
Abstract
A three-stage linear gradient strategy using reverse-phase high-performance liquid chromatography (HPLC) was optimized for rapid, high-quality, and simultaneous purification of the lipopeptide isoforms of iturin, fengycin, and surfactin, which may differ in composition by only a single amino acid and/or the fatty acid residue. Matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry (MALDI-TOF-MS/MS) was applied to detect the lipopeptides harvested from each reversed-phase HPLC peak. Amino acid analysis based on phenyl isothiocyanate derivatization was further used for confirmation of the amino acid species and molar ratio in a certain HPLC fraction. By this MALDI-TOF-MS/MS coupled with amino acid analysis, it was revealed that iturin at m/z 1,043 consists of a circular Asn-Tyr-Asn-Gln-Pro-Asn-Ser peptide and C14 β-OH fatty acid. Surfactin homologs from Bacillus subtilis THY-7 at m/z 1,030, 1,044, 1,058, and 1,072 possess a circular Glu-Leu-Leu-Val-Asp-Leu-Leu peptide and the β-OH fatty acid with a different length (C13–C16). Fengycin species at m/z 1,463 and 1,477 are homologs possessing the circular peptide Glu-Orn-Tyr-Thr-Glu-Ala-Pro-Gln-Tyr-Ile linked to a C16 or C17 γ-OH fatty acid, whereas fengycin at m/z 1,505 contains a Glu-Orn-Tyr-Thr-Glu-Val-Pro-Gln-Tyr-Ile sequence with a Val instead of Ala at position 6. The method developed in this work provided an efficient approach for characterization of diverse lipopeptide isoforms from the iturin, fengycin, and surfactin families.
Similar content being viewed by others
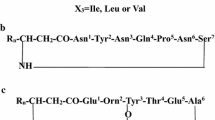

Avoid common mistakes on your manuscript.
Introduction
Lipopeptide biosurfactants produced by members of the genera Bacillus, Pseudomonas, Streptomyces, and Arthrobacter as secondary metabolites [1] are amphiphilic molecules with excellent surface activity and antifungal or antibacterial activities. Bacillus sp., such as Bacillus subtilis [2, 3], Bacillus cereus [4], and Bacillus amyloliquefaciens [5], have been reported as major producers of lipopeptide biosurfactants. Several Bacillus sp. strains have been screened to study their lipopeptide products for potential applications in the last few decades [6–8].
The three most well-known families of lipopeptides are the iturins [9, 10], the fengycins [11, 12], and the surfactins [13]. In addition, some other molecules [14–17], such as kurstarkin, mycosubtilin, bacillomycin, and lichenysin, with similar structures have also been identified. Surfactin contains a cyclic lactone ring consisting of a C12–C16 β-hydroxy fatty acid and a heptapeptide with a variable amino acid at positions 2, 4, and 7 [18]. Iturin is a lactam containing a C14–C17 β-amino fatty acid attached to a heptapeptide with Asp or Asn at position 1 [19]. Fengycin is composed of a β-hydroxy fatty acid chain attached to a decapeptide, which also forms a cyclic lactone ring. Two variants, fengycin A and fengycin B, have been reported with Val or Ala at position 6 [20, 21]. These lipopeptides are synthesized by nonribosomal peptide synthetases [22, 23], which can generate many variants that differ in the fatty acid chain length and amino acid sequence. Some microorganisms can simultaneously produce different families of lipopeptides. These two factors result in the production of complex mixtures of microbial lipopeptide products containing isoforms of iturins, fengycins, and surfactins, among others.
Acid precipitation is the most widely used method to primarily separate lipopeptides from cell-free fermentation broth on a laboratory scale [24]. The long fatty acid chains contribute to high solubility in organic solvents. Therefore, acid precipitation is usually followed by extraction using methanol, dichloromethane, chloroform, or ethyl acetate. Both chloroform–methanol in ratio of 2:1 [25, 26] and methanol alone [20] are commonly used for the extraction of lipopeptides. In consideration of the high toxicity of chloroform, we chose methanol alone in the extraction.
Reverse-phase (RP) high-performance liquid chromatography (HPLC) is highly suitable for the purification of molecules with different hydrophilicity and has been applied to discriminate between homologs of lipopeptides. The different amino acids with diverse hydrophilic characteristics will interact with the mobile phase, and the fraction with higher hydrophilicity in the peptide ring will be eluted at a lower proportion of methanol/acetonitrile. On the other hand, the hydrophobic fatty acid chains will combine with the modified filler of a C18 column; thus, the fraction containing the longer fatty acid will be harder to elute from the column and a higher proportion of methanol/acetonitrile is required. Iturins, fengycins, and surfactins can be eluted by 40–50 % [27], 50–70 % [12], and 85–100 % [1] acetonitrile in water, respectively, on a C18 column in an RP-HPLC system.
Precolumn derivatization of amino acids with phenyl isothiocyanate (PITC) was developed in the 1980s, and proved to have high sensitivity, excellent derivative stability, and high-resolution separation of amino acids [28]. For example, the derivative loss was less than 5 % in 2 days at 4 °C and the response was linear over the range 10–2,000 pmol [29]. The regression equations with a correlation coefficient above 0.999 were obtained from chromatograms of PITC derivatives of 18 standard amino acids [28, 30].
Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) with its high sensitivity and high sample throughput is well suited for the rapid measurement of the molecular masses of intact bioactive peptides. Glycopeptides and lipopeptides (mass below 5,000 Da) containing free amino acid groups can be protonated efficiently using α-cyano-4-hydroxycinnamic acid as the matrix, and these compounds frequently give strong signals in spectra [31]. Surfactins and iturins in the m/z range 1,000–1,100 and fengycins in the m/z range 1,400–1,600 [3, 12, 13, 21, 32] can be detected by MALDI-TOF-MS in single protonated forms or as Na+ or K+ adducts in the m/z range 800–2,000. There is no report about multiple adducts (dimers or trimers) of iturin, fengycin, and surfactin families. Further determination of the amino acid sequence in the peptide relies on collision-induced dissociation (CID) in MS/MS experiments.
To identify different lipopeptide isoforms, effective separation and purification by RP-HPLC in a short time is important. Most studies have focused only on the separation of homologs of a certain family of lipopeptides with a property that meets their requirements. A few studies have detected two or three different lipopeptide families using a linear gradient [3, 12], but the elution time was too long and the isoform fractions were not thoroughly separated.
In this work, we optimized an efficient three-stage RP-HPLC strategy allowing simultaneous good separation of several iturin, fengycinand surfactin isoforms, which may differ in composition by only a single amino acid and/or the fatty acid residues. Subsequently, the identification of the amino acid sequence of peptides from their HPLC fractions was performed using MALDI-TOF-MS/MS assisted by amino acid analysis based on PITC derivatives. This systematic approach facilitates the simultaneous rapid discovery, high-quality purification, and molecular structure identification of isoforms of the iturin, fengycinand surfactin families of molecules from lipopeptide mixtures.
Materials and methods
Identification of the isolated strain
The strain used in this work, B. subtilis THY-7, was previously isolated from the sludge of a lotus pond at Tsinghua University. A 16S ribosomal RNA (rRNA) sequence (1,465 bp) was amplified from the strain by the primer pairs [33] 5′-AGAGTTTGATCCTGGTCAGAACGCT-3′ and 5′-TACGGCTACCTTGTTACGACTTCACCCC-3′. PCR amplification was performed in a total volume of 50 μL containing 0.5 μL template DNA, 5 μL Ex Taq reaction buffer, 4 μL dNTP (each at 2.5 mM), 1.5 U Ex Taq DNA polymerase and 1 μL of each primer (20 μM). The amplified 16S rRNA gene sequence was compared with ribosomal DNA sequences obtained from GenBank (http://www.ncbi.nlm.nih.gov/BLAST/).
Preparation of lipopeptide mixture from B. subtilis
A 5 % inoculum of B. subtilis THY-7 in Luria–Bertani medium was added to the fermentation medium (1 L: 60 g glucose, 1.0 g yeast extract, 25 g NaNO3, 0.333 g KH2PO4, 1.0 g Na2HPO4 · 12H2O, 0.15 g MgSO4 · 7H2O, 0.0075 g CaCl2, 0.006 g MnSO4 · H2O, 0.006 g FeSO4 · 7H2O, pH 7.0), and the culture was incubated for 48 h at 37 °C with shaking at 200 rpm. The fermentation broth was centrifuged at 12,000 g for 20 min. The pH of the supernatant was adjusted to 2.0 with 6 M HCl and was then stored overnight at 4 °C. The sample was centrifuged again at 12,000 g for 20 min to harvest the solid crude lipopeptides and then extracted with methanol. Insoluble impurities were removed by filtration, and the methanol was evaporated under a vacuum at 50 °C using a rotary evaporator to obtain the crude lipopeptides.
RP-HPLC
The lipopeptide mixture was dissolved in an aqueous solution of NaHCO3 at 1 g/L (the lipopeptide mixture at 2 g/L) and filtered through a 0.2 μm membrane filter. A 20 μL aliquot was injected into an InertSustain C18 column (5 μm, 250 mm × 4.6 mm) in the HPLC system to separate the isoforms. The 500 mg/L standard surfactin and iturin (purity of 98 % or greater, Sigma-Aldrich, Shanghai, China) were used to confirm the RP-HPLC fraction groups of three lipopeptide families. The mobile phases were water (A), methanol (B1), and acetonitrile (B2). All of the mobile phases contained 0.1 % trifluoroacetic acid (TFA). Optimization of the RP-HPLC strategy was done stepwise by adjusting the gradient program for the mobile phases, which focused on the concentration range of methanol/acetonitrile and the time of the gradient. The detailed gradient strategies used in the optimization are given in the figure captions. The final gradient strategy for the methanol–water mobile phase system was as follows: 0–3 min, 70 % methanol to 75 % methanol; 3–8 min, 75 % methanol to 85 % methanol; and 8–30 min, 85 % methanol to 95 % methanol. The final optimized gradient strategy for the acetonitrile–water mobile phase system was as follows: 0–3 min, 45 % acetonitrile to 50 % acetonitrile; 3–8 min, 50 % acetonitrile to 80 % acetonitrile; 8–25 min, 80 % acetonitrile to 100 % acetonitrile. The total flow rate of the mobile phases was kept at 0.8 mL/min, and the chromatograms were obtained at 205 nm. The fractions were harvested manually for amino acid analysis and MALDI-TOF mass spectra analysis.
Amino acid analysis
Each fraction was harvested from its HPLC fraction using the vacuum freeze-drying method. Each fraction (3.0 mg) was hydrolyzed with 1 mL of 6 M HCl at 110 °C for 24 h in a sealed tube. The fatty acid residue was extracted three times using 1 mL ether, and the aqueous phase of the hydrolysis solution was dried at 50 °C under a vacuum to remove residual ether and HCl. Then, the dried sample was redissolved in 10 mL double-distilled water.
The analysis of amino acids was based on a precolumn derivatization with PITC [30, 34, 35]. Eighteen amino acids (Sigma-Aldrich, Shanghai, China) were used for standard reference: Asp, Glu, Ser, Gly, His, Arg, Thr, Ala, Pro, Tyr, Val, Met, Cys, Ile, Leu, Phe, Trp, and Lys. A 200 μL aliquot of the 18 standard amino acids (each at 30 mg/L) or the redissolved hydrolysis solution was mixed with 100 μL of 0.1 M PITC and 1 M triethylamine in acetonitrile after 10 μL of a 0.5 g/L solution of norleucine had been added as an internal standard for quantitative calculation. Then, the mixture was vortexed for 1 min and incubated for 30 min at room temperature. After derivatization, 400 μL n-hexane was added and the mixture was vortexed for 1 min and allowed to stand for 5 min. The bottom acetonitrile phase was filtered through a 0.22-μm polytetrafluoroethylene membrane filter before 6 μL (2 μL for the standard amino acid mixture reference) was injected onto an InertSustain C18 column (5 μm, 4.6 mm × 250 mm) at 38 °C. The derivative was stored at 4 °C if it was not analyzed within 2 h when a large number of samples were derived at a time, because the derivative stability is excellent, with less than 5 % loss in 2 days at 4 °C [28]. The mobile phases were as follows: 0.1 M sodium acetate (pH 6.5, adjusted with acetic acid)–acetonitrile (A; 93:7, v/v), and acetonitrile–water (B; 4:1, v/v). The proportion of the mobile phases was controlled using the following gradient program: 0 min, 0 % mobile phase B; 13 min, 7 % mobile phase B; 23 min, 23 % mobile phase B; 29 min, 35 % mobile phase B; 35 min, 40 % mobile phase B; 40 min, 100 % mobile phase B; 45 min, 100 % mobile phase B; 50 min, 0 % mobile phase B; and 60 min, 0 % mobile phase B. The mobile phase was kept at a flow rate of 1.0 mL/min, and the chromatograms were obtained at 254 nm.
MALDI-TOF-MS and MALDI-TOF-MS/MS analysis
MALDI-TOF analysis was used to elucidate the structure of HPLC-purified lipopeptides using a SCIEX 4800 Plus analyzer (Applied Biosystems, Foster City, CA, USA) with a 337-nm nitrogen laser for desorption and ionization. An equal volume of 0.1 % α-cyano-4-hydroxycinnamic acid was used as the matrix in a matrix-to-analyte ratio between 1:1 and 10:1. Mass spectra were accumulated over 50 individual laser shots and were obtained in reflector mode (mass accuracy 0.2 Da or less, sensitivity: 100 amol neurotensin, signal-to-noise ratio of 10:1 or greater) at an initial accelerating voltage of 20 kV [31, 36]. The m/z values were measured in the range from 800 to 2,000.
MALDI-TOF-MS/MS coupled with CID with the same spectrometer was used to analyze the fragment ions of peptides for further characterization of the amino acid sequence. The precursor ions submitted to MS/MS experiments were selected by the MS1 set and then focused in a collision cell. The collision cell was floated at 2 kV to attain a collision energy of 2 keV. Air used as the collision gas was introduced at a pressure leading to an attenuation of the precursor ion beam by almost 70 %.
Results
Optimization of RP-HPLC separation of isoforms from a mixture of three lipopeptide families
The 16S rRNA sequence of B. subtilis THY-7 was submitted and deposited in GeneBank (accession no. KJ777688.1). Comparison of the sequence in GenBank revealed 100 % identity with B. subtilis H12 (accession no. KC441785.1). After fermentation for 48 h, we harvested lipopeptides by acid precipitation, methanol extraction, and rotary evaporation for the further RP-HPLC separation.
Optimization of the RP-HPLC strategy was done stepwise by adjusting the gradient program for the mobile phases (A, water; B1, methanol; B2, acetonitrile; all containing 0.1 % TFA). The flow rate of the mobile phases was kept at 0.8 mL/min.
Methanol and acetonitrile are widely used in RP-HPLC mobile phase systems. In regard to the most important property of polarity, methanol is similar to acetonitrile. In consideration of the toxicity, we firstly chose the methanol–water mobile phase system to separate and purify the lipopeptide isoforms. By adjusting the ratio of methanol in the mobile phase and the gradient program, we were able to control the retention times of the isoforms with different hydrophilic peptide rings and lipophilic fatty acid chains, as displayed in Fig. 1. The optimal chromatogram, displayed in Fig. 1d, demonstrated that the use of a methanol–water system with an appropriate strategy could realize relatively good separation of the iturin and surfactin isoforms, but was inadequate for the fengycin family.

Reverse-phase (RP) high-performance liquid chromatography (HPLC) chromatograms with different elution strategies in the methanol–water system. a 0–3 min, 50 % methanol to 55 % methanol; 3–13 min, 55 % methanol to 85 % methanol; and 13–28 min, 85 % methanol to 100 % methanol. b 0–3 min, 65 % methanol to 75 % methanol; 3–13 min, 75 % methanol to 85 % methanol; and 13–30 min, 85 % methanol to 100 % methanol. c 0–3 min, 75 % methanol to 80 % methanol; 3–10 min, 80 % methanol to 85 % methanol; and 10–30 min, 85 % methanol to 95 % methanol. d 0–3 min, 70 % methanol to 75 % methanol; 3–8 min, 75 % methanol to 85 % methanol; and 8–30 min, 85 % methanol to 95 % methanol
In most cases, the eluting capacity of acetonitrile is better than that of methanol. Therefore, we further explored the acetonitrile–water system as the mobile phase. The initial strategy was a linear gradient that increased from 30 to 100 % acetonitrile in water containing 0.1 % TFA for 60 min at a flow rate of 0.8 mL/min, similar to what was reported in the literature for lipopeptide antibiotics [3, 12]. Although the isoforms were separated (as shown in Fig. 2a), the elution time was too long (about 50 min). There were large time intervals between the groups, especially before the first lipopeptide component of itruin was eluted. Additionally, the second group of fengycin was too flat, indicating insufficient separation. Thus, it was necessary to reduce the retention time and adjust the gradient program systematically.

RP-HPLC chromatograms using different elution strategies in the acetonitrile–water system. a The initial strategy, eluting with a linear gradient increasing from 30 % acetonitrile to 100 % acetonitrile for 60 min. b Strategy I, the gradient program was 50 % acetonitrile to 75 % acetonitrile from 0 to 10 min and then 75 % acetonitrile to 100 % acetonitrile from 10 to 20 min. c Strategy II, the gradient program was as follows: 40 % acetonitrile from 0 to 3 min; 40 % acetonitrile to 50 % acetonitrile from 3 to 8 min; 50 % acetonitrile to 80 % acetonitrile from 8 to 13 min; and 80 % acetonitrile to 100 % acetonitrile from 13 to30 min. d The final optimized efficient three-stage strategy was as follows: 45 % acetonitrile to 50 % acetonitrile, from 0 to 3 min; 50 % acetonitrile to 80 % acetonitrile from 3 to 8 min; and 80 % acetonitrile to 100 % acetonitrile from 8 to 25 min
Strategy I significantly reduced the initial run time by 30 min, and all of the active fractions in the initial chromatogram were observed, as shown in Fig. 2b. However, the first fraction was eluted too early, and thus homologs with shorter fatty acid chains would be eluted earlier and mixed with pigments before 5 min. Furthermore, the second fraction group was still too flat.
The result of strategy II indicated that the elution ability of 40 % acetonitrile during the first 3 min was not enough. However, the second group of fractions were concentrated (Fig. 2c).
The final optimized three-stage elution strategy resulted in a significant reduction of run time, from 60 to 28 min, with good separation of all of the isoforms in the three families. The retention times were in the ranges of 6–10 min, 10–16 min, and 16–25 min, corresponding to the iturin, fengycin, and surfactin families of molecules, respectively, as shown in Fig. 2d. Twelve major fractions, which were eluted at retention times of 7.52, 8.89, 12.40, 12.92, 13.34, 13.84, 18.65, 19.84, 20.12, 21.06, 21.40, and 22.46 min, were marked as fractions 1–12 and were collected for MALDI-TOF-MS and amino acid analysis.
MALDI-TOF-MS of iturin, fengycin, and surfactin isoforms
Intense signals in the m/z ranges 1,000–1,100 and 1,400–1,600 were obtained in the MALDI-TOF-MS spectra of the lipopeptide mixture. In detail, the ions at m/z 1,044 and 1,058 and at m/z 1,065 and 1,079 were hypothesized to be surfactins and iturins, respectively, whereas the ions at m/z 1,485, 1,499, and 1,505 belonged to the fengycin family, as illustrated in Fig. 3a. MALDI-TOF-MS of the RP-HPLC fractions revealed a series of ions in the m/z range from 1,000 to 1,600. Peaks 1 and 2 in the first HPLC group were standard iturins containing C14 and C15 fatty acid chains, with series of H+, Na+, and K+ adduct ions at m/z 1,043, 1,065, and 1,081 (Fig. 3b) and at m/z 1,057, 1,079, and 1,095 (Fig. 3c). The mass spectra of HPLC peaks 3–6 in the subsequent group mainly exhibited the [fengycin + H]+ ions containing C16 and C 17 fatty acid chains at m/z 1,495, 1,463, 1,477 (Fig. 3d), and 1,505 (Fig. 3e). The mass spectra of HPLC peaks 7 and 9 showed the [surfactin + Na]+ ions at m/z 1,030 (Fig. 3f), the mass spectra of HPLC peak 8 and 11 showed the [surfactin + Na]+ ions at m/z 1,044, and the mass spectra of HPLC peaks 10 and 12 showed the [surfactin + Na]+ ions at m/z 1,058 (Fig. 3g) and 1,072, respectively. The MALDI-TOF-MS spectra of the other fractions are presented in Figs. S1–S6.

Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) spectra of the lipopeptide mixture and isoforms in HPLC peaks. a High mass intensity in the m/z range 1,000–1,100 proposed to contain surfactin and iturin ions, and in the m/z range 1,400–1,600 proposed to contain fengycin ions. b, c Iturins related to fractions 1 and 2 displayed a high mass intensity at m/z 1,043 and 1,057 in their protonated forms and at m/z 1,065 and 1,079 as the sodium adducts. d, e Protonated fengycins containing a C17 fatty acid chain with Ala or Val at position 6 related to fractions 5 and 6, displaying a high mass intensity at m/z 1,477 and 1,505. f, g Surfactin isoforms containing C14 and C15 fatty acid chains related to fractions 9 and 10, at m/z 1,030 and 1,058, respectively, as [M + Na]+
MALDI-TOF-MS/MS analysis of the HPLC fraction structures
Some of the isoforms in different HPLC fractions detected by MALDI-TOF-MS had the same m/z. Thus, it is essential to precisely identify their structure by MS/MS techniques, especially for the amino acid sequence of the peptide portion of the molecules.
Figure 4 displays the MALDI-TOF-MS/MS spectrum of the precursor ion at m/z 1,043 of HPLC fraction 1. Nucleophilic attack by the amide oxygen of the Gln side chain on the carbon center of the protonated amide bond leads to the formation of cyclic derivatives [37, 38]; thus, the C-terminal fragment of Gln leaves the parent ion. The MSn experiments studying the His effect by Wysocki et al. [39] demonstrated that the peptide tended to break at the N terminus of Pro. Therefore, the peptide ring mainly cleaved at Gln-Pro, between the C terminus of Gln and the N terminus of Pro. The series of b+ ions at m/z 1,043 → 915 → 801 → 638 → 524 → 299 → 212 represented cleavage along the peptide bonds by the loss of Gln, Asn, Tyr, Asn, C14 β-OH fatty acid, and Ser from the C terminus of Gln, whereas the y+ fragment ions at m/z 832 → 745 and 520 → 406 showed the loss of Ser and Asn. The MS/MS spectrum exhibiting b+ and y+ fragment ions confirmed the sequence of Pro-Asn-Ser–C14 β-OH fatty acid–Asn-Tyr-Asn-Gln.

MALDI-TOF-MS/MS spectrum of the iturin precursor ion at m/z 1,043. The spectrum exhibits b+ fragments of the precursor ion at m/z 212, 299, 524, 638, 801, and 915 and y+ fragments of the precursor ion at m/z 406, 520, 745, and 832. The precursor ion at m/z 1,043 was confirmed as an iturin consisting of Asn-Tyr-Asn-Gln-Pro-Asn-Ser and a C14 fatty acid chain
Figure 5c displays the MS/MS spectrum of the precursor ion at m/z 1,058 in HPLC fraction 10. The series of b+ fragment ions at m/z 1,058 → 945 → 832(−H2O, 814) → 717 revealed the loss of Leu-Leu-Asp from the C terminus. Furthermore, another typical set of y+ fragment ions at m/z 707 → 594 → 481 → 382 → 267 suggested the loss of the Leu-Leu-Val-Asp in the middle of the peptide chain. The peak at m/z 832 in Fig. 5c was formed by loss of the Leu-Leu from molecule, and the peak of m/z 814 resulted from the loss of Leu-Leu–OH2, which confirmed that the ester structure has been formed by the carboxyl group of Leu and the hydroxyl group of the aliphatic part in the surfactin molecule. The precursor ion at m/z 1,058 was demonstrated to be the sodium adduct of a surfactin containing a C15 β-OH fatty acid, whose peptide sequence was Glu-Leu-Leu-Val-Asp-Leu-Leu. Additionally, the MS/MS spectra of sodium adducted ions found at m/z 1,030 (Fig. 5a), 1,044 (Fig. 5b), and 1,072 (Fig. 5d) with a difference of 14 Da (−CH2–) proved to be homologs possessing the same peptide sequence but different C13, C14, and C16 β-OH fatty acids, respectively.

MALDI-TOF-MS/MS spectra of surfactin precursors containing C13–C16 fatty acid chains. a Surfactin precursor ion [M + Na]+ at m/z 1,030, containing a Glu-Leu-Leu-Val-Asp-Leu-Leu peptide and a C13 β-hydroxy fatty acid chain. b Surfactin precursor ion [M + Na]+ at m/z 1,044, containing a C14 β-hydroxy fatty acid chain. c Surfactin precursor ion [M + Na]+ at m/z 1,058, containing a C15 β-hydroxy fatty acid chain. d Surfactin precursor ion [M + Na]+ at m/z 1,072, containing a C16 β-hydroxy fatty acid chain
Another peptide sequence slightly different from the typical surfactin was discovered in fraction 9 (Fig. 6a) and fraction 11 (Fig. 6b). For the precursor ion at m/z 1,030 in HPLC fraction 9 (Fig. 6a), the significant ion series at m/z 1,030 → 931 → 818(−H2O,800) → 703 → 590 and 693 → 594 → 481 → 368 → 253 represented the sequence of Val-Leu-Asp-Leu from the C terminus and Val-Leu-Leu-Asp in the middle, suggesting the precursor ion possessed a peptide sequence of Glu-Val-Leu-Leu-Asp-Leu-Val and a C14 fatty acid.

Isoforms of surfactin containing a Glu-Val-Leu-Leu-Asp-Leu-Val peptide. a Surfactin precursor ion [M + Na]+ at m/z 1,030, containing a C14 β-hydroxy fatty acid chain. b Surfactin precursor ion [M + Na]+ at m/z 1,044, containing a C15 β-hydroxy fatty acid chain
Figure 7 displays the MALDI-TOF-MS/MS spectra of the precursor ions at m/z 1,463 in fraction 4, m/z 1,477 in fraction 5, and m/z 1,505 in fraction 6. The γ-OH fatty acyl moiety had the effect of stabilizing the positive charge on the N terminus. It encouraged proton migration to the amide bonds in the first two positions of the peptide chain, making these two positions more labile to bond cleavage. Thus, the precursor ions mainly cleaved at the Glu–Orn and Orn–Tyr bonds, resulting in the specific octapeptide ring ions at m/z 966 and 1,080 (Fig. 7a, b) and at m/z 994 and 1,108 (Fig. 7c), respectively [40]. The extra stability of the charged octapeptide ring system suppressed further fragmentation along the peptide backbone and thus minimized the amount of sequence information. We analyzed some other ionic species for further analytical information and found some ions derived from the octapeptide ring. Significant ion series at m/z 665 → 520(+H2O) → 389 → 226 → 115(+H2O) in Fig. 7b represented the sequence of Pro-Gln-Tyr-Ile-Tyr from the N terminus after the first cleavage of bonds between Ala/Val and Pro in the specific octapeptide ring ions. Besides, we also found z8 and z9 ions corresponding to the y8 and y9 ions with a loss of NH (15 Da). The ions in Fig. 7a and b agreed with fengycin containing the same peptide ring, but with a different length of fatty acid chain, C16 or C17. The ions in Fig. 7b and c agreed with fengycin containing a C17 fatty acid chain, with the only difference being Ala or Val at position 6. The amino acid sequence of the lactone ring could not be perfectly identified. In this situation, amino acid analysis based on PITC derivatives verified the identities of the amino acids and their molar ratios.

MALDI-TOF-MS/MS spectra of the fengycin precursor ions. a Precursor ion at m/z 1,463 with Ala at position 6, containing a C16 γ-hydroxy fatty acid chain. b Precursor ion at m/z 1,477 with Ala at position 6, containing a C17 γ-hydroxy fatty acid chain. c Precursor ion at m/z 1,505 with Val at position 6, containing a C17 γ-hydroxy fatty acid chain
Amino acid analysis of isoforms from the HPLC fractions
Fractions harvested from the HPLC fractions were hydrolyzed with 6 M HCl and derivatized with PITC after norleucine had been added as an internal standard. Figure 8 displays the chromatograms of the PITC derivatives of 18 standard amino acids (Fig. 8a) and amino acid residues from samples in fractions 5 and 6, which showed m/z values of 1,477 and 1,505, respectively, in their protonated forms. Figure 8b demonstrates that the hydrolyzed fraction of m/z 1,477 contained Glu (Gln), Thr, Ala, Pro, Tyr, Ile, and Orn in a molar ratio of 2.90:1.00:1.01:1.10:2.04:1.10:1.03, and Fig. 8c demonstrates that the hydrolyzed fraction of m/z 1,505 contained Glu (Gln), Thr, Pro, Tyr, Val, Ile, and Orn in a molar ratio of 3.06:1.00:1.16:2.17:1.10:1.15:1.07, which just differed in the substitution of Val for Ala. The amino acid analysis results were in perfect agreement with the amino acid sequence of the fengycin molecules, as illustrated in Fig. 7d. The amino acids of the major fraction at m/z 1,058 in fraction 10 identified by MALDI-TOF-MS/MS were also verified by amino acid analysis, as shown in Fig. S10.

Chromatograms of phenyl isothiocyanate derivatives of amino acids obtained by C18-RP-HPLC. a Standard amino acids: Asp, Glu, Ser, Gly, His, Arg, Thr, Ala, Pro, Tyr, Val, Met, Cys, Ile, Leu, Phe, Trp, and Lys. b The hydrolyzed sample of m/z 1,477 in fraction 5, with Ala at position 6. c The hydrolyzed sample of m/z 1,505 in fraction 6, with Val at position 6
Finally, the structures of 12 lipopeptide isoforms detected in 12 fractions from the RP-HPLC were all identified and are summarized in Table 1.
Discussion
Lipopeptide biosurfactants have the advantages of low toxicity, high efficiency, high biodegradability, and high sustainability compared with chemical surfactants. There are a wide range of potential applications in agriculture and industry [41–46], such as microbial-enhanced oil recovery and cosmetics. The iturin and fengycin families display strong antifungal and antibacterial activity [47, 48], and the surfactin family also displays hemolytic, antiviral, antimycoplasma, and antibacterial activities [49, 50].
For research in the fields of cosmetics and medicine, RP-HPLC is widely used for high-quality purification. Literature reports on lipopeptide separation usually focus on a certain family, such as the report by Yuan et al. [27], who eluted iturin using 40 % acetonitrile in water for 20 min. Additionally, Villegas-Escobar et al. [12] eluted fengycin between retention times of 26 and 50 min (48–60 % acetonitrile in water) and Liu et al. [35] eluted surfactin with 90 % methanol containing 0.05 % TFA for 24 min. Some studies have detected more than one family of molecules when the sample was eluted with a rather long gradient. Chen et al. [3] detected iturin, fengycin, and surfactin at the same time with acetonitrile increasing from 30 to 80 % for 60 min, but the fractions were not perfectly separated. With use of our three-stage gradient strategy, isoforms of iturin, fengycin, and surfactin were separated in groups at retention times in range of 6–10 min, 10–16 min, and 16–25 min, respectively. The purity of the RP-HPLC fractions was very high, and thus the MALDI-TOF-MS spectra exhibited only one series of [M + H]+, [M + Na]+, and [M + K]+ ions corresponding to a lipopeptide isoform in a fraction. Good purification of the isoforms is crucial for the structural identification and for some applications with strict demands of purity. An aim would be to optimize further the separation of molecules of the fengycin family.
MS/MS coupled with CID is a powerful tool for identifying the amino acid sequence of peptides. The mobile proton model provides a solid background for qualitatively understanding the fragmentation pathways of protonated peptides. The model states that protonated peptides activated under low-energy collision conditions fragment mainly by charge-directed reactions at the amide bonds along the backbone [38], forming structurally informative sequence ions, especially the b and y ions containing the N terminus and the C terminus, respectively. In double hydrogen transfer, the cleavage happens between alpha and beta atoms and two hydrogens are transferred to the alkoxy part including alpha atoms [51]. As a result, for the ester structure the H2O would be added to the C-terminal part and its mass would be increased by 18. MALDI-TOF-MS/MS affords high sensitivity and exhibits abundant singly charged sequence ions of the cyclic peptides without the need for hydrolytic treatment. However, partial base hydrolysis treatment (such as treatment with 1 M KOH at room temperature for 1 h [12, 14]) can contribute to produce adequate sequence information for the peptide, especially the fengycin family.
The precolumn derivatization method with PITC was proved to have enough derivative stability, high sensitivity, and high-resolution separation of 18 amino acids [28, 29]. The results of amino acid analysis further confirmed the amino acid species and molar ratio in a certain peptide, especially for the two isobaric amino acids Leu and Ile. Since the lipopeptide hydrolysate samples were treated with 6 M HCl, the Asn and Gln would become Asp and Glu. Thus, this analysis method could not distinguish Asn and Gln from Asp and Glu [52]. The peaks of Asp and Glu were considered as Asx and Glx, which could be verified in combination with the mass loss in the MS/MS spectra [14].
Conclusion
An efficient three-stage gradient RP-HPLC strategy was optimized for the rapid and high-quality purification of a lipopeptide mixture produced by B. subtilis. The methanol–water mobile phase system realized a relatively good separation of the iturin and surfactin isoforms. Further optimization in an acetonitrile–water mobile phase system resulted in a better simultaneous separation of the iturin, fengycin and surfactin families in groups with retention time ranges of 6–10 min, 10–16 min, and 16–25 min. The isoforms obtained using the optimized acetonitrile–water strategy showed a high level of purity in MALDI-TOF-MS. MALDI-TOF-MS/MS analysis precisely elucidated the amino acid sequence of the cyclic isoforms of iturin and surfactin harvested from RP-HPLC fractions without hydrolytic treatment. The typical cyclic fengycins mainly cleaved at Glu–Orn and Orn–Tyr bonds, and amino acid analysis based on PITC derivatization was used to verify the identities and molar ratios of the amino acids present. To sum up, we have presented a systematic approach for the simultaneous rapid discovery, high-quality purification, and characterization of lipopeptide isoforms from the iturin, fengycin, and surfactin families, which may differ in composition by only a single amino acid and/or the fatty acid residue.
References
Liu XY, Yang SZ, Mu BZ (2009) Production and characterization of a C-15-surfactin-O-methyl ester by a lipopeptide producing strain Bacillus subtilis HSO121. Process Biochem 44(10):1144–1151. doi:10.1016/j.procbio.2009.06.014
Arima K, Kakinuma A, Tamura G (1968) Surfactin, a crystalline peptidelipid surfactant produced by Bacillus subtilis: isolation, characterization and its inhibition of fibrin clot formation. Biochem Biophys Res Commun 31(3):488–494. doi:10.1016/0006-291x(68)90503-2
Chen H, Wang L, Su CX, Gong GH, Wang P, Yu ZL (2008) Isolation and characterization of lipopeptide antibiotics produced by Bacillus subtilis. Lett Appl Microbiol 47(3):180–186. doi:10.1111/j.1472-765X.2008.02412.x
Wakayama S, Ishikawa F, Oishi K (1984) Mycocerein, a novel antifungal peptide antibiotic produced by Bacillus cereus. Antimicrob Agents Chemother 26(6):939–940
Lee SC, Kim SH, Park IH, Chung SY, Choi YL (2007) Isolation and structural analysis of bamylocin A, novel lipopeptide from Bacillus amyloliquefaciens LP03 having antagonistic and crude oil-emulsifying activity. Arch Microbiol 188(4):307–312. doi:10.1007/s00203-007-0250-9
Desai JD, Banat IM (1997) Microbial production of surfactants and their commercial potential. Microbiol Mol Biol Rev 61(1):47–64
Banat IM, Makkar RS, Cameotra SS (2000) Potential commercial applications of microbial surfactants. Appl Microbiol Biotechnol 53(5):495–508
Das P, Mukherjee S, Sen R (2008) Antimicrobial potential of a lipopeptide biosurfactant derived from a marine Bacillus circulans. J Appl Microbiol 104(6):1675–1684. doi:10.1111/j.1365-2672.2007.03701.x
Hiradate S, Yoshida S, Sugie H, Yada H, Fujii Y (2002) Mulberry anthracnose antagonists (iturins) produced by Bacillus amyloliquefaciens RC-2. Phytochemistry 61(6):693–698. doi:10.1016/s0031-9422(02)00365-5
Yu GY, Sinclair JB, Hartman GL, Bertagnolli BL (2002) Production of iturin A by Bacillus amyloliquefaciens suppressing Rhizoctonia solani. Soil Biol Biochem 34(7):955–963. doi:10.1016/s0038-0717(02)00027-5
Vanittanakom N, Loeffler W, Koch U, Jung G (1986) Fengycin - a novel antifungal lipopeptide antibiotic produced by Bacillus subtilis F-29-3. J Antibiot 39(7):888–901
Villegas-Escobar V, Ceballos I, Mira JJ, Argel LE, Peralta SO, Romero-Tabarez M (2013) Fengycin C Produced by Bacillus subtilis EA-CB0015. J Nat Prod 76(4):503–509. doi:10.1021/np300574v
Bonmatin JM, Laprevote O, Peypoux F (2003) Diversity among microbial cyclic lipopeptides: Iturins and surfactins. Activity-structure relationships to design new bioactive agents. Comb Chem High Throughput Screen 6(6):541–556
Hathout Y, Ho YP, Ryzhov V, Demirev P, Fenselau C (2000) Kurstakins: a new class of lipopeptides isolated from Bacillus thuringiensis. J Nat Prod 63(11):1492–1496. doi:10.1021/np000169q
Peypoux F, Pommier MT, Marion D, Ptak M, Das BC, Michel G (1986) Revised structure of mycosubtilin, a peptidolipid antibiotic from Bacillus subtilis. J Antibiot 39(5):636–641
Volpon L, Besson F, Lancelin JM (1999) NMR structure of active and inactive forms of the sterol-dependent antifungal antibiotic bacillomycin L. Eur J Biochem 264(1):200–210. doi:10.1046/j.1432-1327.1999.00605.x
Li YM, Yang SZ, Mu BZ (2010) The surfactin and lichenysin isoforms produced by Bacillus licheniformis HSN 221. Anal Lett 43(6):929–940. doi:10.1080/00032710903491047
Bonmatin JM, Labbe H, Grangemard I, Peypoux F, Magetdana R, Ptak M, Michel G (1995) Production, isolation and characterization of [Leu4]- and [Ile4]surfactins from Bacillus subtilis. Lett Pept Sci 2(1):41–47. doi:10.1007/bf00122922
Volpon L, Tsan P, Majer Z, Vass E, Hollosi M, Noguera V, Lancelin JM, Besson F (2007) NMR structure determination of a synthetic analogue of bacillomycin Lc reveals the strategic role of L-Asn1 in the natural iturinic antibiotics. Spectrochim Acta Part A 67(5):1374–1381. doi:10.1016/j.saa.2006.10.027
Vater J, Kablitz B, Wilde C, Franke P, Mehta N, Cameotra SS (2002) Matrix-assisted laser desorption ionization-time of flight mass spectrometry of lipopeptide biosurfactants in whole cells and culture filtrates of Bacillus subtilis C-1 isolated from petroleum sludge. Appl Environ Microbiol 68(12):6210–6219. doi:10.1128/aem. 68.12.6210-6219.2002
Sun LJ, Lu ZX, Bie XM, Lu FX, Yang SY (2006) Isolation and characterization of a co-producer of fengycins and surfactins, endophytic Bacillus amyloliquefaciens ES-2, from Scutellaria baicalensis Georgi. World J Microbiol Biotechnol 22(12):1259–1266. doi:10.1007/s11274-006-9170-0
Finking R, Marahiel MA (2004) Biosynthesis of nonribosomal peptides. Annu Rev Microbiol 58:453–488. doi:10.1146/annurev.micro.58.030603.123615
Marahiel MA (2009) Working outside the protein-synthesis rules: insights into non-ribosomal peptide synthesis. J Pept Sci 15(12):799–807. doi:10.1002/psc.1183
Chen CY, Baker SC, Darton RC (2006) Batch production of biosurfactant with foam fractionation. J Chem Technol Biotechnol 81(12):1923–1931. doi:10.1002/jctb.1625
Mata-Sandoval JC, Karns J, Torrents A (1999) High-performance liquid chromatography method for the characterization of rhamnolipid mixtures produced by Pseudomonas aeruginosa UG2 on corn oil. J Chromatogr A 864(2):211–220. doi:10.1016/s0021-9673(99)00979-6
Haddad NIA, Wang J, Mu BZ (2008) Isolation and characterization of a biosurfactant producing strain, Brevibacilis brevis HOB1. J Ind Microbiol Biotechnol 35(12):1597–1604. doi:10.1007/s10295-008-0403-0
Yuan J, Raza W, Huang QW, Shen QR (2011) Quantification of the antifungal lipopeptide iturin A by high performance liquid chromatography coupled with aqueous two-phase extraction. Jo Chromatogr B 879(26):2746–2750. doi:10.1016/j.jchromb.2011.07.041
Bidlingmeyer BA, Cohen SA, Tarvin TL (1984) Rapid analysis of amino acids using pre-column derivatization. J Chromatogr 336(1):93–104. doi:10.1016/s0378-4347(00)85133-6
Cohen SA, Bidlingmeyer BA, Tarvin TL (1986) PITC derivatives in amino acid analysis. Nature 320(6064):769–770. doi:10.1038/320769a0
Campanella L, Crescentini G, Avino P (1999) Simultaneous determination of cysteine, cystine and 18 other amino acids in various matrices by high-performance liquid chromatography. J Chromatogr A 833(2):137–145. doi:10.1016/s0021-9673(98)01023-1
Harvey DJ (2003) Matrix-assisted laser desorption/ionization mass spectrometry of carbohydrates and glycoconjugates. Int J Mass Spectrom 226(1):1–35. doi:10.1016/s1387-3806(02)00968-5
Sivapathasekaran C, Mukherjee S, Samanta R, Sen R (2009) High-performance liquid chromatography purification of biosurfactant isoforms produced by a marine bacterium. Anal Bioanal Chem 395(3):845–854. doi:10.1007/s00216-009-3023-2
Yanagi M, Yamasato K (1993) Phylogenetic analysis of the family Rhizobiaceae and related bacteria by sequencing of 16S rRNA gene using PCR and DNA sequencer. FEMS Microbiol Lett 107(1):115–120
Yang J, Sun L, Bai X, Zhou H (2002) Simultaneous determination of 18 amino acids by reversed-phase high performance liquid chromatography with precolumn phenylisothiocyanate derivatization. Se Pu 20(4):369–371
Liu X-Y, Yang S-Z, Mu B-Z (2008) Isolation and characterization of a C(12)-lipopeptide produced by Bacillus subtilis HSO 121. J Pept Sci 14(7):864–875. doi:10.1002/psc.1017
Zabet-Moghaddam M, Heinzle E, Tholey A (2004) Qualitative and quantitative analysis of low molecular weight compounds by ultraviolet matrix-assisted laser desorption/ionization mass spectrometry using ionic liquid matrices. Rapid Commun Mass Spectrom 18(2):141–148. doi:10.1002/rcm.1293
Harrison AG (2003) Fragmentation reactions of protonated peptides containing glutamine or glutamic acid. J Mass Spectrom 38(2):174–187. doi:10.1002/jms.427
Paizs B, Suhai S (2005) Fragmentation pathways of protonated peptides. Mass Spectrom Rev 24(4):508–548. doi:10.1002/mas.20024
Wysocki VH, Tsaprailis G, Smith LL, Breci LA (2000) Special feature: commentary - mobile and localized protons: a framework for understanding peptide dissociation. J Mass Spectrom 35(12):1399–1406. doi:10.1002/1096-9888(200012)35:12<1399::aid-jms86>3.0.co;2-r
Madonna AJ, Voorhees KJ, Taranenko NI, Laiko VV, Doroshenko VM (2003) Detection of cyclic lipopeptide biomarkers from Bacillus species using atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Anal Chem 75(7):1628–1637. doi:10.1021/ac020506v
Sen R (2008) Biotechnology in petroleum recovery: the microbial EOR. Prog Energy Combust 34(6):714–724. doi:10.1016/j.pecs.2008.05.001
Bachmann RT, Johnson AC, Edyvean RGJ (2014) Biotechnology in the petroleum industry: an overview. Int Biodeterior Biodegrad 86:225–237. doi:10.1016/j.ibiod.2013.09.011
Sachdev DP, Cameotra SS (2013) Biosurfactants in agriculture. Appl Microbiol Biotechnol 97(3):1005–1016. doi:10.1007/s00253-012-4641-8
Mandal SM, Barbosa A, Franco OL (2013) Lipopeptides in microbial infection control: scope and reality for industry. Biotechnol Adv 31(2):338–345. doi:10.1016/j.biotechadv.2013.01.004
Bockmuhl D (2012) Biosurfactants as antimicrobial ingredients for cleaning products and cosmetics. Tenside Surfactants Deterg 49(3):196–198
Whang LM, Liu PWG, Ma CC, Cheng SS (2009) Application of rhamnolipid and surfactin for enhanced diesel biodegradation-effects of pH and ammonium addition. J Hazard Mater 164(2–3):1045–1050. doi:10.1016/j.jhazmat.2008.09.006
Ongena M, Jacques P, Toure Y, Destain J, Jabrane A, Thonart P (2005) Involvement of fengycin-type lipopeptides in the multifaceted biocontrol potential of Bacillus subtilis. Appl Microbiol Biotechnol 69(1):29–38. doi:10.1007/s00253-005-1940-3
Ongena M, Jacques P (2008) Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol 16(3):115–125. doi:10.1016/j.tim.2007.12.009
Kracht M, Rokos H, Ozel M, Kowall M, Pauli G, Vater J (1999) Antiviral and hemolytic activities of surfactin isoforms and their methyl ester derivatives. J Antibiot 52(7):613–619
Dufour S, Deleu M, Nott K, Wathelet B, Thonart P, Paquot M (2005) Hemolytic activity of new linear surfactin analogs in relation to their physico-chemical properties. Biochimi Biophys Acta 1726(1):87–95. doi:10.1016/j.bbagen.2005.06.015
Yang SZ, Wei DZ, Mu BZ (2006) Determination of the amino acid sequence in a cyclic lipopeptide using MS with DHT mechanism. J Biochem Biophys Methods 68(1):69–74. doi:10.1016/j.jbbm.2006.03.008
Kwanyuen P, Burton JW (2010) A modified amino acid analysis using PITC derivatization for soybeans with accurate determination of cysteine and half-cystine. J Am Oil Chem Soc 87(2):127–132. doi:10.1007/s11746-009-1484-2
Acknowledgments
This work was supported by the 973 National Key Basic Research Project (2013CB733600), the National Natural Science Foundation (no. 21176143), and the Tsinghua University Initiative Scientific Research Program (no. 20111081120).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 435 kb)
Rights and permissions
About this article

Cite this article
Yang, H., Li, X., Li, X. et al. Identification of lipopeptide isoforms by MALDI-TOF-MS/MS based on the simultaneous purification of iturin, fengycin, and surfactin by RP-HPLC. Anal Bioanal Chem 407, 2529–2542 (2015). https://doi.org/10.1007/s00216-015-8486-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8486-8