
Abstract
Seven quick, easy, cheap, effective, rugged and safe (QuEChERS)-based procedures, differing in both the extraction and clean-up steps, were investigated for the recovery of bentazone (BTZ), atrazine (ATZ), carbamazepine (CBZ), phenytoin (PNT) and its metabolite 5-(p-hydroxyphenyl-),5-phenylhydantoin (HPPH) from soil. Target analytes were chosen for their extensive use and/or occurrence in soil, as well as for their medium-high polarity characteristics (log K OW values in the range 0.88–2.80), which have been reported as a critical parameter for the recovery from soil with QuEChERS approaches. Liquid chromatography coupled with UV and pulsed amperometric (PA) detection at a glassy carbon electrode was used as instrumental technique. The recovery data obtained within each tested procedure were discussed for each compound investigated, highlighting different behaviour depending on the specific physicochemical characteristics of the analytes. The optimized QuEChERS conditions consisted of the extraction of analytes with CH3CN:H2O 70:30, 5 % CH3COOH, followed by a dispersive solid-phase extraction (d-SPE) clean-up step with C18 sorbent. This method, in which water is directly added to the soil together with acetonitrile and salts, allowed the rehydration step to be avoided, which can be as long as 30 min. Matrix effects were evaluated for both the detection techniques at different concentration levels, and they were below 24 % for both the detection technique used. The recoveries were evaluated at three concentration levels by a matrix-matched calibration and were in the ranges of 83–113 % (relative standard deviations (RSD) ≤ 14 %) and 88–109 % (RSD ≤ 11 %) for UV and PA detection, respectively, highlighting very good performances of the method, even for the more polar analytes. Method detection limits ranged from 4 μg/kg (BTZ) to 493 μg/kg (PNT) and from 4 μg/kg (HPPH) to 11 μg/kg (BTZ) for UV and PA detection, respectively. The method was finally compared with a microwave-assisted extraction procedure which provided less satisfactory extraction performances than the optimized QuEChERS procedure.

ᅟ
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soil is increasingly subject to both inorganic and organic chemical contamination, owing to either diffuse or point-source input of contaminants. Soil contamination is a problem of high environmental concern, especially considering the low renewal capacity of this matrix, compared to atmosphere and water bodies. Moreover, the presence of contaminants in soil represents a serious environmental risk for their potential accumulation in the food chain [1] and the capacity to migrate in underground waters [2, 3].
The extraction of contaminant residues from soil is commonly performed by classical approaches such as Soxhlet [4] or ultrasonic-assisted techniques [5]. Pressurized liquid extraction [6] and microwave-assisted extraction (MAE) [7, 8] have been also used as sample preparation techniques for the recovery of many classes of compounds from soil.
The application of these extraction methods to the recovery of pesticides [9, 10] and pharmaceuticals [11] from soil samples has been recently reviewed.
An alternative approach for the extraction of organic compounds from soils and sediments is the quick, easy, cheap, effective, rugged and safe (QuEChERS) method, originally developed by Anastassiades et al. [12] and subsequently validated by Lehotay et al. [13] and Anastassiades et al. [14], as a rigid analytical protocol for the recovery of pesticide residues from fruits and vegetables.
As highlighted by Bruzzoniti and colleagues [15], the validated QuEChERS methods have been used only in a few cases for environmental analysis, whereas most studies have adopted more or less modified procedures, according to the matrix and solute characteristics. The environmental applications of the QuEChERS approach mainly cover the determination of pesticides, while, according to our literature survey, only two papers regarding the analysis of pharmaceuticals have been published till now [15].
Within the different types of pesticides commonly applied on soil, herbicides represent the most used class in European countries [16] and their presence has been frequently reported in soil [17–19]. More in detail, bentazone has been recently reported as one of the herbicides most frequently found in European groundwater [3], including Italy, where its presence in aquifers and surface water intended for human consumption has been found of high concern [20]. As regards atrazine, even though this herbicide was banned in Europe since 2004, it is still one of the top 30 most frequently detected compounds in aquifers, according to the Environment Agency groundwater organic micropollutant database [3]. It should also be mentioned that bentazone recovery from soil matrices has been investigated by QuEChERS methods in only one study [21], whereas for atrazine, few papers using this sample preparation approach are reported in the literature [22, 23, 19].
Among contaminants that greatly affect the soil quality, pharmaceuticals are doubtless an organic compound class frequently determined in soil, not only as a consequence of direct excretion from animals [24], but also as a result of land application of biosolids [25], reclaimed water [26] and also urine [27], as fertilizing agents. Within this group of pollutants, carbamazepine and phenytoin are both on the World Health Organization’s List of Essential Medicines, as two of the most important anticonvulsant drugs needed in a basic health system [28]. Carbamazepine is excreted by humans as conjugated and hydroxylated metabolites and with a much lower extent (about 3 %) as unchanged molecule [29]. Nevertheless, carbamazepine has been frequently determined in the effluent of wastewater treatment plants (WTPs) [30–32] and especially in biosolids [33], where it accumulates preferentially compared to its metabolites [32]. Hence, carbamazepine has been found in sludge-amended soil [2], also because of its persistence in this matrix [34]. In this regard, carbamazepine has been also proposed as a possible marker of anthropogenic contamination [30].
Phenytoin is present in human excreta mainly as unmetabolized drug or free and conjugated 5-(4-hydroxyphenyl)-5-phenylhydantoin (HPPH) [35]; furthermore, it has been reported as quite recalcitrant to aerobic degradation in WTPs [36]. Accordingly, phenytoin is expected to be present in biosolids and sludge-amended soils, even though no studies are reported in the literature on this topic according to our literature investigation.
It should also be remarked that the recoveries from soil of phenytoin and HPPH have not yet been investigated by QuEChERS methods, whereas for carbamazepine, only one paper is published in the literature [24].
Based on the aforementioned considerations, this study focused on the evaluation of seven different QuEChERS-based methods for the recovery of bentazone, atrazine, carbamazepine, phenytoin and HPPH. The investigated QuEChERS approaches differed in either extraction or clean-up conditions, allowing for evaluating the behaviour of target analytes in both the analytical steps. It is also remarkable that the analyte group under investigation covered a medium (log K OW = 2.80) to high (log K OW = 0.82) polarity range (see Electronic Supplementary Material (ESM) Fig. S1), making their study even more interesting, since the recovery of very polar compounds may be critical with QuEChERS approaches [15].
Liquid chromatography was employed as instrumental analytical technique, coupling it with both spectrophotometric and amperometric detectors, the latter never investigated in combination with QuEChERS sample treatment. It should also be remarked that amperometry detection, performed in the pulsed mode, was applied in an innovatively way since glassy carbon was used as working electrode, rather than noble metals which were traditionally designed for this kind of detection system.
The figures of merit of the optimized QuEChERS method were determined and compared, when possible, with the available literature data. Finally, the performances of the optimized procedure were also comparatively evaluated with those provided by a MAE procedure previously developed [8], replacing cyclohexane with acetonitrile, which is the typical solvent used for QuEChERS procedure.
Experimental
Chemicals and reagents
Chromasolv gradient-grade acetonitrile, ACS reagent methanol and acetic acid (99.8 % w/w) were from Sigma-Aldrich (St. Louis, MO, USA). Sodium hydroxide (50 % w/w), used to control pH, was purchased from J.T. Baker (Center Valley, PA, USA). Sodium chloride and magnesium sulphate, ACS reagent salts, were supplied by Riedel-de Haën (Seelze, Germany). End-capped C18 and primary secondary amine bulk sorbent (PSA), both used for the clean-up step, were from Agilent Technologies (Santa Clara, CA, USA). Bentazone (BTZ), 5-(4 hydroxyphenyl)-5-phenylhydantoin (HPPH), phenytoin (PHT), carbamazepine (CBZ) and atrazine (ATZ), all of analytical grade, were purchased from Sigma-Aldrich. Purified water used to prepare eluents and standard solutions was obtained from a Millipore (Billerica, MA, USA) Milli-Q Plus ultrapure water system.
Preparation of standard solutions
The stock solution of BTZ was prepared in water at a concentration of 100 mg/L. Stock solutions of HPPH, CBZ, PHT and ATZ (100 mg/L each) were prepared in methanol. All the stock solutions were stored in the dark at +4 °C.
From the individual stock standard solutions, working solutions were prepared by proper dilutions with water, or with the post-extracted solutions, for matrix effect (ME) evaluation and matrix-matched calibration (MMC).
Sample collection and preparation
The soil used in this work (about 100 g) was collected in Bruino (Piedmont, Italy) at a depth of approximately 10–15 cm. After homogenization, one sample aliquot of approximately 10 g was taken for physicochemical characterization of the soil, whereas the remaining part was packed airtight in a low-density polyethylene bag and stored in the dark at +4 °C until its treatment.
The soil was characterized for selected physicochemical parameters, according to the Italian Official Methods of Soil Chemical Analysis (1999) [37], obtaining the following results: texture = sandy loam; pH = 5.7; organic carbon = 13.7 g/kg d.w.; cation exchange capacity = 7.855 cmol(+)/kg d.w.
The sample was freeze dried until a constant weight was achieved, homogenized, finely ground using a mortar and passed through a 2-mm sieve to remove coarse particles and to increase sample homogeneity.
In order to verify the presence/absence of target drugs and herbicides in the soil, a soil batch was firstly extracted using the QuEChERS protocols identified in Fig. 1 as version 1 and version 4. None of the analytes under investigation were determined in the soil extract.

Optimization of the QuEChERS procedure: scheme and details of the conditions used
For the method optimization and validation, the soil was spiked according to the following procedure: 5 g of soil was introduced into a tube and spiked with target analytes at proper concentrations (i.e. 2 mg/kg for the optimization phase or other desired concentration for the method validation, see Table 1) by the addition of 2 mL of an aqueous solution containing the analytes at a suitable initial concentration, in order to obtain the final desired concentration in the soil. The tube was then vortex and the soil sample left to soak for 72 h at room temperature, to promote the interaction of target compounds with the soil matrix. In parallel, an aliquot of sieved and homogenized soil was treated in the same way with 2 mL of purified water, to obtain the “blank soil sample.” Such a term is therefore used in this manuscript to mean a soil sample which was not spiked with target analytes.
Spiked and blank soil samples were freeze dried and stored at −20 °C until their use.
Modified QuEChERS method
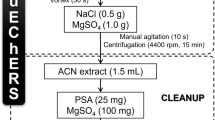
Target analytes were recovered from soil samples using a modified QuEChERS procedure which was optimized by evaluating different extraction and clean-up conditions (versions 1–7, Fig. 1), following the precautions reported by Bruzzoniti and co-workers [15]. All the QuEChERS versions were comparatively evaluated using high-performance liquid chromatography (HPLC)-UV as instrumental technique.
Note that version 1 of Fig. 1 differed from the original QuEChERS procedure [12] only for the use of a solvent excess in respect to the soil amount (1:2 w/v sample: solvent ratio); this modification was necessary for ensuring a supernatant volume high enough to perform the various analytical steps [38]. In order to assess the influence of different clean-up approaches on apparent recovery [39], versions 1 (conventional dispersive solid-phase extraction (d-SPE) with PSA), 2 (d-SPE with C18) and 3 (no clean-up) were compared one with the others.
The evaluation of the influence of sample rehydration was highlighted by the comparison of versions 2 and 4.
The effects of extractant acidification were evaluated either in the presence (version 4 vs. version 5) or in the absence (version 2 vs. version 6) of water in the extraction mixture.
Finally, the comparison between versions 5 and 7 allowed for evaluating the effect of reducing the volume of the extracting solution, keeping constant the salt ratios.
According to the experimental results obtained, version 5 was the best compromise for recovering target analytes and was therefore selected for the evaluation of ME and validation parameters. Briefly, the optimized extraction conditions were the following. Freeze-dried soil aliquots (5 g) were weighted into a 50-mL centrifuge tube, together with 4 g of anhydrous MgSO4 and 1 g of NaCl; 10 mL of the extraction solution (CH3CN:H2O 70:30, 5 % CH3COOH) was then added, and the tube was immediately hand shaken (1 min) in order to prevent agglomeration of salts; afterwards, the tube was centrifuged at 2000×g for 10 min at room temperature. The clean-up step was performed by d-SPE on 4-mL aliquots of supernatant into a d-SPE clean-up minitube containing 0.25 g of C18 and 0.5 g of anhydrous MgSO4. The tube was hand shaken (1 min) and centrifuged (2000×g, 10 min); 2-mL supernatant aliquots were diluted 1:1 (v/v) with MilliQ water, filtered and analyzed by HPLC.
MAE
MAE extraction was performed on 0.4 g soil aliquots spiked with 2 mg/kg of each analyte using 5 mL of a 3:2 (v/v) acetone/acetonitrile mixture as extractant. The following program was set as follows: t = 0–10 min temperature is increased up to the desired temperature (T = 130–150 °C) with a holding time of 5 min (t = 10–15 min), keeping a maximum power of 300 W, accordingly to Bruzzoniti et al. [8]. Under these conditions, no escape of solvent was observed, thus ensuring that losses of analytes did not occur. Three replicated extractions were performed. The extracts were then centrifuged (2000×g, 5 min). An aliquot of 4.5 mL of supernatant was evaporated to dryness under a gentle nitrogen stream, and the residual was reconstituted with 2 mL H2O. Before injection into HPLC, all samples were filtered with Nylon 0.45-μm filters. A blank was run in parallel.
In order to assess the stability of the target compounds under the instrumental conditions above-reported, 5 mL of 1 mg/L individual standard solutions in 3:2 (v/v) acetone/acetonitrile mixture was treated in the microwave digestor following as described. HPLC-UV analysis of individual standard solutions before and after the microwave treatment was performed.
Instrumental conditions
For chromatographic measurements, a Dionex ICS-3000 chromatograph (Thermo Scientific, Sunnyvale, CA, USA), equipped with a reversed-phase Lichrospher C-18 analytical column (125 × 3.0 mm, 5 μm, Merck, Darmstadt, Germany), was used. As previously optimized [40], the mobile phase was a 72:28 (v/v) mixture of aqueous 50 mM sodium acetate (pH = 5) and CH3CN. Elutions were performed under isocratic conditions (flow rate 0.5 mL/min). For each analysis, 10 μL of sample was injected. Two detection systems, namely a UV-VIS spectrophotometer (AD25 Absorbance Detector, Dionex Thermofisher, Sunnyvale, CA, USA) and an amperometric detector (AD40 Electrochemical Detector Dionex, Thermofisher, Sunnyvale, CA, USA) coupled in series were used.
The UV-VIS detection conditions were optimized with the aim of obtaining the maximum signal-to-noise ratio (S/N), using a HPLC system Shimadzu (Kyoto, Japan), consisting of two pumps LC-10ADVP, an autoinjector SIL-10AD VP and a diode array detector (DAD) SPD-M10A VP. To this aim, a portion of soil was extracted according to version 5 of Fig. 1 and the extract was spiked with known amounts of target analytes. According to the results obtained, a wavelength of 252 nm (band width = 1 nm) was selected.
For the amperometric detection, measurements were performed using a glassy carbon working electrode (Ag/AgCl reference electrode). Detection was performed by pulsed amperometry (PA), setting the detection conditions as optimized for the selected compounds throughout a recent study by Rivoira et al. [40]. PA parameters are briefly recalled in Table S1 in the ESM.
For MAE extraction, a Discover SP-D (CEM, Bergamo, Italy) microwave digestor provided with autosampler was used throughout this work. The digestor has a focused single-mode cavity designed to maximize the microwave energy input to the sample in a high-density field and to reduce solvent consumption.
A Jouan B4i centrifuge (Thermo Scientific Inc., USA) was used to separate the supernatant from the solid components. Econofilter Nylon 25/45 (CPS Analitica, Milan, Italy) 0.45-μm syringe filters were used to filter solutions before HPLC analysis.
ME evaluation
To evaluate the ME, three blank soil aliquots were extracted by version 5 as previously illustrated (see the paragraphs “Sample collection and preparation” and “Modified QuEChERS method” within this section). These post-extracted blank soil solutions were spiked with a mixture of the selected analytes at two concentration levels—(i) 1 mg/L (4 mg/kg) each, and (ii) 15 μg/L (60 μg/kg) BTZ, 10 μg/L (40 μg/kg) HPPH, 25 μg/L (100 μg/kg) CBZ, 250 μg/L (1000 μg/kg) PNT and 50 μg/L (200 μg/kg) ATZ—and injected in the HPLC system. ME was evaluated for both UV and PA detection techniques. The chromatographic area obtained (A std,matrix) was compared with that attained by spiking the same amount of analytes in the extraction solvent CH3CN:H2O 70:30, 5 % CH3COOH (A std,solvent), to calculate ME, according to the following Eq. 1:
It should be noted that a ME> |20 % | is commonly considered to have a significant impact on the performance of the method [23, 41, 42].
Analytical performance and method validation
Specificity of the method was assessed by the analysis of three blank soil samples, extracted by the optimized QuEChERS method (version 5). The method optimized was finally validated for linearity, recovery, intraday precision, limits of detection (MDLs) and quantification (MQLs), using spiked soil samples (see the paragraph “Sample collection and preparation” within this section) according to the indications provided in SANCO/12571/2013 procedure [43].
Matrix-matched calibration curves
To compensate any matrix effect, a matrix-matched calibration (MMC) was used for both UV and PA detection modes. In particular, the extracted solution from blank soil aliquots were spiked at five concentration levels. Peak area was used as analyte response, and calibration curves were constructed by plotting the peak areas versus the concentration of analytes and performing calculations on the average peak areas of four repeated injections. Coefficients of determination (R 2) were also calculated for each analyte. The concentration ranges used for UV detection were 3.5–1500 μg/L (BTZ), 44–1000 μg/L (HPPH), 54–2500 μg/L (CBZ), 360–5000 μg/L (PNT) and 70–2500 μg/L (ATZ), which, considering the overall sample preparation procedure, correspond to a concentration range of 0.014–20 mg/kg in the soil. For PA detection, the following concentration ranges were studied—9.5–1500 μg/L (BTZ), 3.5–1000 μg/L (HPPH), 7.5–2500 μg/L (CBZ)—corresponding to a concentration range of 0.014–10 mg/kg in the soil.
Limits of detection and quantification of the method
Instrumental limits of detection (LODmatrix) and quantification (LOQmatrix) in matrix were calculated according to the below-reported Eqs. 2 and 3 [44], where σ is the standard deviation (n = 8) of the signal registered at the retention time of the selected analyte for the blank soil extract and S is the slope of the calibration curve in matrix.
MDLs and MQLs were then estimated on the basis of LODmatrix and LOQmatrix, taking into account the soil amount and solvent volumes involved in the extraction and clean-up processes.
Recovery and precision
Method optimization was performed evaluating the apparent recovery of each analyte from a fortified soil sample (2 mg/kg for each analyte). For each QuEChERS version, three replicated extractions were performed and the apparent recovery was calculated comparing the HPLC-UV chromatographic areas of target analytes in the extracted samples with those found in the standard solution of analytes prepared in ultrapure water, at the same nominal concentration of the sample after extraction (0.5 mg/L).
Validation of the optimized method (version 5) was performed using MMC, evaluating the extraction recovery as defined by IUPAC [39], in order to compensate ME [45]. The percentage extraction recovery (ER%) was calculated according to Eq. 4:
where A spiked soil is the chromatographic area of a certain analyte spiked in the soil at a given concentration and A std,matrix is the area obtained by spiking the same analyte at the same concentration in the post-extracted blank soil.
For each analyte, recoveries and corresponding intraday precisions were evaluated at three specific spiking levels (see Table 1 for details) by a MMC curve on four independent replicated experiments.
Statistical evaluations
Linear univariate correlations between variables were investigated by the least square method, using Microsoft® Office Excel 2007 (Microsoft Corporation, Redmond, WA, USA).
One-way analysis of variance and Dunnett T3 nonparametric test were performed on the original data, at the 95 % probability level (p ≤ 0.05), by using the statistical package SPSS, version 17.0 for Windows (SPSS Inc., Chicago, IL, USA).
Results and discussion
QuEChERS method development
In order to investigate the influence on apparent recovery of some key parameters affecting extraction and clean-up steps, polarity of the extraction solvent, pH conditions and the type of sorbent used in the clean-up were changed (Fig. 1), according to the considerations reported in the paragraph “Modified QuEChERS method” within the section “Experimental.” During optimization, each QuEChERS procedure was tested in triplicate. The results obtained and hereafter discussed are reported in Fig. 2.

Mean values (n = 3) of apparent recoveries and standard deviations obtained for target compounds after soil spiking at 2 mg/kg each, using different QuEChERS versions. For details of the different QuEChERS procedures, see Fig. 1. Bars with different letters refer to mean values statistically different according to the Dunnett T3 nonparametric contrast test (p ≤ 0.05). Conversely, bars provided with at least one common letter refer to mean values not statistically different from one to the other
Versions 1–3 were characterized by a common extraction procedure based on the original QuEChERS method without sample rehydration and differed only in the clean-up step that was performed with PSA and C18 in versions 1 and 2, respectively, and was omitted in version 3 (Fig. 1). With this last version, chromatograms of lesser quality were obtained compared to those deriving from versions 1 and 2, owing to the presence of a noisy baseline and peaks deriving from co-extracted species (data not shown). Nevertheless, with version 3, all target analytes were detected, even though with very different apparent recoveries. More in detail, ATZ showed the highest value (74.6 ± 2.6 %), followed by CBZ (44.0 ± 2.8 %), BTZ (29.8 ± 2.9 %), PNT (11.3 ± 1.7 %) and HPPH (7.8 ± 0.8 %).
As shown in Fig. 2, the only analytes detected using version 1 were ATZ and CBZ, with apparent recoveries (78.8 ± 9.2 and 36.0 ± 5.2 %, respectively) not statistically different from the ones achieved with version 3, according to the Dunnett T3 contrast test. The complete failure in the recovery of BTZ, PNT and HPPH with version 1 can be attributed to their adsorption by PSA, probably due to hydrogen bonding interactions. In fact, all these compounds contain a hydroxyl group, belonging to a phenolic moiety (HPPH) or deriving from keto-enol (BTZ) or imino-imide (PNT and HPPH) tautomerisms (see chemical structures and tautomery in ESM Fig. S1) [46, 47]. Strong hydrogen bonds may therefore occur between the hydrogen-donor hydroxyl group of the analyte and the hydrogen-acceptor nitrogen of PSA, together with other weaker hydrogen-bonding interactions. It should be also underlined that these interactions are enhanced in the aprotic solvent acetonitrile.
The use of C18 as d-SPE sorbent (version 2) allowed to recover all the target analytes, with apparent recoveries not statistically different compared to the ones achieved with version 3. These results were in agreement with the poor sorption properties expected for C18 sorbent in the partition process of relatively polar compounds, such as co-extracted matrix components and target analytes (see log K OW values in ESM Fig. S1), with organic aprotic solvents like acetonitrile.
As usually performed for dry food samples, water can be added to the soil before the analysis, with the aim of reconstituting a matrix with a high water percentage, for which the QuEChERS method was originally designed [15]. In this work, we chose to add water directly in the extracting phase, thus reducing the time of sample preparation due to soil rehydration, which can be as high as 30 min [48, 19]. Accordingly, in version 4, water was added to CH3CN (30/70 v/v water/CH3CN ratio). To evaluate the effect of water addition, the results obtained should be directly compared with those obtained by version 2. As observed in Fig. 2, the addition of water significantly enhanced the recovery of the analytes, with the only exception of ATZ, which was however well recovered, even in the absence of rehydration. The improved results in terms of apparent recovery should be linked to a more efficient extraction of analytes from soil; in fact, it should be noted that water can successfully compete with analytes for adsorption sites of soil humic substance, promoting their desorption; moreover, the soil rehydration step allows acetonitrile to gain better access into the soil pores, thus improving the partitioning process between aqueous and organic phases [15].
The influence of acidification of the extraction mixture was evaluated by adding 5 % (w/w) CH3COOH, both in the presence (version 5) or in the absence (version 6) of water in the extraction mixtures. This concentration is higher than the one usually proposed for buffered extractions (1 % CH3COOH) [49, 15] and was chosen because it allowed for reducing the pH value of the aqueous phase after equilibration with soil, from approximately 5.8 to about 2.7, which is intermediate between pKa values of ATZ and BTZ (see ESM Fig. S1). As illustrated in Fig. 2 (version 5 vs. version 4), the acid addition to the water/acetonitrile mixtures produced a significant increase in the apparent recovery of BTZ, PNT and HPPH; an augment of the mean value of recovery, even though not statistically significant, was also observed for CBZ, whereas for ATZ, the recovery showed a statistically significant decrease (from 77.3 ± 1.7 % in version 4 to 68.6 ± 1.8 % in version 5). These changes can be interpreted on the basis of different partition properties of target analytes between aqueous and organic phases at pH ≈ 2.7, compared to pH ≈ 5.8. In fact, at pH ≈ 2.7, HPPH, PNT and BTZ should be present as uncharged species and are therefore expected to be better extracted in the organic solvent. Conversely, for ATZ, which is significantly present as positively charged form under these pH conditions, the partition into the organic solvent is less efficient.
The acid addition to the extraction mixture in the absence of water (version 6 vs. version 2) showed a limited influence on the apparent recoveries of BTZ, CBZ PNT and HPPH, in accordance with the very similar polarities of the two solvents [50]. Conversely, for the recovery of ATZ, a statistically significant reduction (approximately equal to 20 %) was observed. In this regard, it should be noted that the presence of CH3COOH in a protophobic aprotic solvent like CH3CN may give rise to the charged form of ATZ, which partitioned differently between soil and extraction mixture, compared to the sole CH3CN.
With the purpose of increasing sensitivity, a reduced volume of the extraction mixture (7.5 mL instead of 10 mL) was employed in version 7, compared to version 5. This approach produced chromatograms with a higher background noise than that obtained with version 5, suggesting the presence of greater concentrations of co-extracted matrix components in the final solution injected. However, for most analytes, the recovery was not statistically affected by the increase of the sample/solvent ratio (PNT and HPPH) or showed a modest reduction (BTZ and CBZ). A very different behaviour was highlighted for ATZ, for which a drastic recovery decrease was observed, probably due to its aforementioned acid-base properties, which are peculiar, compared to those of the other investigated molecules. In this case, stronger interactions between ATZ and humic substances and a lower partition into the organic solvent can be hypothesized.
According to the comprehensive discussion reported above, version 5 represented the best compromise and was therefore chosen to evaluate the figures of merit of the whole procedure.
ME evaluation
Soil is a very complex matrix, and its extraction by aqueous-organic solvent mixtures often leads to the presence of co-extracted matrix components in the final extract to be injected, thus decreasing or increasing the instrumental response factors of target analytes, compared to those observed in solvent.
In this work, ME was evaluated at two concentration levels (see the paragraph “ME evaluation” within the section “Experimental” and Fig. 3 for full details) by comparing the signals of target analytes in the soil extracts obtained by version 5 with the ones determined in solvent for the same concentration, according to the equation reported in the paragraph “ME evaluation” within the section “Experimental.” ME was evaluated with both UV and PA detection, the latter being investigated in a previous study, highlighting the hydrodynamic electroactivity for BTZ, CBZ and HPPH at a glassy carbon working electrode [40].

Mean values (n = 3) of matrix effects obtained for target compounds using different detection modes (UV and pulsed amperometry), at two different spiking levels: a 1 mg/L each and b 15 μg/L BTZ, 50 μg/L ATZ, 25 μg/L CBZ, 250 μg/L PNT and 10 μg/L HPPH. Error bars refer to standard deviation as calculated by error propagation rules. The matrix is obtained by the soil extraction with version 5
As illustrated in Fig. 3, for all the tested compounds, suppressive effects were observed for either UV or PA detection modes, even though with different extent, depending on both the analyte and the concentration level investigated.
For both UV and PA detection modes, a higher ME was observed when lower analyte concentrations were determined, probably due to the higher co-extracted compounds/analytes concentration ratio.
As regards UV detection, at the highest concentration level investigated (1 mg/L, see Fig. 3a), ME ranged from +0.8 % (PNT) to −12.1 % (CBZ) and was therefore well below the threshold limit of 20 %, commonly considered low enough to avoid the use of the MMC approach [23, 41, 42]. As above-mentioned, the signal suppression due to the matrix increased, when the 10–250-μg/L concentration levels were tested (see Fig. 3b); however, ME remained quite low, being it slightly higher than 20 % only for BTZ (21.2 %) and PNT (−23.7 %) and included between −13.7 and −20.1 % for the other analytes. It is noteworthy to mention that the literature dealing with the use of QuEChERS procedures coupled with UV detection for soil analysis of organic residues is focused to compounds different from our target analytes [51–53], and most important, in these papers, ME evaluation was not provided, thus hampering any comparison between our results and other published studies. Our findings are in agreement with the modest ME usually experienced in HPLC-UV detection, since neither the transmission nor the extinction coefficient of analytes is likely to be changed by the presence of co-extracted matrix components [54]. It should also be underlined that the lack of signal enhancement in the analysis of spiked matrix extracts, compared to standard in solvent, is in agreement with the absence of significant co-elutions among target analytes and matrix components, which represents the only important source of the positive matrix effect with UV detection [55].
For PA detection, mean values of ME were generally lower than or comparable with those found with UV, being them less than 10 and 20 % for the highest (1 mg/L) and the lowest (10–25 μg/L) analyte concentrations tested, respectively (see Fig. 3b). The lack of signal enhancement in matrix again supported the good chromatographic separations, as well as the selectivity of the detection system. The presence of suppressive effects, even if limited, might be ascribed to a decrease of the density of active site groups on the surface of glassy carbon electrode surface (typically carboxylic and hydroxyl groups), caused by interactions of the electrode with the co-extracted species, at the detriment of the electron transfer of the oxidation reaction and hence of the reactivity of the glassy carbon electrode, as highlighted in our previous study [40]. Since, based on our literature survey, the present work represents the first one coupling PA detection mode with a QuEChERS extraction, further comparisons with literature data are not possible.
According to the results above-discussed, although the ME was quite low, in order to avoid any underestimation of target analytes, a MMC was used for the method validation.
Method validation
For both UV and PA detection modes, the method validation was performed evaluating specificity, linearity, MDLs, MQLs, recovery and precision.
The specificity of the method was assessed through the analysis of blank soil samples extracted by the optimized QuEChERS method (version 5), ascertaining that no other significant peak (S/N > 3) was present at the retention times of target compounds, in agreement with the ME results previously discussed. Typical UV and PA chromatograms of QuEChERS extracts from spiked soil and blank soil samples are shown in Fig. 4.

Typical chromatograms obtained for a “blank soil” (dashed line) and a fortified soil (solid line) sample after QuEChERS extraction (version 5) and HPLC-UV (a) and HPLC-pulsed amperometry (b) analysis. Concentration levels: a 200 μg/kg bentazone (BTZ), 200 μg/kg 5-(4 hydroxyphenyl)-5-phenylhydantoin (HPPH), 400 μg/kg carbamazepine (CBZ), 2 mg/kg phenytoin (PNT) and 600 μg/kg atrazine (ATZ); b 60 μg/kg BTZ, 40 μg/kg HPPH and 100 μg/kg CBZ
The chromatographic response of target analytes as a function of their spiked concentrations in matrix extracts was linear for either UV or PA detectors. More in detail, R 2 values ranged from 0.9954 for ATZ to 0.9993 for PNT for UV and were in all cases higher than 0.999 for PA.
For the HPLC-UV method, the recoveries achieved using the proposed QuEChERS procedure felt within the range of 83–113 % with their relative standard deviations (RSD) in all cases ≤14 %. For PA detection, recoveries were included between 88 and 109 %, with RSD ≤ 11 %. As regards the three analytes that could be determined with both UV and PA, the recoveries found with the two detection modes were quite in accordance, with the main exceptions of HPPH at the intermediate calibration level and, above all, BTZ at the lowest one (see Table 1).
In the case of BTZ, ATZ and CBZ, the recoveries obtained in our study can be compared to literature data obtained with various QuEChERS methods; conversely, for the more polar PNT and HPPH, no comparison with literature data is available.
The recovery of BTZ from soil using the QuEChERS approach was recently investigated by Fuhrmann and co-workers [21], evidencing an extraction efficiency comparable to that observed in our investigation.
For ATZ, data shown in Table 1 highlighted recoveries remarkably higher than those found by Lesueur et al. [22], who applied the original QuEChERS procedure on dried soil samples. This finding is in accordance with the lower recovery efficiency, already assessed for soil extraction in the absence of water [15]. As a further confirmation of the importance of water addition during the QuEChERS extraction, our recoveries were very similar with the ones achieved by Yang et al. [19] and Mei et al. [23] who adopted the sample rehydration technique with different QuEChERS procedures. It should, however, be noted that with the QuEChERS method herein proposed, an actual rehydration step, which entails a time consumption as high as 30 min, is not required.
Also for CBZ, the recoveries found in this work were comparable with those elsewhere obtained [24], using a quite complicated clean-up step, based on SPE coupled with strong anion-exchange (to remove matrix) and polymeric reversed-phase (to retain the analytes of interest) cartridges in series before instrumental analysis.
For UV detection, MDLs ranged from 4 μg/kg (BTZ) to 493 μg/kg (PNT). As regards the analytes detectable with PA, significantly lower MDLs were observed for CBZ and HPPH, whereas a slightly higher limit of detection was found for BTZ (Table 1).
The sensitivity of this method was obviously influenced by the kind of detectors coupled with LC; in fact, UV and PA generally exhibit lower performances compared to mass spectrometry. More specifically, for BTZ, ATZ and CBZ, tandem mass spectrometry was found to be 2–3 magnitude orders more sensitive than the detectors used in this work [21, 23, 24, 56]. However, it should be remarked that MDLs and MQLs obtained herein for ATZ with UV detection were comparable to those achieved by Lesueur et al. and Brondi et al. by GC-MS [22, 57].
QuEChERS versus MAE
The results obtained by the QuEChERS approach were compared with those achieved by MAE, which is extensively used for the recovery of organic micropollutants from soil and sediments [9, 11]. It should be highlighted that, according to our literature survey, no recovery data for BTZ, PNT and HPPH by MAE from either environmental or food matrices are present in the literature. Conversely, ATZ and CBZ were successfully recovered by MAE from soil [58] and sludge [59] using acetonitrile and acetone, respectively. It is noteworthy to mention that the intrinsic characteristics of these solvents, such as mass heat capacity and microwave power absorption, make them two valuable options for MAE procedures [60]. It should be also underlined that acetonitrile and acetone are recommended for multi-residue extraction of pesticides by the Canadian Pest Management Regulatory Agency and US Food and Drug Administration [61], respectively.
According to the above-mentioned considerations, a 3:2 (v/v) acetone/acetonitrile mixture was chosen as an extractant for the target analytes, and two different extraction temperatures (130 and 150 °C) were tested.
The stability test performed on standard solutions (1 mg/L) of target analytes evidenced a quantitative recovery with the only exception of BTZ for which a loss as high as 70 % was found.
Data reported in Table 2, concerning the determination of the mean apparent recoveries after MAE on three replicated tests, evidenced that only ATZ and CBZ could be detected with this method, even though with unsatisfying recovery percentages.
CBZ was recovered at mean values of 38–51 %, which were about half than that found by Mohapatra et al. for MAE from sludge of this drug [59]. The significant differences between these recoveries could be ascribed to a very different ME occurring in the two cases; in fact, in the study of Mohapatra and colleagues, a clean-up step based on C18 SPE and tandem mass spectrometric detection were adopted.
Also, for ATZ, the results obtained in this work (44–46 %) were lower than those achieved by Shah and co-workers (about 60 %), who apparently worked under more drastic extraction conditions (microwave potency of about 680 W), using only acetonitrile as extraction solvent and UV detection, without any clean-up step [58].
Conclusions
This work provided an in-depth investigation of different extraction and clean-up conditions within the QuEChERS approach for the recovery from soil of bentazone (BTZ), atrazine (ATZ), carbamazepine (CBZ), phenytoin (PNT) and its metabolite 5-(p-hydroxyphenyl-),5-phenylhydantoin (HPPH).
In this regard, it is noteworthy to mention that very few literature is available on the recovery of BTZ, ATZ and CBZ from soil samples by QuEChERS, and that this study represents the first application of the QuEChERS techniques for PNT and HPPH.
The interpretation of the analyte behaviour under the different extraction and clean-up QuEChERS conditions was attempted, evidencing the importance of evaluating the physicochemical characteristics of target molecules to drive a correct selection of the parameters involved in the recovery by QuEChERS methods.
Very good recoveries were obtained for all the target analytes by adding water directly in the extraction mixture, without adopting the preliminary sample rehydration step, usually recommended when the QuEChERS technique is employed; this modification allowed for greatly reducing the analysis time.
This work demonstrates the suitability of the QuEChERS method also for the recovery of molecules characterized by high polarity such as PNT and, above all, HPPH.
Pulsed amperometry at a glassy carbon electrode was used for the first time together with the QuEChERS methodology, providing successful results in terms of both sensitivity and matrix effect, when compared to UV.
References
Wu C, Spongberg AL, Witter JD, Fang M, Czajkowski KP (2010) Uptake of pharmaceutical and personal care products by soybean plants from soils applied with biosolids and irrigated with contaminated water. Environ Sci Technol 44:6157–6161
Li WC (2014) Occurrence, sources, and fate of pharmaceuticals in aquatic environment and soil. Environ Pollut 187:193–201
Stuart M, Lapworth D, Crane E, Hart A (2012) Review of risk from potential emerging contaminants in UK groundwater. Sci Total Environ 416:1–21
Jurado-Sánchez B, Ballesteros E, Gallego M (2013) Comparison of microwave assisted, ultrasonic assisted and Soxhlet extractions of N-nitrosamines and aromatic amines in sewage sludge, soils and sediments. Sci Total Environ 463–464:293–301
Huang Y, Cheng M, Li W, Wu L, Chen Y, Luo Y, Christie P, Zhang H (2013) Simultaneous extraction of four classes of antibiotics in soil, manure and sewage sludge and analysis by liquid chromatography-tandem mass spectrometry with the isotope-labelled internal standard method. Anal Methods U K 5:3721–3731
Abou-Elwafa Abdallah M, Drage D, Harrad S (2013) A one-step extraction/clean-up method for determination of PCBs, PBDEs and HBCDs in environmental solid matrices. Environ Sci Process Impacts 15:2279–2287
Merdassa Y, Liu JF, Megersa N (2013) Development of a one-step microwave-assisted extraction method for simultaneous determination of organophosphorus pesticides and fungicides in soils by gas chromatography mass spectrometry. Talanta 114:227–234
Bruzzoniti MC, Maina R, Tumiatti V, Sarzanini C, Rivoira L, De Carlo RM (2012) Fast low-pressure microwave assisted extraction and gas chromatographic determination of polychlorinated biphenyls in soil samples. J Chromatogr A 1265:31–38
Tadeo JL, Pérez RA, Albero B, García-Valcárcel AI, Sánchez-Brunete C (2012) Review of sample preparation techniques for the analysis of pesticide residues in soil. J AOAC Int 95:1258–1271
Andreu V, Picó Y (2004) Determination of pesticides and their degradation products in soil: critical review and comparison of methods. TRAC Trend Anal Chem 23:772–789
Pavlović DM, Babić S, Horvat AJM, Kaštelan-Macan M (2007) Sample preparation in analysis of pharmaceuticals. TRAC Trend Anal Chem 26:1062–1075
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Lehotay SJ, De Kok A, Hiemstra M, Van Bodegraven P (2005) Validation of a fast and easy method for the determination of residues from 229 pesticides in fruits and vegetables using gas and liquid chromatography and mass spectrometric detection. J AOAC Int 88(2):595–614
Anastassiades M, Scherbaum E, Taşdelen B, Štajnbaher D (2007) Recent developments in QuEChERS methodology for pesticide multiresidue analysis. In: Pesticide chemistry: crop protection, Public Health, Environmental Safety. pp 439–458
Bruzzoniti MC, Checchini L, De Carlo RM, Orlandini S, Rivoira L, Del Bubba M (2014) QuEChERS sample preparation for the determination of pesticides and other organic residues in environmental matrices: a critical review. Anal Bioanal Chem 406:4089–4116
Eurostat database (2014) European Commission, Bruxelles, Belgium. http://epp.eurostat.ec.europa.eu/portal/page/portal/product_results/search_results?mo=containsall&ms=herbicides&saa=&p_action=SUBMIT&l=us&co=equal&ci=,&po=equal&pi=. Accessed 7 Nov 2014
Field JA, Reed RL, Sawyer TE, Griffith SM, Wigington PJ Jr (2003) Diuron occurrence and distribution in soil and surface and ground water associated with grass seed production. J Environ Qual 32:171–179
Pateiro-Moure M, Arias-Estévez M, López-Periago E, Martínez-Carballo E, Simal-Gándara J (2008) Occurrence and downslope mobilization of quaternary herbicide residues in vineyard-devoted soils. Bull Environ Contam Toxicol 80:407–411
Yang XB, Ying GG, Kookana RS (2010) Rapid multiresidue determination for currently used pesticides in agricultural drainage waters and soils using gas chromatography-mass spectrometry. J Environ Sci Health B 45(2):152–161
Fava L, Orrù MA, Scardala S, Alonzo E, Fardella M, Strumia C, Martinelli A, Finocchiaro S, Previtera M, Franchi A, Calà P, Dovis M, Bartoli D, Sartori G, Broglia L, Funari E (2010) Pesticides and their metabolites in selected Italian groundwater and surface water used for drinking. Ann Ist Super Sanita 46:309–316
Fuhrmann A, Gans O, Weiss S, Haberhauer G, Gerzabek MH (2014) Determination of bentazone, chloridazon and terbuthylazine and some of their metabolites in complex environmental matrices by liquid chromatography-electrospray ionization-tandem mass spectrometry using a modified QuEChERS method: an optimization and validation study. Water Air Soil Pollut 225(5):1–15
Lesueur C, Gartner M, Mentler A, Fuerhacker M (2008) Comparison of four extraction methods for the analysis of 24 pesticides in soil samples with gas chromatography-mass spectrometry and liquid chromatography-ion trap-mass spectrometry. Talanta 75(1):284–293
Mei M, Du Z-X, Cen Y (2011) QuEChERS-ultra-performance liquid chromatography tandem mass spectrometry for determination of five currently used herbicides. Chin J Anal Chem 39(11):1659–1664
Salvia MV, Vulliet E, Wiest L, Baudot R, Cren-Olive C (2012) Development of a multi-residue method using acetonitrile-based extraction followed by liquid chromatography-tandem mass spectrometry for the analysis of steroids and veterinary and human drugs at trace levels in soil. J Chromatogr A 1245:122–133
Kinney CA, Furlong ET, Kolpin DW, Burkhardt MR, Zaugg SD, Werner SL, Bossio JP, Benotti MJ (2008) Bioaccumulation of pharmaceuticals and other anthropogenic waste indicators in earthworms from agricultural soil amended with biosolid or swine manure. Environ Sci Technol 42(6):1863–1870
Chefetz B, Mualem T, Ben-Ari J (2008) Sorption and mobility of pharmaceutical compounds in soil irrigated with reclaimed wastewater. Chemosphere 73(8):1335–1343
Winker M, Clemens J, Reich M, Gulyas H, Otterpohl R (2010) Ryegrass uptake of carbamazepine and ibuprofen applied by urine fertilization. Sci Total Environ 408(8):1902–1908
WHO Model List of Essential Medicines (2013) World Health Organisation, Geneva, Switzerland. http://www.who.int/medicines/publications/essentialmedicines/en/. Accessed 7 Nov 2014
RxList The Internet Drug Index (2014) RxList Inc. San Clemente, CA, USA. http://www.rxlist.com/tegretol-drug/clinical-pharmacology.htm. Accessed 7 Nov 2014
Clara M, Strenn B, Kreuzinger N (2004) Carbamazepine as a possible anthropogenic marker in the aquatic environment: investigations on the behaviour of Carbamazepine in wastewater treatment and during groundwater infiltration. Water Res 38(4):947–954
Al-Rifai JH, Gabelish CL, Schäfer AI (2007) Occurrence of pharmaceutically active and non-steroidal estrogenic compounds in three different wastewater recycling schemes in Australia. Chemosphere 69(5):803–815
Miao XS, Yang JJ, Metcalfe CD (2005) Carbamazepine and its metabolites in wastewater and in biosolids in a municipal wastewater treatment plant. Environ Sci Technol 39(19):7469–7475
Lapen DR, Topp E, Metcalfe CD, Li H, Edwards M, Gottschall N, Bolton P, Curnoe W, Payne M, Beck A (2008) Pharmaceutical and personal care products in tile drainage following land application of municipal biosolids. Sci Total Environ 399(1–3):50–65
Li YB, Dong FS, Liu XG, Xu J, Chen X, Han YT, Liang XY, Zheng YQ (2013) Development of a multi-residue enantiomeric analysis method for 9 pesticides in soil and water by chiral liquid chromatography/tandem mass spectrometry. J Hazard Mater 250:9–18
Yamanaka H, Nakajima M, Hara Y, Katoh M, Tachibana O, Yamashita J, Yokoi T (2005) Urinary excretion of phenytoin metabolites, 5-(4′-hydroxyphenyl)-5-phenylhydantoin and its O-glucuronide in humans and analysis of genetic polymorphisms of UDP-glucuronosyltransferases. Drug Metab Pharmacokinet 20:135–143
Yu JT, Bouwer EJ, Coelhan M (2006) Occurrence and biodegradability studies of selected pharmaceuticals and personal care products in sewage effluent. Agric Water Manag 86(1–2):72–80
Republic of Italy (1999). Italian official methods of soil chemical analysis. Gazzetta Ufficiale della Repubblica Italiana, Supplemento ordinario No. 248, 21/10/1999
Asensio-Ramos M, Hernandez-Borges J, Ravelo-Perez LM, Rodriguez-Delgado MA (2010) Evaluation of a modified QuEChERS method for the extraction of pesticides from agricultural, ornamental and forestal soils. Anal Bioanal Chem 396(6):2307–2319
Burns DT, Danzer K, Townshend A (2003) Use of the terms “recovery” and “apparent recovery” in analytical procedures (IUPAC Recommendations 2002). Pure Appl Chem 74:2201–2205
Rivoira L, De Carlo RM, Cavalli S, Bruzzoniti MC (2015) Simple SPE-HPLC determination of some common drugs and herbicides of environmental concern by pulsed amperometry. Talanta 131:205–212
Fernandes VC, Domingues VF, Mateus N, Delerue-Matos C (2013) Multiresidue pesticides analysis in soils using modified QuEChERS with disposable pipette extraction and dispersive solid-phase extraction. J Sep Sci 36:376–382
Frenich AG, Gómez-Pérez R, Gómez-Pérez ML, Martínez Vidal JL (2011) Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J Chromatogr A 1218:4349–4356
European-Commission (2013) Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. European Commission Health & Consumer Protection Directorate General, SANCO/12571/2013
International Conference on Harmonisation (2009) Validation of analytical procedures: text and methodology Q2(R1). Fed Regist 62:27463–27467
Sulyok M, Berthiller F, Krska R, Schuhmacher R (2006) Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun Mass Spectrom 20:2649–2659
Ania CO, Beguin F (2007) Mechanism of adsorption and electrosorption of bentazon on activated carbon cloth in aqueous solutions. Water Res 41:3372–3380
Cairns D (2012) Essentials of pharmaceutical chemistry, 4th edn. Pharmaceutical, London
Rashid A, Nawaz S, Barker H, Ahmad I, Ashraf M (2010) Development of a simple extraction and clean-up procedure for determination of organochlorine pesticides in soil using gas chromatography-tandem mass spectrometry. J Chromatogr A 1217:2933–2939
AOAC Official Method 2007.01 (2007) Pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate.
Snyder LR, Kirkland JJ (1979) Introduction to modern liquid chromatography, 2nd edn. Wiley-Interscience, New York
Abd-Alrahman SH, Ahmed NS (2012) Dissipation of penconazole in tomatoes and soil. Bull Environ Contam Toxicol 89:873–876
Hong H, Yang Y, Huang Q, Miao X, Yuan Y, Yang X (2014) Analysis of residue dynamics of clofentezine in tangerine and field soil by QuEChERS and HPLC-UV methods. Int J Environ Anal Chem 94:639–651
Wang Y-H, Li X, Zhou X-M, Bai L-Y (2012) Simplified approach for the extraction of quinclorac from soils. Asian J Chem 24:2042–2044
Fernandez-Alba AR (2005) Chromatographic-mass spectrometric food analysis for trace determination of pesticide residues. In: Compr Anal Chem 43. Elsevier Science B.V.
Verdu CF, Gatto J, Freuze I, Richomme P, Laurens F, Guilet D (2013) Comparison of two methods, UHPLC-UV and UHPLC-MS/MS, for the quantification of polyphenols in cider apple juices. Molecules 18:10213–10227
Berlioz-Barbier A, Vauchez A, Wiest L, Baudot R, Vulliet E, Cren-Olive C (2014) Multi-residue analysis of emerging pollutants in sediment using QuEChERS-based extraction followed by LC-MS/MS analysis. Anal Bioanal Chem 406:1259–1266
Brondi SH, de Macedo AN, Vicente GH, Nogueira AR (2011) Evaluation of the QuEChERS method and gas chromatography-mass spectrometry for the analysis pesticide residues in water and sediment. Bull Environ Contam Toxicol 86:18–22
Shah J, Rasul Jan M, Ara B, Shehzad F-u-N (2011) Quantification of triazine herbicides in soil by microwave-assisted extraction and high-performance liquid chromatography. Environ Monit Assess 178:111–119
Mohapatra DP, Brar SK, Tyagi RD, Picard P, Surampalli RY (2012) Carbamazepine in municipal wastewater and wastewater sludge: ultrafast quantification by laser diode thermal desorption-atmospheric pressure chemical ionization coupled with tandem mass spectrometry. Talanta 99:247–255
Kormin F, Abdurahman NH, Yunus RM, Rivai M (2013) Study the heating mechanisms of temperature controlled microwave closed system (TCMCS). Int J Eng Sci Innov Technol 2:417–429
Schenck FJ, Lehotay SJ, Vega V (2002) Comparison of solid-phase extraction sorbents for cleanup in pesticide residue analysis of fresh fruits and vegetables. J Sep Sci 25:883–890
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 34 kb)
Rights and permissions
About this article

Cite this article
De Carlo, R.M., Rivoira, L., Ciofi, L. et al. Evaluation of different QuEChERS procedures for the recovery of selected drugs and herbicides from soil using LC coupled with UV and pulsed amperometry for their detection. Anal Bioanal Chem 407, 1217–1229 (2015). https://doi.org/10.1007/s00216-014-8339-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8339-x