
Abstract
In a study on the behaviour of pesticides in a soil–plant–water system, the quick, easy, cheap, effective, rugged and safe (QuEChERS) method for analysing pesticide or metabolite residues in soil and maize (leaves, roots and kernels) was optimized and validated. The pesticides bentazone, chloridazon and terbuthylazine and their metabolites bentazone-methyl, chloridazon-desphenyl, chloridazon-methyl-desphenyl, terbuthylazine-desethyl and terbuthylazine-2-hydroxy were selected in this study. The QuEChERS extracts obtained from soil and maize matrices and the collected leachate were analysed by liquid chromatography–electrospray ionization–tandem mass spectrometry (LC–ESI–MS/MS) using a high-performance liquid chromatography and an ultra-high-performance liquid chromatography (UHPLC) analytical column. As expected, shorter run times and higher sensitivity were achieved with the UHPLC column. Validation studies focused on recovery, repeatability, matrix effects, limits of detection and quantification. Recoveries (and repeatability relative standard deviation (RSD)) of the spiked samples were in the range of 55 to 98 % (7.4–18) in soil, 23 to 101 % (1.7–20) in maize and 82 to 105 % (4.4–25) in leachate. Quantification limits were lower than 3.0 μg kg−1 in soil, 7.3 μg kg−1 in maize and 0.080 μg l−1 in leachate.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Pesticides are ingredients of plant protection products used in agriculture to increase productivity. The use of pesticides for weed control on agricultural fields often leads to the contamination of soil, plants and water. Residues of commonly used pesticides and their metabolites can be detected in the environment for years. Following their application, pesticides undergo a variety of transformations that give rise to a complex pattern of metabolites. The presence of metabolites raises particular concern, as they can exist at higher levels than the parent pesticides (Andreu and Picó 2004). A good example is the parent compound atrazine and its associated metabolites (Kolpin et al. 1998).
To monitor pesticides in soil and plant material, an appropriate sample preparation method is required which assures the comprehensive extraction of the pesticides of interest. Traditionally, soxhlet extraction (Prados-Rosales et al. 2002; US EPA 1996) or alternatively pressurized liquid extraction (PLE) (Henriksen et al. 2002; Dagnac et al. 2005) is used to analyse pesticides in soils. Quick, easy, cheap, effective, rugged and safe (QuEChERS) is a sample preparation method based in dispersive liquid–liquid partitioning with acetonitrile (ACN) followed by a dispersive solid-phase-extraction (SPE) cleanup, first introduced by Anastassiades et al. (2003) for a broad range of pesticide residues in fruits and vegetables. Since then, the acetate buffering version has gained the distinction of becoming the AOAC Official Method 2007.01 (Lehotay 2007), and the citrate buffering version was released by the European Committee for Standardization as Standard Method EN 15662 (CEN 2008). The QuEChERS multiresidue procedure replaces previously complicated analytical steps, increasing sample throughput and reducing material costs. The method is frequently used for the extraction of a wide variety of compounds in different matrices as modifications can be implemented easily. Lehotay (2007) stated that, except those relatively few that contain carboxylic acid groups, nearly all pesticides can be monitored by the QuEChERS method. The effectiveness of the method for extracting pesticides from different food matrices is well documented (Cunha et al. 2007; Garrido-Frenich et al. 2008; Lehotay et al. 2005; Lehotay 2007; Lesueur et al. 2008a; Payá et al. 2007). The QuEChERS method has also been applied to the analysis of veterinary drugs (Stubbings and Bigwood 2009), mycotoxins (Sospedra et al. 2010; Vaclavik et al. 2010; Zachariasova et al. 2010) plus soil analysis for pesticides (Lesueur et al. 2008b; Rashid et al. 2010; Zhang et al. 2012), phenols (Padilla-Sánchez et al. 2010) and chlorinated compounds (Pinto et al. 2010).
Analytical methods to determine pesticides and/or metabolites have improved, making it possible to detect low residue levels in complex environmental matrices. In water analysis, liquid–liquid extraction (LLE) (Galeano-Díaz et al. 2008) or the alternative SPE (Kuster et al. 2006) is widely used as a pre-concentration step to provide the sensitivity required for liquid chromatography–tandem mass spectrometry (LC–MS/MS) or gas chromatography (GC)–MS/MS. Due to the high sensitivity and selectivity of tandem mass spectrometers, various classes of pesticides can be determined by direct injection. The use of direct injection LC–MS/MS is now widely accepted for pesticide analysis in water (Kuster et al. 2006; Reemtsma et al. 2013). Recently, an attractive alternative to using high-performance liquid chromatography (HPLC) known as ultra-high-performance liquid chromatography (UHPLC) has been developed, whereby the diameter and the particle size of the chromatographic columns are decreased, the run time reduced and the resolution enhanced. Compared to conventional HPLC, the instrumentation is operated at high pressures, and mobile phases at high velocities are used (Kmellár et al. 2011; Kowal et al. 2009; Wode et al. 2012). The potential for matrix effects when using HPLC/UHPLC connected to a tandem mass spectrometer via an electrospray ionization (ESI) interface should be considered. Matrix effects induce the suppression or enhancement of the analyte response due to co-eluting compounds. The influence of the co-eluting compounds occurs during the analyte ionization process, before the analyte ion reaches the high vacuum of the mass analyser (Kruve et al. 2008; Niessen et al. 2006).
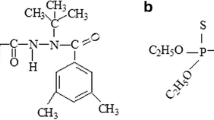
The pesticides selected for this study were bentazone, chloridazon and terbuthylazine as well as their metabolites bentazone-methyl, chloridazon-methyl-desphenyl, chloridazon-desphenyl, terbuthylazine-desethyl and terbutylazine-2-hydroxy. Bentazone, chloridazon and terbuthylazine are in widespread agricultural use. Their metabolites are usually more polar and thus pose a greater potential risk of groundwater contamination (Loss et al. 2010). The selection has a wide range of physicochemical properties (Table 1), and some of them are particularly challenging to analyse (e.g. chloridazon-desphenyl).
The present study focuses on the optimization and validation of the QuEChERS method for the determination of the pesticides and metabolites described above in soil and maize. The quantification of the pesticide and metabolite residues in QuEChERS extracts and leachate samples was performed using HPLC–MS/MS and UHPLC–MS/MS with ESI. To the best of our knowledge, QuEChERS has not previously been used to extract the selected pesticides or metabolites from the extremely pertinent matrices of soil and maize.
2 Material and Methods
2.1 Chemicals and Standards
Pesticide standards (bentazone, bentazone-methyl, chloridazon, chloridazon-desphenyl, chloridazon-methyl-desphenyl, terbuthylazine, terbuthylazine-desethyl, terbuthylazine-2-hydroxy) and isotopically labelled internal standards (bentazone-d6, chloridazon-d5, chloridazon-desphenyl-15N2, terbuthylazine-d5, terbuthylazine-desethyl-d9) were obtained from Dr. Ehrenstorfer (Augsburg, Germany). All organic solvents were of HPLC grade. ACN and water (HPLC) were obtained from LGC PromoChem (Wesel, Germany). Methanol (MeOH) was purchased from VWR (Vienna, Austria). Formic acid (98–100 %), acetic acid and ammonium acetate, all of analytical grade, were obtained from Merck (Darmstadt, Germany). Ultra-pure water was produced in the laboratory with a Milli-Q gradient system produced by Millipore (Vienna, Austria). Anhydrous magnesium sulphate and sodium citrate dibasic sesquihydrate in powder form were obtained from Sigma Aldrich (Vienna, Austria). Sodium citrate dehydrate, sodium chloride and calcium chloride were purchased from Merck (Darmstadt, Germany). Primary secondary amine (PSA) sorbent was obtained from Varian (Palo Alto, CA, USA), and C18 from J.T. Baker was purchased from Bartelt (Vienna, Austria). Disposable syringe filters (Chromafil PTFE 0.45 μm) were purchased from Macherey-Nagel (Düren, Germany) and syringes (2 ml) from B. Braun (Melsungen, Germany).
A solution of 5 % formic acid (v/v) was prepared in ACN. The salts used for the initial extraction step were prepared by mixing 4 g anhydrous MgSO4, 1 g NaCl, 1 g trisodium citrate dihydrate (Na3Citrate × 2H2O) and 0.5 g disodium hydrogencitrate sesquihydrate (Na2HCitrate × 1.5 H2O). Several sorbent combinations were filled in 15 ml centrifuge tubes for the cleanup: 150 mg PSA and 950 mg anhydrous MgSO4, 300 mg PSA and 300 mg CaCl2, 150 mg PSA, 900 mg anhydrous MgSO4, and 150 mg C18.
Stock solutions of the individual standards were prepared in MeOH. Solutions were stored in 4 ml amber glass vials at 4 °C. A working standard solution in MeOH containing all target pesticides at a concentration of 1 μg ml−1 was prepared from stock solutions. An internal standard mixture solution was made at a concentration of 1 μg ml−1 in MeOH. These solutions were used for fortification of the samples and for the preparation of the analytical calibration curves. Calibration solutions ranging from 0.075 to 20 ng ml−1 were prepared by adding equal aliquots of working standard solution (100 ng ml−1 in H2O or ACN) and internal standard mixture (100 ng ml−1 in H2O or ACN) into individual vials for the analysis of soil and maize extracts. Each solution was made up to a final volume of 1,500 μl. For the analysis of the leachate, calibration solutions with analyte concentrations ranging from 0.01 to 20 ng ml−1 were obtained by adding aliquots of working standard solution into individual vials. An internal standard mixture (15 μl) was added to each vial to a final volume of 1,500 μl.
2.2 Sample Sources and Preparation
Samples were taken from an experimental site located in Wagna (Styria, Austria). The soil is classified as sandy loam Dystric Cambisol with 51.8 % sand, 33.5 % silt and 14.6 % clay. Further characteristics are a pH of 6.6 (CaCl2), an organic carbon content (OC) of 2.7 % and a cation exchange capacity (CEC) of 11.53 cmolc kg−1 at a depth of 0–25 cm. Soil samples were air-dried, sieved (<2 mm), and stored at room temperature until required. Maize samples were divided into the green part (leaves and stems), roots and kernels. Samples were lyophilized, homogenised and ground using a cutting mill (SM 2000, Retsch, Haan, Germany) for the leaves and stems and a centrifugal mill (ZM 200, Retsch, Haan, Germany) for root samples. Maize kernels were milled to a flour consistency using a vibratory tungsten carbide disc mill (KHD Humboldt Wedag, Germany). Leachate samples were collected in flasks and were stored at −18 °C prior to analysis.
2.3 Liquid Chromatography–Tandem Mass Spectrometry
Analyses were performed on an HP1200 HPLC system and an HP1290 UHPLC system (Agilent Technologies, Vienna, Austria) connected to a 4000 QTRAP triple-stage quadrupole mass spectrometer (Applied Biosystems, Darmstadt, Germany) and controlled by Analyst 1.6.1 software. Qualification and quantification data was obtained with the electrospray probe operated in the positive and negative ion mode. The HPLC and UHPLC systems were equipped with a membrane degasser, a binary high-pressure pump, an automatic sampler and a column heater. Different gradient methods for the positive and negative ion mode of the mass spectrometer were used. Soil and leachate samples were analysed with both the HPLC and UHPLC systems. All maize extracts were analysed using the UHPLC system. When working with the HPLC system, the eluents for the positive ion mode were water-modified with 0.01 % formic acid (A) and MeOH comprising 2 mM ammonium acetate (B). Separation was performed on a 2 × 150-mm Luna C18 column (Phenomenex, Aschaffenburg, Germany) with 5 μm particle size attached to a security guard cartridge C18 4 × 2 mm (Phenomenex, Aschaffenburg, Germany) at 30 °C. Gradient elution was used starting with 2 % B at 0 min, held for 3 min, increased to 18 % B within 2 min, increased to 35 % B within 1 min, increased to 98 % B within 18 min, held for 10 min and decreased to 2 % B within 1 min. After 46 min, the system was ready for injection again. Flow was set to 200 μl/min. The injection volume was 50 μl for soil samples and 100 μl for leachate samples. The gradient method for the negative ion mode was operated with the eluents water-modified with 0.2 % acetic acid (A) and MeOH comprising 0.2 % acetic acid (B). The compounds were separated with a 2.1 × 150-mm Zorbax Eclipse Plus C18 column (Agilent Technologies, Vienna, Austria) with 3.5 μm particle size attached to a security guard cartridge C18 4 × 2 mm (Phenomenex, Aschaffenburg, Germany) at 40 °C. Gradient elution was used starting with 20 % B at 0 min, held for 2 min, increased to 98 % B within 16 min, held for 3 min and decreased to 20 % B within 1 min. After 30 min, the system was ready for injection again. Flow was set to 300 μl/min. The injection volume was 50 μl for soil and leachate samples.
When working with the UHPLC system, the eluents for the positive ion mode composed of 0.01 % formic acid and 2 mM ammonium acetate in water (A) and 2 mM ammonium acetate in methanol (B) for soil and maize samples. Analysing leachate samples, water modified with 0.01 % formic acid was used as eluent (A). Separation was achieved on a Kinetex column (C8, 2.6 μm particle size, 2.1 × 100 mm, Phenomenex, Aschaffenburg, Germany) with a security guard cartridge (C8, 2.1 × 4.6 mm, Phenomenex, Aschaffenburg, Germany) at 30 °C. For soil and leachate samples, gradient elution was used starting with 2 % B at 0 min, held for 2 min, increased to 40 % B within 2 min, increased to 95 % B within 4 min, held for 3 min and decreased to 2 % B within 1 min. For the maize samples, gradient elution was used starting with 5 % B at 0 min, held for 2 min, increased to 40 % B within 2 min, increased to 95 % B within 4 min, held for 3 min and decreased to 5 % B within 1 min.
After 15 min, the system was ready for injection again. Flow was set to 400 μl/min. The injection volume was 40 μl for soil and leachate samples and 3 μl for maize samples. The gradient method for the negative run was operated with the eluents water-modified with 0.04 % acetic acid (A) and ACN (B). The UHPLC column Zorbax Eclipse Plus C18 2.1 × 50 mm (Agilent Technologies, Vienna, Austria) with 1.8 μm particle size at 40 °C was used for separation. Gradient elution was used starting with 20 % B at 0 min, held for 2 min, increased to 50 % B within 1 min, increased to 95 % B within 1 min, held for 1 min and decreased to 20 % B within 1 min. After 9 min, the system was ready for injection again. Flow was set to 300 μl/min. The injection volume was 10 μl for soil samples, 20 μl of leachate samples and 5 μl for maize samples.
When working with the HPLC system, an ionization voltage of 5,500 and a temperature of 700 °C were used in the positive ion mode. In the negative ion mode, an ionization voltage of −4,500 and a temperature of 500 °C were operated. For the UHPLC system, an ionization voltage of 4,200 and a temperature of 700 °C were used in the positive ion mode. In the negative ion mode, an ionization voltage of −4,200 and a temperature of 500 °C were operated. Nitrogen was provided by a nitrogen generator (CMC instruments, Eschborn, Germany) and used as nebulizer, curtain and collision cell gas. Numerous experiments using solutions of the individual analytes were performed to determine the optimal MRM transition, collision energies and declustering potentials for each individual compound. A syringe at constant flow was used to infuse the standard solutions directly into the instrument.
2.4 Leachate
Samples were analysed by direct injection after the addition of an internal standard mixture as injection standard. Ten microlitres of internal standard mixture (100 ng ml−1 in MeOH) containing bentazone-d6, chloridazon-d5, chloridazon-desphenyl-15N2, terbuthylazine-d5 and terbuthylazine-desethyl-d9 was added to 1 ml leachate sample.
2.5 QuEChERS Procedures
The original QuEChERS method, according to Anastassiades et al. (2003) and CEN EN 15662 (2008), was developed for the extraction of samples with more than 75 % water content and consists of the following steps: (1) weigh 10 g sample into 50 ml centrifuge tubes; (2) add 10 ml ACN and shake the sample vigorously for 1 min; (3) add 4 g MgSO4, 1 g NaCl, 1 g Na3Citrate × 2H2O and 0.5 g Na2HCitrate × 1.5 H2O and shake immediately for 1 min; (4) centrifuge the extract for 5 min at 3,000 U/min; (5) take an aliquot into a 15-ml centrifuge tube containing MgSO4 and sorbent; (6) shake the sample for 30 s and centrifuge for 5 min at 3,000 U/min and (7) take an aliquot and add 5 % formic acid in ACN prior to the determination by GC–MS and LC–MS.
In this study, the original QuEChERS procedure was adapted for the dry matrices of soil and maize (leaf/stem, root and kernel). Several procedures and QuEChERS compositions were tested through recovery studies. The studied steps were: (i) the addition of water for matrix swelling and the acidification of the extraction solvent, (ii) the extraction time and (iii) different cleanup procedures.
An experiment to compare three acid variations, namely 1 % (v/v) acetic acid, 1 % (v/v) formic acid or 5 % (v/v) formic acid, in combination with ACN as an extraction solvent was investigated. The extraction solvent experiment was carried out in triplicate using 5 g soil and 5 ml of water added for swelling.
The influence of extraction time was evaluated in maize kernels testing. Samples of maize kernels (2.5 g) were spiked with the target compounds (40 μg kg−1) and either shaken for 1 min on the vortex, or placed for 1 h on a wrist shaker after the addition of 10 ml water and 10 ml ACN containing 5 % (v/v) formic acid. The presence of fats requires the additional cleanup of freezing out to obtain appropriate extracts. Briefly, after the initial extraction step with 4 g MgSO4, 1 g NaCl, 1 g Na3Citrate × 2H2O and 0.5 g Na2HCitrate × 1.5H2O, aliquots of 8 ml were taken from the ACN phase, placed into 15 ml centrifuge tubes and stored for 2 h in a freezer (−20 °C). Three replicates were analysed at each extraction condition.
To effectively remove co-extracts and to identify interactions between the pesticides and sorbents, a comparison of different sorbents for the dispersive SPE cleanup for maize samples of leaves and stems was performed. After the first centrifugation, 6 ml of the upper ACN extract was transferred into 15 ml centrifuge tubes containing either 150 mg PSA, 900 mg MgSO4 and 150 mg C18 or 300 mg PSA and 300 mg CaCl2 (CVUA 2009). Three replicates for each sorbent mixture were tested.
After QuEChERS extraction, solvent exchange of the ACN extracts to water were examined for all soil and maize (leaf/stem, root and kernel) matrices. One millilitre of the ACN extract was transferred into an auto-sampler vial. The extract was evaporated to 0.5 ml under a stream of nitrogen at a temperature of 30 °C. Following the addition of 0.5 ml HPLC–water, the extracts were again reduced to 0.5 ml at 30 °C. The extract obtained was filled up with HPLC water to 1 ml. In the case of the maize matrices, extracts in ACN and water were measured.
2.6 Method Validation
A validation study of the optimized extraction procedures was carried out in terms of recovery, repeatability, matrix effects and analytical limits including method limits of quantification (LOQs) and instrument limits of detection (LODs). Basic validation for each pesticide was carried out with SQS 2000 to determine the instrumental LOQ. Solvent-based calibration standards were measured three times at each concentration level to provide data for the basic validation.
For leachate, method LOQ in micrograms per litre was equivalent to the instrumental LOQ resulting from the basic validation. The method LOD was determined by dividing the method LOQ by 2. The internal standard mixture was added prior to the instrumental analysis to compensate for matrix and instrument variations. To determine the recovery rates, the peak areas of the isotopically labelled internal standards (bentazone-d6, chloridazon-d5, chloridazon-desphenyl-15N2, terbuthylazine-d5 and terbuthylazine-desethyl-d9) and those obtained from the solvent-based standards were used.
The method LOQ for soil and maize was calculated from the instrumental LOQ values of the mass spectrometer, multiplied by the extraction factor, divided by the lowest weighed sample and corrected with the mean recovery, minus the standard deviation of the corresponding deuterated internal standards. In the case of chloridazon-desphenyl in soil, the value was multiplied by the dilution factor. The method LOD was determined by dividing the method LOQ by 2.
Extractions from non-spiked soil and maize samples were performed to check the absence of the selected pesticides and the chromatographic interferences that precluded the correct detection and quantification of the analytes. The internal standard mixture was added (at the same concentration level as the pesticide standard) before extraction to keep track of possible losses occurring during the sample preparation and chromatographic analysis. Recoveries were determined at the concentration levels of 3 μg kg−1 for soil, 5 μg kg−1 for root, 10 μg kg−1 for leaf/stem and 40 μg kg−1 for maize kernel. The pesticide concentration measured by performing the complete procedure was compared with the pesticide concentration initially added to the individual blank matrices. The overall recovery of each pesticide was calculated as the mean recovery of the spiked samples extracted on different days using the same method and the same equipment. Repeatability is expressed as % RSD. Matrix effects in soil were examined by comparing the concentration derived from standard additions into sample extract to concentrations in pure aqueous solution. A post-extraction spiked experiment was carried out in four replicates by adding 10 μl of appropriate standard solution to 200 μl of soil extract and 200 μl of water.
3 Results and Discussion
3.1 Chromatographic Optimization
The optimized conditions of the selected (MRM) transitions of the eight pesticides are summarised in Table 2. In comparison with HPLC, the application of UHPLC–MS/MS improved the quantitative response and reduced the analysis time. The flow rate was increased and the injection volume reduced. Under the chromatographic conditions described above, the total analytical time for instrumentation using UHPLC was reduced from 46 to 15 min in the positive mode and from 30 to 9 min in the negative mode. Figure 1 presents a typical ion chromatogram of the eight pesticides or metabolites (all of them at 1 ng ml−1 concentrations) which were obtained from a standard sample in the positive mode using the UHPLC method. The chloridazon metabolites chloridazon-desphenyl and chloridazon-methyl-desphenyl did not allow proper peak recognition when the extracts of soil and maize samples were injected in ACN. The solvent exchange of QuEChERS extracts from ACN to water and additionally, a dilution of 1:5 (v/v), improved the detection of chloridazon-desphenyl in soil. Chromatograms of chloridazon-desphenyl in soil matrix are given in Fig. 2.

Extracted ion chromatogram from the positive mode UHPLC–MS/MS of 1 ng/ml (1) chloridazon-desphenyl, (2) chloridazon-methyl-desphenyl, (3) chloridazon, (4) terbuthylazine-2-hydroxy, (5) terbuthylazine-desethyl, (6) bentazone-methyl and (7) terbuthylazine from a standard sample in water

Examples of ion suppression on chloridazon-desphenyl in water: a a control soil matrix, b soil matrix at 3 μg kg−1 and c 1:5 (v/v) dilution of B from HPLC and d a control soil matrix, e soil matrix at 3 μg kg−1 and f 1:5 (v/v) dilution of e from UHPLC
3.2 Matrix Effect
The effect of ion suppression, or in rare cases, enhancement from using an ESI source is well-known, and environmental samples contain a large amount of compounds that can interfere with the analytical signal, producing matrix effects. In leachate, matrix effects for chloridazon-desphenyl were evaluated using the method of standard addition at five concentration levels. Results indicated that no significant suppression or enhancement was observed for chloridazon-desphenyl in leachate (1.2 %). An isotopically labelled internal standard was added just before instrumental analysis, thereby compensating run-to-run variation in instrument response and improving the precision. However, isotopically labelled internal standards are often not commercially available for recently found metabolites.
In soil, matrix effects were determined by a post-extraction spiked experiment. Results indicated a strong ionization suppression of chloridazon-desphenyl in soil extracts. A possible explanation is that chloridazon-desphenyl is the first eluting compound in the chromatogram where interference of the sample matrix with the solvent can occur. Kruve et al. (2008) documented that the ionization efficiency of polar pesticides is more affected by co-eluting compounds. Soil extracts were diluted with water (1:5 and 1:10 v/v) to reduce the amount of matrix components introduced into the LC–MS/MS system. Soil samples diluted 1:5 (v/v) provided an overall recovery of 60 % for chloridazon-desphenyl, whereas a complete elimination of matrix effects was observed when analysing the samples with a 1:10 (v/v) dilution. The main drawback of using the dilution approach to minimize matrix effects is the increase of the detection limit (Niessen et al. 2006; Sancho et al. 2002). Thus, all soil extracts were diluted (1:5, v/v) prior to injection to overcome the matrix effect of chloridazon-desphenyl (Fig. 2). No visible precipitation of matrix compounds was noticed, and the peak shapes of polar pesticides (chloridazon-desphenyl and chloridazon-methyl-desphenyl) were clearly improved. In addition, the isotopically labelled internal standard chloridazon-desphenyl-15N2 was added in every sample before the extraction and cleanup stages to compensate matrix effects and thus improve the accuracy and precision of the method.
In maize, direct injection of small volumes (3 and 5 μl) of the crude QuEChERS extracts in ACN and water were used to avoid matrix effects. Choi et al. (2001), Niessen et al. (2006) and Lacina et al. (2010) documented the effect of injection volume on matrix signal suppression. The risk of a rapid contamination of the sample cone of the mass spectrometer resulting in a significant decrease in the sensitivity of the analyte detection is also obviated by using small injection volumes.
3.3 Extraction Procedure
Several parameters were studied to optimize the performance of the extraction methods, such as the ratio of sample mass to extraction solvent volume, the extraction solvent, the extraction time and different cleanup procedures before the validation experiments.
3.3.1 Soil
The first optimization experiments used parts of the original QuEChERS and CEN method for soil. The best overall results were achieved using 5 g soil, 5 ml water, and 10 ml ACN including 5 % formic acid for the first extraction step. Recoveries increased to 24, 30 and 67 % for chloridazon-desphenyl and 70, 78 and 85 % for chloridazon-methyl-desphenyl using 1 % acetic acid, 1 % formic acid and 5 % formic acid. Chloridazon, terbuthylazine and terbuthylazine-desethyl achieved recoveries of 91, 92, and 91; 88, 89 and 84; and 92, 95 and 85 % for 1 % acetic acid, 1 % formic acid and 5 % formic acid, respectively. Based on these results, all subsequent experiments were carried out with 5 g soil, 5 ml water and 10 ml ACN including 5 % formic acid.
The freezing out step was not included in the final procedure as no precipitation of co-extracts and no improvement of recoveries were observed. The extraction method was further optimized by using sorbent combination of 150 mg PSA and 950 mg anhydrous MgSO4 in the dispersive SPE cleanup. The resulting soil extracts were taken for the solvent exchange prior to the LC–MS/MS analysis. Good recoveries were achieved for this optimized method (Table 3).
3.3.2 Root
The optimized method used followed the main steps and proportions of the original QuEChERS and CEN methods together with the optimizations already investigated for soil. To obtain the best homogenization and dispersion between the root and the extraction solvent, the ratio of 2 g sample, 8 ml of water for swelling and 10 ml ACN (5 % formic acid) was used for all further root extractions. The resulting root extracts were ready for injection into UHPLC–MS/MS, and an aliquot of 1 ml from the ACN extract was taken for the solvent exchange. The recovery results (Table 3) indicate that further investigation will be necessary to achieve better pesticide recoveries from root matrices.
3.3.3 Leaf and Stem
The extraction method previously used for soil and root was further optimized for maize leaves and stems. The volume of the leaves and stems necessitated a reduction of the sample amount to ensure sufficient ACN for the collection of the supernatant that followed. In addition, the effect of different sorbents in the dispersive SPE cleanup was investigated to improve purification and recoveries for maize leaves and stems. Recovery yields for the pesticides studied were satisfactory with each sorbent combination used. The recoveries obtained in water extracts were 26 and 41 % for chloridazon-desphenyl, 82 % for chloridazon-methyl-desphenyl, 76 % for chloridazon, 28 and 24 % for terbuthylazine, 66 to 45 % for terbuthylazine-desethyl, 84 and 83 % for terbuthylazine-2-hydroxy and 100 and 88 % for bentazone-methyl using 150 mg PSA, 900 mg MgSO4 and 150 mg C18 or 300 mg PSA and 300 mg CaCl2. A visual observation of the initial and final extracts showed less coloured extracts using 300 mg PSA and 300 mg CaCl2, and therefore, these sorbents were selected for subsequent validation experiments.
The resulting extracts were ready for injection into UHPLC–MS/MS, and an aliquot of 1 ml from the ACN extract was taken for the solvent exchange. Satisfactory recoveries were achieved for leaves and stems using the optimized method (Table 3).
3.3.4 Maize Kernel
The extraction method was further optimized for maize kernels by reducing the sample amount, examining the appropriate extraction time, performing the freezing-out step and using a different sorbent combination in the dispersive SPE cleanup. The sample amount was reduced to a 2.5-g sample, following Mastovska et al. (2010). The samples were extracted with 10 ml water and 10 ml ACN (5 % formic acid).
The influence of extraction time was studied for maize kernels using both 1 min and 1 h. In the same experiment, the freezing-out step for maize kernels was also examined in order to reduce the intrusive effect of the maize starch in the initial extracts. Recoveries showed no differences between the extraction times. Results indicated cleaner extracts using the freezing-out step with no significant effects on pesticide recoveries. Recoveries obtained in ACN extracts were 65 and 69 % for chloridazon, 67 and 66 % for terbuthylazine, 70 and 75 % for terbuthylazine-desethyl, 67 and 70 % for terbuthylazine-2-hydroxy and 87 and 82 % for bentazone-methyl, with or without the freeze-out step.
As a result, an extraction time of 1 min was chosen in order to simplify the optimized method as far as possible. A freeze-out step of 2 h was carried out, and C18 associated to PSA and MgSO4 in the dispersive SPE was used to minimize the presence of interfering compounds in the extract.
The resulting extracts were ready for injection into UHPLC–MS/MS, and an aliquot of 1 ml from the ACN extract was taken for the solvent exchange. Adequate recoveries were obtained using the optimized method shown in Table 3.
3.4 Method Performance
Recovery and repeatability were determined for each pesticide or metabolite in different environmental matrices using the above described methods. In leachate, results reveal that the recoveries for all compounds were satisfactory, ranging from 82 to 105 % with RSD values lower than 25 % in all cases (Table 4). The method LOD and LOQ values obtained for the selected pesticides are shown in Table 4.
The extractions carried out with the non-spiked samples detected a contamination of the metabolite terbuthylazine-2-hydroxy in soil (0.73 μg kg−1) and maize leaves and stems (7.5 μg kg−1), and root samples contained concentrations of chloridazon (62 μg kg−1) and chloridazon-desphenyl (51 μg kg−1). The initial pesticide concentrations were considered in the calculation of the recoveries. Detailed recovery and repeatability data for all pesticides and metabolites analysed in soil and maize are given in Table 3. All recoveries given were overall recoveries including matrix effects.
In soil, the compounds bentazone, bentazone-methyl and chloridazon gave excellent recoveries in the range of 80–87 %, and a good repeatability of less than 10 % was obtained with the RSDs. The recoveries of terbuthylazine (64 %) and terbuthylazine-desethyl (55 %) were lower in the validation experiments compared with those obtained in the method development stage. Results of the post-extraction spiked experiment showed matrix effects of 19 % for terbuthylazine and 26 % for terbuthylazine-desethyl. The large RSD values of terbuthylazin-2-hydroxy can be linked to the initial concentrations of the pesticide in blank samples.
The metabolites chloridazon-desphenyl and chloridazon-methyl-desphenyl were the most problematic compounds, due to their polar characteristics. These polar transformation products required the solvent exchange of the extracts to water to improve retention on the HPLC column as well as the peak shape. Thus, the matrix effects are minimized with overall recoveries of 67 and 73 % in soil (Table 3).
The final solvent used led to differing matrix effects of the selected pesticides in maize and thus influenced the overall recoveries. Some pesticides indicated less matrix effects in the ACN extracts, whereas others had better overall recoveries in water extracts. Bentazone-methyl, chloridazon and terbuthylazine indicated better recoveries in ACN extracts for all maize matrices. Recoveries obtained for terbuthylazine-desethyl in the ACN extracts were higher in roots and maize kernels and slightly lower in leaves and stems. Higher recoveries for bentazone in the ACN extracts were only achieved in maize kernels. In roots, and leaves and stems, the overall recoveries of bentazone increased to 60 and 68 % using the water extracts. Comparing the results of terbuthylazine-2-hydroxy, the water extracts provided the better recoveries for all maize matrices.
As described above, chloridazon-desphenyl and chloridazon-methyl-desphenyl were obtained in water extracts due to their polarity. Good recovery values were found for chloridazon-methyl-desphenyl, ranging from 74 % in roots, 82 % in leaf and stem and 96 % in maize kernels, whereas chloridazon-desphenyl only reached 41 % in leaf and stem as well as in maize kernels. Recoveries for chloridazon and chloridazon-desphenyl in root samples could not be calculated because the root samples used were highly contaminated with these compounds. Thus, the optimized method for root samples could not be properly evaluated.
This illustrates the difficulties in developing a single method for the determination of compounds with a wide range of physical–chemical properties. It can be observed that RSD values were lower than 20 % for all the compounds investigated in water and ACN of all maize samples. The detection limits for the majority of the pesticides indicates that the optimized HPLC and UHPLC–MS/MS method is capable of sensitive quantitation of pesticides from environmental samples (Tables 3 and 4).
High LOQ values were found for chloridazon-desphenyl in comparison to the other selected pesticides. This is due to the stronger matrix effects and difficulties in chromatographic separation, resulting in a higher uncertainty of measurements. Despite the described difficulties, the performance of the optimized methods was found to be useful for the investigation of pesticide behaviour in a lysimeter experiment. The proposed methods were applied for research into the transfer of pesticides in soil, water and plants.
4 Conclusion
The proposed methods were optimized and validated for the determination of bentazone, chloridazon, terbuthylazine and their known main metabolites in different environmental samples by LC–MS/MS. The extraction procedures described showed sufficient recoveries and precision. The different properties of the selected pesticides were challenging especially the chloridazon metabolites. A solvent exchange in water was necessary to ensure the correct quantification of the chloridazon metabolites in soil and maize. An LC–MS/MS method using HPLC and UHPLC in both positive and negative mode is available for the quantitative determination of the eight selected pesticides in soil, maize and water. The shorter injection cycle time and the improved sensitivity have led to the increasing adoption of UHPLC–MS/MS. The validated methods were successfully applied to determine the behaviour of bentazone, chloridazon, terbuthylazine and some of their metabolites in the complex system soil, plant and water using lysimeter experiments. Results will be published separately.
References
Anastassiades, M., Lehotay, S. J., Stajnbaher, D., & Schenck, F. J. (2003). Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. Journal of AOAC International, 86, 412–431.
Andreu, V., & Picó, Y. (2004). Determination of pesticides and their degradation products in soil: critical review and comparison of methods. Trends in Analytical Chemistry, 23, 772–789.
Choi, B. K., Hercules, D. M., & Gusev, A. I. (2001). Effect of liquid chromatography separation of complex matrices on liquid chromatography–tandem mass spectrometry signal suppression. Journal of Chromatography A, 907, 337–342.
Cunha, S. C., Lehotay, S. J., Mastovska, K., Fernandes, J. O., & Oliveira, B. M. P. P. (2007). Evaluation of the QuEChERS sample preparation approach for the analysis of pesticide residues in olives. Journal of Separation Science, 30, 620–632.
CVUA Stuttgart. (2009). Available at: http://quechers.cvua-stuttgart.de/pdf/q-tea.pdf.
Dagnac, T., Bristeau, S., Jeannot, R., Mouvet, C., & Baran, C. (2005). Determination of chloroacetanilides, triazines and phenylureas and some of their metabolites in soils by pressurised liquid extraction, GC–MS/MS, LC–MS and LC–MS/MS. Journal of Chromatography A, 1067, 225–233.
European Committee for Standardization (CEN). (2008). Standard method EN 15662. Available at: http://www.cen.eu.
Galeano-Díaz, T., Guiberteau-Cabanillas, A., López-Soto, M. D., & Ortiz, J. M. (2008). Determination of fenthion and fenthion-sulfoxide, in olive oil and in river water, by square-wave adsorptive-stripping voltammetry. Talanta, 76, 809–814.
Garrido-Frenich, A., Martínez-Vidal, J. L., Pastor-Montoro, E., & Romero-González, R. (2008). High-throughput determination of pesticide residues in food commodities by use of ultra-performance liquid chromatography–tandem mass spectrometry. Analytical and Bioanalytical Chemistry, 390, 947–959.
Henriksen, T., Svensmark, B., & Juhler, R. K. (2002). Analysis of metribuzin and transformation products in soil by pressurized liquid extraction and liquid chromatographic–tandem mass spectrometry. Journal of Chromatography A, 957, 79–87.
Kaune, A., Bruggeman, R., & Kettrup, A. (1998). High performance liquid chromatographic measurement of the octanol–water partition coefficient of s-triazine herbicides and some of their degradation products. Journal of Chromatography A, 805, 119–126.
Kmellár, B., Pareja, L., Ferrer, C., Fodor, P., & Fernández-Alba, A. (2011). Study of the effect of operational parameters on multiresidue pesticide analysis by LC-MS/MS. Talanta, 84, 262–273.
Kolpin, D. W., Thurman, E. M., & Linhart, S. M. (1998). The environmental occurrence of herbicides: the importance of degradates in ground water. Archives of Environmental Contamination and Toxicology, 35, 385–390.
Kowal, S., Balsaa, P., Werres, F., & Schmidt, T. (2009). Determination of the polar pesticide degradation product N, N-dimethylsulfamide in aqueous matrices by UPLC–MS/MS. Analytical and Bioanalytical Chemistry, 395, 1787–1794.
Kruve, A., Künnapas, A., Herodes, K., & Leito, I. (2008). Matrix effects in pesticide multi-residue analysis by liquid chromatography–mass spectrometry. Journal of Chromatography A, 1187, 58–66.
Kuster, M., López de Alda, M. L., & Barceló, D. (2006). Analysis of pesticides in water by liquid chromatography–tandem mass spectrometric techniques. Mass Spectrometry Reviews, 25, 900–916.
Lacina, O., Urbanova, J., Poustka, J., & Hajslova, J. (2010). Identification/quantification of multiple pesticide residues in food plants by ultra-high-performance liquid chromatography–time-of-flight mass spectrometry. Journal of Chromatography A, 1217, 648–659.
Lehotay, S. J. (2007). Determination of pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate: collaborative study. Journal of AOAC International, 90, 485–520.
Lehotay, S. J., De Kok, A., Hiemstra, M., & Van Bodegraven, P. (2005). Validation of a fast and easy method for the determination of residues from 229 pesticides in fruits and vegetables using gas and liquid chromatography and mass spectrometric detection. Journal of AOAC International, 88, 595–614.
Lesueur, C., Knittl, P., Gartner, M., Mentler, A., & Fuerhacker, M. (2008a). Analysis of 140 pesticides from conventional farming foodstuff samples after extraction with the modified QuEChERS method. Food Control, 19, 906–914.
Lesueur, C., Gartner, M., Mentler, A., & Fuerhacker, M. (2008b). Comparison of four extraction methods for the analysis of 24 pesticides in soil samples with gas chromatography–mass spectrometry and liquid chromatography–ion trap-mass spectrometry. Talanta, 25, 284–293.
Loss, R., Locoro, G., Comero, S., Contini, S., Schwesig, D., Werres, F., et al. (2010). Pan-European survey on the occurrence of selected polar organic persistent pollutants in ground water. Water Research, 44, 4115–4126.
Mastovska, K., Dorweiler, K. J., Lehotay, S. J., Wegscheid, J. S., & Szpylka, K. A. (2010). Pesticide multiresidue analysis in cereal grains using modified QuEChERS method combined with automated direct sample introduction GC–TOFMS and UPLC–MS/MS techniques. Journal of Agricultural and Food Chemistry, 58, 5959–5972.
Niessen, W. M. A., Manini, P., & Andreoli, R. (2006). Matrix effects in quantitative pesticide analysis using liquid chromatography–mass spectrometry. Mass Spectrometry Reviews, 25, 881–899.
Padilla-Sánchez, J. A., Plaza-Bolaños, P., Romero-González, R., Garrido-Frenich, A., & Martínez-Vidal, J. L. (2010). Application of a quick, easy, cheap, effective, rugged and safe-based method for the simultaneous extraction of chlorophenols, alkylphenols, nitrophenols and cresols in agricultural soils, analyzed by using gas chromatography–triple quadrupole-mass spectrometry/mass spectrometry. Journal of Chromatography A, 1217, 5724–5731.
Payá, P., Anastassiades, M., Mack, D., Sigalova, I., Tasdelen, B., Oliva, J., et al. (2007). Analysis of pesticide residues using the quick, easy, cheap, effective, rugged and safe (QuEChERS) pesticide multiresidue method in combination with gas and liquid chromatography and tandem mass spectrometric detection. Analytical and Bioanalytical Chemistry, 389, 1697–1714.
Pesticide Properties DataBase (PPDB). (2013). Agriculture & Environment Research Unit (AERU). University of Hertfordshire 2006–2013. Available at: http://sitem.herts.ac.uk/aeru/ppdb/en/index.htm.
Pinto, C. G., Laespada, M. E. F., Martín, S. H., Ferreira, A. M. C., Pavón, J. L. P., & Cordero, B. M. (2010). Simplified QuEChERS approach for the extraction of chlorinated compounds from soil samples. Talanta, 81, 385–391.
Prados-Rosales, R. C., Herrera, M. C., Luque-García, J. L., & Luque de Castro, M. D. (2002). Study of the feasibility of focused microwave-assisted Soxhlet extraction of N-methylcarbamates from soil. Journal of Chromatography A, 953, 133–140.
Rashid, A., Nawaz, S., Barker, H., Ahmad, I., & Ashraf, M. (2010). Development of a simple extraction and clean-up procedure for determination of organochlorine pesticides in soil using gas chromatography–tandem mass spectrometry. Journal of Chromatography A, 1217, 2933–2939.
Reemtsma, T., Alder, L., & Banasiak, U. (2013). A multimethod for the determination of 150 pesticide metabolites in surface water and groundwater using direct injection liquid chromatography–mass spectrometry. Journal of Chromatography A, 1271, 95–104.
Sancho, J. V., Pozo, O. J., López, F. J., & Hernández, F. (2002). Different quantitation approaches for xenobiotics in human urine samples by liquid chromatography/electrospray tandem mass spectrometry. Rapid Communications in Mass Spectrometry, 16, 639–645.
Sospedra, I., Blesa, J., Soriano, J. M., & Mañes, J. (2010). Use of the modified quick easy cheap effective rugged and safe sample preparation approach for the simultaneous analysis of type A- and B-trichothecenes in wheat flour. Journal of Chromatography A, 1217, 1437–1440.
Stubbings, G., & Bigwood, T. (2009). The development and validation of a multiclass liquid chromatography tandem mass spectrometry (LC–MS/MS) procedure for the determination of veterinary drug residues in animal tissues using a QuEChERS (quick, easy, cheap, effective rugged and safe) approach. Analytica Chimica Acta, 637, 68–78.
US Environmental Protection Agency. (1996). Method 3540C. Available at: http://www.epa.gov/wastes/hazard/testmethods/sw846/pdfs/3540c.pdf.
Vaclavik, L., Zachariasova, M., Hrbek, V., & Hajslova, J. (2010). Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta, 82, 1950–1957.
Wode, F., Reilich, C., Van Baar, P., Dünnbier, U., Jekel, M., & Reemtsma, T. (2012). Multiresidue analytical method for the simultaneous determination of 72 micropollutants in aqueous samples with ultra high performance liquid chromatography–high resolution mass spectrometry. Journal of Chromatography A, 1270, 118–126.
Zachariasova, M., Lacina, O., Malachova, A., Kostelanska, M., Poustka, J., Godula, M., et al. (2010). Novel approaches in analysis of Fusarium mycotoxins in cereals employing ultra performance liquid chromatography coupled with high resolution mass spectrometry. Analytica Chimica Acta, 662, 51–61.
Zhang, F., Wang, L., Zhou, L., Wu, D., Pan, H., & Pan, C. (2012). Residue dynamics of pyraclostrobin in peanut and field soil by QuEChERS and LC–MS/MS. Ecotoxicology and Environmental Safety, 78, 116–122.
Acknowledgments
This work was financed by the European Regional Development Fund and the national public funding, project MURMAN (4300-762/2010/7). The authors would like to thank Johann Fank and Barbara Zirngast from Joanneum Research, Graz for providing samples from the research station in Wagna.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fuhrmann, A., Gans, O., Weiss, S. et al. Determination of Bentazone, Chloridazon and Terbuthylazine and Some of Their Metabolites in Complex Environmental Matrices by Liquid Chromatography–Electrospray Ionization–Tandem Mass Spectrometry Using a Modified QuEChERS Method: an Optimization and Validation Study. Water Air Soil Pollut 225, 1944 (2014). https://doi.org/10.1007/s11270-014-1944-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-014-1944-7