFMS instruments are known to produce essentially the same resolution across different scan rates. Therefore, to achieve UPLC separations, the acquisition rate of Orbitrap MS needs to be decreased at the expense of resolving power. This disadvantage is particularly important when the minimum number of points per peak cannot be achieved. However, the scan rate for Orbitrap technology is faster and attempts are being made to provide a sufficient number of scans (≥10) across the chromatographic peak in full-scan mode. As regards mass accuracy, whereas most TOFMS instruments require internal calibration for continuous correction of the mass axis, the Orbitrap analyzer is capable of maintaining mass accuracy over several days after initial calibration. With internal calibration in (Q)TOFMS, the ionization of the calibrant(s) may interfere with the ionization of the analytes. Moreover, the calibrant signal may collapse in the presence of high matrix loads because of ion suppression, and its intensity may vary with changing mobile-phase composition during gradient elution. For obvious reasons, the choice of an appropriate internal calibrant has to be such that spectral interferences with the analytes of interest can be ruled out.
Regarding qualitative analysis, LC–HRMS provides more information about each sample than LRMS instruments, enabling suspect and non-target screening without retrospective analysis. Different workflows based on their accurate mass measurements have been used to reduce the number of false-positive findings. New commercial software and the availability of public HRMS libraries of environmental contaminants and their TPs could help mine the data from controlled laboratory experiments (conducted at high concentrations) in an efficient and comprehensive fashion. Regarding non-target analysis, more studies on the prediction of retention times, ionization behavior, and MS–MS fragmentation are still needed and are expected to eventually deliver the computational tools essential for easier identification of unknowns.
References
Eichhorn P, Pérez S, Barceló D (2012) Time-of-flight mass spectrometry versus orbitrap-based mass spectrometry for the screening and identification of drugs and metabolites: is there a winner? Compr Anal Chem 58:217–272
J. Aceña, D. Rivas, B. Zonja, S. Péreza, D. Barcelóa, Liquid Chromatography–Mass Spectrometry: Quantification and Confirmation Aspects
Kaufmann A, Butcher P, Maden K, Walker S, Widmer M (2011) Quantitative and confirmative performance of liquid chromatography coupled to high‐resolution mass spectrometry compared to tandem mass spectrometry. Rapid Commun Mass Spectrom 25:979–992
Hernández F, Sancho J, Ibáñez M, Abad E, Portolés T, Mattioli L (2012) Current use of high-resolution mass spectrometry in the environmental sciences. Anal Bioanal Chem 403:1251–1264
Krauss M, Singer H, Hollender J (2010) LC–high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal Bioanal Chem 397:943–951
Hernández F, Ibáñez M, Bade R, Bijlsma L, Sancho J (2014) Investigation of pharmaceuticals and illicit drugs in waters by liquid chromatography-high-resolution mass spectrometry. TrAC Trends Anal Chem 63:140–157
Agüera A, Martínez Bueno M, Fernández-Alba A (2013) New trends in the analytical determination of emerging contaminants and their transformation products in environmental waters. Environ Sci Pollut Res 20:3496–3515
Kaufmann A, Butcher P, Maden K, Walker S, Widmer M (2010) Comprehensive comparison of liquid chromatography selectivity as provided by two types of liquid chromatography detectors (high resolution mass spectrometry and tandem mass spectrometry):“Where is the crossover point?”. Anal Chim Acta 673:60–72
Ferrer I, Thurman EM (2012) Analysis of 100 pharmaceuticals and their degradates in water samples by liquid chromatography/quadrupole time-of-flight mass spectrometry. J Chromatogr A 1259:148–157
Shrivastava A, Gupta VB (2011) Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chronicles of Young Sci 2:21
Farré M, Picó Y, Barceló D (2014) Application of ultra-high pressure liquid chromatography linear ion-trap orbitrap to qualitative and quantitative assessment of pesticide residues. J Chromatogr A 1328:66–79
Wille K, Claessens M, Rappé K, Monteyne E, Janssen CR, De Brabander HF, Vanhaecke L (2011) Rapid quantification of pharmaceuticals and pesticides in passive samplers using ultra high performance liquid chromatography coupled to high resolution mass spectrometry. J Chromatogr A 1218:9162–9173
Padilla-Sánchez JA, Plaza-Bolaños P, Romero-González R, Grande-Martínez Á, Thurman EM, Garrido-Frenich A (2012) Innovative determination of polar organophosphonate pesticides based on high‐resolution Orbitrap mass spectrometry. J Mass Spectrom 47:1458–1465
Vergeynst L, Haeck A, De Wispelaere P, Van Langenhove H, Demeestere K (2015) Multi-residue analysis of pharmaceuticals in wastewater by liquid chromatography–magnetic sector mass spectrometry: Method quality assessment and application in a Belgian case study. Chemosphere 119:S2–S8
Commission E (2002) Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Comm L 221:8–36
Nurmi J, Pellinen J (2011) Multiresidue method for the analysis of emerging contaminants in wastewater by ultra performance liquid chromatography–time-of-flight mass spectrometry. J Chromatogr A 1218:6712–6719
Wille K, Bussche JV, Noppe H, De Wulf E, Van Caeter P, Janssen C, De Brabander H, Vanhaecke L (2010) A validated analytical method for the determination of perfluorinated compounds in surface-, sea-and sewagewater using liquid chromatography coupled to time-of-flight mass spectrometry. J Chromatogr A 1217:6616–6622
Lara-Martín PA, González-Mazo E, Brownawell BJ (2011) Multi-residue method for the analysis of synthetic surfactants and their degradation metabolites in aquatic systems by liquid chromatography–time-of-flight-mass spectrometry. J Chromatogr A 1218:4799–4807
Farré M, Gros M, Hernandez B, Petrovic M, Hancock P, Barceló D (2008) Analysis of biologically active compounds in water by ultra‐performance liquid chromatography quadrupole time‐of‐flight mass spectrometry. Rapid Commun Mass Spectrom 22:41–51
Nielen M, Van Engelen M, Zuiderent R, Ramaker R (2007) Screening and confirmation criteria for hormone residue analysis using liquid chromatography accurate mass time-of-flight, Fourier transform ion cyclotron resonance and orbitrap mass spectrometry techniques. Anal Chim Acta 586:122–129
Makarov A, Scigelova M (2010) Coupling liquid chromatography to Orbitrap mass spectrometry. J Chromatogr A 1217:3938–3945
K. Comstock, Y (2011) Huang, Multiple fragmentation methods for small molecule characterization on a dual pressure linear ion trap Orbitrap hybrid mass spectrometer, Application Note, 540
Coscollà C, León N, Pastor A, Yusà V (2014) Combined target and post-run target strategy for a comprehensive analysis of pesticides in ambient air using liquid chromatography-Orbitrap high resolution mass spectrometry. J Chromatogr A 1368:132–142
Cahill MG, Dineen BA, Stack MA, James KJ (2012) A critical evaluation of liquid chromatography with hybrid linear ion trap—Orbitrap mass spectrometry for the determination of acidic contaminants in wastewater effluents. J Chromatogr A 1270:88–95
Negreira N, López de Alda M, Barceló D (2015) Degradation of the cytostatic etoposide in chlorinated water by liquid chromatography coupled to quadrupole-Orbitrap mass spectrometry: Identification and quantification of by-products in real water samples. Sci Total Environ 506–507:36–45
Chalcraft KR, Lee R, Mills C, Britz-McKibbin P (2009) Virtual Quantification of Metabolites by Capillary Electrophoresis-Electrospray Ionization-Mass Spectrometry: Predicting Ionization Efficiency Without Chemical Standards. Anal Chem 81:2506–2515
Kruve A, Kaupmees K, Liigand J, Leito I (2014) Negative electrospray ionization via deprotonation: predicting the ionization efficiency. Anal Chem 86:4822–4830
Oss M, Kruve A, Herodes K, Leito I (2010) Electrospray ionization efficiency scale of organic compounds. Anal Chem 82:2865–2872
B. Zonja, J. Aceña, S. Pérez, D. Barceló (2013) Chapter 16 - Methods for Elucidation of Transformation Pathways: Identification of Intermediate Products, Chiral, and Isotope-Ratio Mass Spectrometry Analysis, in: D.B. Mira Petrovic, P. Sandra (Eds.) Comprehensive Analytical Chemistry, Elsevier, pp. 593–610
Eichhorn P, Ferguson PL, Pérez S, Aga DS (2005) Application of Ion Trap-MS with H/D Exchange and QqTOF-MS in the Identification of Microbial Degradates of Trimethoprim in Nitrifying Activated Sludge. Anal Chem 77:4176–4184
Pérez S, Eichhorn P, Celiz MD, Aga DS (2006) Structural Characterization of Metabolites of the X-ray Contrast Agent Iopromide in Activated Sludge Using Ion Trap Mass Spectrometry. Anal Chem 78:1866–1874
Pérez S, Barceló D (2008) First Evidence for Occurrence of Hydroxylated Human Metabolites of Diclofenac and Aceclofenac in Wastewater Using QqLIT-MS and QqTOF-MS. Anal Chem 80:8135–8145
Aceña J, Pérez S, Gardinali P, Abad JL, Eichhorn P, Heuett N, Barceló D (2014) Structure elucidation of phototransformation products of unapproved analogs of the erectile dysfunction drug sildenafil in artificial freshwater with UPLC-Q Exactive-MS. J Mass Spectrom 49:1279–1289
Zonja B, Gonçalves C, Pérez S, Delgado A, Petrovic M, Alpendurada MF, Barceló D (2014) Evaluation of the phototransformation of the antiviral zanamivir in surface waters through identification of transformation products. J Hazard Mater 265:296–304
Rousu T, Herttuainen J, Tolonen A (2010) Comparison of triple quadrupole, hybrid linear ion trap triple quadrupole, time-of-flight and LTQ-Orbitrap mass spectrometers in drug discovery phase metabolite screening and identification in vitro – amitriptyline and verapamil as model compounds. Rapid Commun Mass Spectrom 24:939–957
Moschet C, Piazzoli A, Singer H, Hollender J (2013) Alleviating the reference standard dilemma using a systematic exact mass suspect screening approach with liquid chromatography-high resolution mass spectrometry. Anal Chem 85:10312–10320
Hug C, Ulrich N, Schulze T, Brack W, Krauss M (2014) Identification of novel micropollutants in wastewater by a combination of suspect and nontarget screening. Environ Pollut 184:25–32
Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J (2014) Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ Sci Technol 48:2097–2098
Wode F, van Baar P, Dünnbier U, Hecht F, Taute T, Jekel M, Reemtsma T (2015) Search for over 2000 current and legacy micropollutants on a wastewater infiltration site with a UPLC-high resolution MS target screening method. Water Res 69:274–283
Bushee JL, Argikar UA (2011) An experimental approach to enhance precursor ion fragmentation for metabolite identification studies: application of dual collision cells in an orbital trap. Rapid Commun Mass Spectrom 25:1356–1362
Zonja B, Delgado A, Pérez S, Barceló D (2015) LC-HRMS Suspect Screening for Detection-Based Prioritization of Iodinated Contrast Media Photodegradates in Surface Waters. Environ Sci Technol 49:3464–3472
Kern S, Fenner K, Singer HP, Schwarzenbach RP, Hollender J (2009) Identification of transformation products of organic contaminants in natural waters by computer-aided prediction and high-resolution mass spectrometry. Environ Sci Technol 43:7039–7046
Kern S, Baumgartner R, Helbling DE, Hollender J, Singer H, Loos MJ, Schwarzenbach RP, Fenner K (2010) A tiered procedure for assessing the formation of biotransformation products of pharmaceuticals and biocides during activated sludge treatment. J Environ Monit 12:2100–2111
Wang J, Gardinali PR (2014) Identification of phase II pharmaceutical metabolites in reclaimed water using high resolution benchtop Orbitrap mass spectrometry. Chemosphere 107:65–73
A.A. Bletsou, J. Jeon, J. Hollender, E. Archontaki, N.S. Thomaidis (2015) Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment, TrAC Trends Anal Chem
Jeon J, Kurth D, Hollender J (2013) Biotransformation Pathways of Biocides and Pharmaceuticals in Freshwater Crustaceans Based on Structure Elucidation of Metabolites Using High Resolution Mass Spectrometry. Chem Res Toxicol 26:313–324
Gómez-Ramos MM, Pérez-Parada A, García-Reyes JF, Fernández-Alba AR, Agüera A (2011) Use of an accurate-mass database for the systematic identification of transformation products of organic contaminants in wastewater effluents. J Chromatogr A 1218:8002–8012
Zedda M, Zwiener C (2012) Is nontarget screening of emerging contaminants by LC-HRMS successful? A plea for compound libraries and computer tools. Anal Bioanal Chem 403:2493–2502
Kind T, Fiehn O (2007) Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinforma 8:105
Chiaia-Hernandez AC, Schymanski EL, Kumar P, Singer HP, Hollender J (2014) Suspect and nontarget screening approaches to identify organic contaminant records in lake sediments. Anal Bioanal Chem 406:7323–7335
Hogenboom A, Van Leerdam J, de Voogt P (2009) Accurate mass screening and identification of emerging contaminants in environmental samples by liquid chromatography–hybrid linear ion trap Orbitrap mass spectrometry. J Chromatogr A 1216:510–519
Schymanski EL, Singer HP, Longrée P, Loos M, Ruff M, Stravs MA, Ripollés Vidal C, Hollender J (2014) Strategies to characterize polar organic contamination in wastewater: exploring the capability of high resolution mass spectrometry. Environ Sci Technol 48:1811–1818
Weiss J, Simon E, Stroomberg G, de Boer R, de Boer J, van der Linden S, Leonards PG, Lamoree M (2011) Identification strategy for unknown pollutants using high-resolution mass spectrometry: Androgen-disrupting compounds identified through effect-directed analysis. Anal Bioanal Chem 400:3141–3149
Vergeynst L, Van Langenhove H, Joos P, Demeestere K (2014) Suspect screening and target quantification of multi-class pharmaceuticals in surface water based on large-volume injection liquid chromatography and time-of-flight mass spectrometry. Anal Bioanal Chem 406:2533–2547
Martínez Bueno MJ, Ulaszewska MM, Gomez MJ, Hernando MD, Fernández-Alba AR (2012) Simultaneous measurement in mass and mass/mass mode for accurate qualitative and quantitative screening analysis of pharmaceuticals in river water. J Chromatogr A 1256:80–88
Hernández F, Ibáñez M, Portolés T, Cervera MI, Sancho JV, López FJ (2015) Advancing towards universal screening for organic pollutants in waters. J Hazard Mater 282:86–95
Acknowledgments
S. Pérez acknowledges the contract from the Ramon y Cajal Program of the Spanish Ministry of Economy and Competitiveness. This work has been financially supported by the Generalitat de Catalunya (Consolidated Research Groups “2014 SGR 418 - Water and Soil Quality Unit”, 2014 SGR 291.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection High-Resolution Mass Spectrometry in Food and Environmental Analysis with guest editor Aldo Laganà.
Rights and permissions
About this article

Cite this article
Aceña, J., Stampachiacchiere, S., Pérez, S. et al. Advances in liquid chromatography–high-resolution mass spectrometry for quantitative and qualitative environmental analysis. Anal Bioanal Chem 407, 6289–6299 (2015). https://doi.org/10.1007/s00216-015-8852-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8852-6