
Abstract
This paper presents a current theoretical study on structural, electronic, vibrational, dielectric and energetic properties of the pristine cubic and tetragonal barium strontium titanate (BSTc and BSTt), as well as Sn-doped cubic and tetragonal barium strontium titanate (BSTSc and BSTSt) crystals. For this purpose, first-principle calculations within the Density Functional Theory method at the B3LYP level are implemented in the CRYSTAL14 code. Structural and thermodynamic analysis indicates that the change in geometry, as well as the Sn-doping process in BST samples, induces structural defects, which govern its electronic structure, generating singular bandgap values attributed to the perturbation of electronic levels in the vicinity of the Fermi level. Moreover, the vibration analysis was helpful to identify how the Sr and Sn doping process along with the A- and B-site of perovskite structure can affect the structural disorder, mainly for the tetragonal phase. Besides, electron density maps showed that the electronic properties were associated with the presence of [AO12] (A=Ba and Sr) and [MO6] (M=Ti and Sn) clusters with distinct bonding character. Furthermore, our structural, vibrational, and electronic calculations are in good agreement with the available experimental data and pave the avenue towards the complete understanding of the overall properties of perovskite materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Perovskite oxides with general formula ABO3 have attracted much attention due to their excellent physical and chemical properties across a wide range of technological fields, including electronics, optics, optoelectronics, etc. Different A-site and B-site dopants (where A=Sr, Ba; B=Ti, Sn) are used to modify the electrical properties of perovskite [1,2,3,4,5,6,7,8,9,10,11,12]. A solid solution of both BaTiO3 and SrTiO3 produces the barium strontium titanate (BaxSr1−xTiO3, BST) [3, 13,14,15], or as mentioned by Yustanti et al. [16] and Pasha et al. [17] with the presence of Sr ions as a dopant in BaTiO3 formed BST crystal structure. BST is a promising material, due to its dielectric properties, adjusting the mole ratio of Ba/Sr to meet a wide variety of applications in electronics, such as microwave phase shifters, dielectric capacitors, dynamic random-access memories (DRAM), etc. Various preparation methods for BST have been investigated, as conventional solid-state reaction [15, 18,19,20], solvothermal [21], sol–gel [22, 23], coprecipitation [24], spray pyrolysis[25], organic precursor method [26] and hydrothermal methods [13, 27].
The pure compounds, BaTiO3 and SrTiO3, have the same cubic perovskite structure in the high-temperature phase. Barium titanate is a typical ferroelectric, which undergoes three consecutive phase transitions from a cubic Pm3m to a tetragonal P4mm phase at 403 K, then to an orthorhombic Amm2 phase at 278 K, and, finally, to a rhombohedral R3m phase at 183 K. Studies on the BaxSr1-xTiO3 report the dependence of the temperatures of such phase transitions versus Sr content (x) [16, 18, 28, 29].
The ideal perovskite has a cubic space group Pm-3 m that contains one unit formula per cell. In particular, the A-site cation is 12-fold coordinated, while the B-site cation is coordinated by six O anions in a body-face-centered cubic structure[30]. Furthermore, most of the perovskite materials can present a slightly distorted structure obtained from the cubic structure by different ways, such as the displacement of ions from the ideal position, tilting the BO6 octahedral, or defects in the structure that can break the cubic symmetry resulting in a “non-cubic” structure [10, 31].
Theoretical studies of perovskite compounds are of particular significance because they can be performed by different methods helping to explain the experimental results [32]. However, if a dopant is introduced in the compound, its properties are changed, and the ab initio simulations become far more complicated. In this case, the doping of titanate-based perovskites with several kinds of metals cations is widely performed because of the vast range of peculiar properties it allows to tune in fields as diverse as structural, electrical, and optical properties [33,34,35].
In this study, DFT calculations were applied to rationalize the effects of local structural changes induced by the introduction of Sn4+ in the crystal lattice and on the change in geometry of BST structure. We also demonstrate that the electronic properties are associated with the presence of different clusters with a unique bonding environment from electron density distribution. In particular, the formation of intermediate levels in the bandgap (Egap) region was discussed following the structural disorders. Also, the thermodynamic stability of the pristine BST (cubic and tetragonal; BSTc and BSTt) and Sn-doped (BSTSc and BSTSt) structures were analyzed to discuss the primary role of doping on the geometry stabilization. Notably, our models support the available experimental results, showing an excellent agreement. The manuscript is organized into three other sections in the following manner: (i) the next Sect. (2) describes in details the employed computational methodologies; (ii) Sect. 3 contains the results for structural, electronic, vibrational, and energetic properties; (iii) at last, the Sect. 4 contains the main conclusions of our work.
2 Computational details
The computational methods and theoretical procedures have been employed to study the bulk related to pristine cubic and tetragonal BST, as well as Sn-doped BST models with a doping amount of 50%, respectively. Calculations were carried out using the periodic ab initio CRYSTAL14 package [36] based on Density Functional Theory (DFT) using hybrid functional B3LYP [37, 38]. This computational technique has been successfully applied for the study of the electronic and structural properties of various materials, including perovskite and several other oxides[39,40,41,42,43,44,45,46,47,48,49]. In all calculations, the atomic centers were described by standard all-electron basis set 6–31G*, 976–41(d51)G, 9763–311(d631)G, 86–411(d31)G, and 9,763,111,631 for the O, Sr, Ba, Ti, and Sn atoms, respectively[40, 41, 50, 51].
Further, the electronic properties were analyzed in terms of the Density of States (DOS), Band Structure profiles, and the charge density map. Besides, Visualization for Electronic and Structural Analysis (VESTA) [52] and X-Window Crystalline Structures and Densities (XCrySDen) [53] software were used for visualization and representation of supercell models, X-ray diffraction (XRD) pattern calculation and the charge density maps construction. Electronic integration was performed using a dense 8 × 8 × 8 Monkhorst–Pack [54] k-mesh for the pristine and doped cells, containing 75 k-points for both BST (cubic and tetragonal) and Sn-doped BST models. The accuracy of the Coulomb and exchange integral calculations were controlled by five thresholds set to 8, 8, 8, 8, and 16. The converge criterion for mono- and bi-electronic integrals were set to 10–8 Ha, while the root-mean-square (RMS) gradient, RMS displacement, maximum gradient, and maximum displacement were set to 9.4 × 10–5, 7.3 × 10–4, 1.4 × 10–4, and 1.2 × 10–3 a.u. for BSTc, 1.9 × 10–4, 8.3 × 10–4, 3.4 × 10–4, and 1.7 × 10–3 a.u. for BSTSc, and 6.1 × 10–5, 4.7 × 10–4, 1.3 × 10–4, and 8.2 × 10–4 a.u., for BSTt respectively. In all cases, both lattice parameters and atomic positions were relaxed.
In this work, a cubic and tetragonal supercell model of 10 atoms, which corresponds to a 1 × 1 × 2 conventional cell, was used to simulate both pristine and Sn-doped BST structures. In particular, for the Sn-doped BST model, a 1 × 1 × 2 supercell (10 atoms) was considered, where one Ti4+ (ionic radius 0.605 Å) cations were replaced by Sn4+ (ionic radius 0.690 Å)[55] corresponding to a doping concentration of 50%.
3 Results and discussion
3.1 Structural properties
The initial lattice parameters and atomic position used in the optimization process were obtained from the BSTc, BSTt, BSTSc, and BSTSt, results of the Rietveld refinement, according to Souza et al. [56] and Chihaoui et al. [57]. The calculated values for the pristine barium strontium titanate (Ba,Sr)TiO3 (BST) (cubic and tetragonal; BSTc and BSTt) and Sn-doped (BSTSc and BSTSt) structures are collected in Table 1 and compared with experimental results.
As can be observed in Table 1, the crystal structure and the lattice parameters of theoretical results exhibit a small mean percentage error in comparison with the experimental results, evidencing that our calculations are in agreement with the experiments. Moreover, the unit-cell volume was underestimated by 1.41% and 3.01% for pristine BSTc, and BSTt, while the obtained results for BSTSc and BSTSt indicate an overestimation of 2.88% and 1.83%, respectively. Furthermore, comparing the crystalline parameters for both pristine and Sn-doped BST models, it was observed that for cubic polymorphs (BSTc and BSTSc) an expansion of cell parameters of ~ 2.13% was found after the Sn doping, as well as for BSTt and BSTSt models with a variation of 5.52%. This behavior can be explained by the differences between the ionic radius of Sn4+ (0.690 Å) and Ti4+ ions (0.605 Å).
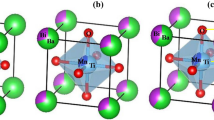
From a theoretical point of view, three models were constructed using a conventional 1 × 1x2 supercell containing 10 atoms. First, structural and electronic properties were calculated for a perfect bulk of BSTc, and BSTt lattice, and BSTSc, posteriorly. The representation of the BSTc, BSTt, and BSTSc bulk structures is shown in Fig. 1. This Fig. 1(a-c) illustrates the green, blue, gray, and red balls correspond to Ba, Sr, Ti, Sn, and O atoms, respectively. In this case, the Ti and Sn atoms are coordinated by six O atoms, producing octahedral [TiO6] and [SnO6] clusters. Correspondingly, the Ba and Sr atoms are coordinated to twelve O atoms, resulting in [BaO12] and [SrO12] clusters.

Conventional unit cell for pristine (Ba,Sr)TiO3 and Sn-doped structures a cubic, b tetragonal polymorphs, c [Ba,SrO12], [TiO6] and [SnO4] clusters
Furthermore, in order to analyze the structural disorder caused by the change in geometry, as well as the role of the Sn-doping in BST matrix, the bonding environment of both [TiO6], [SnO6], [BaO12] and [SrO12] clusters was investigated in details. Table S1 (Supplementary Information) shows the calculated B3LYP values of bond distances (M–O) and the atomic coordinates in Cartesian coordinates (Å) along the 1 × 1 × 2 supercell used for BSTc, BSTc, BSTSc, BSTt, and BSTSt, respectively.
It is important to note that atomic coordinates and M–O bond distances are stretched and shortened in the cubic, tetragonal and doped structures, respectively (see Table S1). This effect can be related to the structural order–disorder effect associated with the change in geometry and the insertion of the Sn dopant. Besides, the bond distances (M–O) undergo a slight increase in comparing Sn4+ and Ti4+ centers. The increase in bond distances (M–O) can be due to three reasons: (i) the distortion of the environment of the A ion during to tilting, (ii) the Sr atom is partially substituted by an atom (Ba) of higher ionic radios and (iii) the Jahn–Teller effect in by the partial removal of the eg and t2g degeneracies from cubic to tetragonal models. In previous studies, the same behavior has been observed by Joseph et al. [58].
Additionally, the structural study of the pristine BSTc and BSTt, BSTSc, and BSTSt crystals was performed by X-ray diffraction (XRD) pattern using VESTA software. The XRD pattern for the BSTc, BSTSc, BSTt, and BSTSt structures is shown in Fig. 2(a-d), and indicates a single-phase cubic and tetragonal perovskite with the Pm-3 m and P4mm space group, respectively.

Simulated X-ray diffraction patterns of a BSTc, b BSTSc, c BSTt, and d BSTSt models by VESTA software
The simulation of XRD patterns for the cubic BST and BSTS (Fig. 2a-b) is in good agreement with the crystallographic report JCPDS 39–1395, according to experimental data reported by Souza et al. [56]. In both crystals, the strongest peak around 2θ = 32°corresponds to the (110) crystalline plane; however, doping with Sn4+ cations induces a shift of the main reflections toward 2θ values with the formation of clusters of [SnO6], from 32.11 to 31.42(°), due to the different electronic density to Ti4+ ions, which can be seen from the strongest diffraction peak (Fig. 2a–b). The same was also observed for the following diffraction peaks changing from 39.60 to 38.87(°); 46.05 to 45.03(°), 57.24 to 55.96 (°), continuously for BSTc and BSTSc. Moreover, in the doped system, some crystallographic planes were barely noticeable due to the low intensity as (100), (300), (212), (026), and (304). This observation is a strong indication of the successful substitution of Ti4+ by Sn4+ in the B sites of the perovskite BSTc material. Our observation for the simulation of XRD patterns of BSTt (Fig. 2c–d) follows the previous study reported by Chihaoui et al. [57]. The tetragonal phase and space group P4mm is confirmed by the presence of peaks (001), (100), (101), (110), (012), (111), (004), (200), (002), (102), (210), (211), (112), (220), (202), (204), (300), (212), (103), (310), (032), (106), (016), ((132), (312), (116), (224) and (133). In this case, similar behavior was observed for the doped system, as mentioned in the cubic system. In pristine BSTc and BSTt crystals structures, the main observation was the appearance of crystallographic plans, as shown in Fig. 2a and c.
3.2 Electronic properties
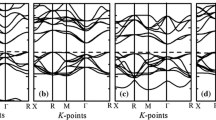
In this section, we present the theoretical results for the electronic properties of the investigated perovskite compounds. The electron distribution plays a fundamental role in determining the band structure, the density of states (DOS), and the charge density. The band structures plotted along the path Γ (0,0,0), X (½,0,0), M (½,½,0), Z (0,0, ½), R (½,0, ½), and A (½,½,½), as well as the calculated DOS projected for the atoms of the BSTc, BSTt, BSTSc, and BSTSt crystals are displayed in Fig. 3a–b. These figures show that the shaded region in the band structure indicates the bandgap region, while pink bands indicate the valence band maximum (VBM) and conduction band minimum (CBM).

a Band structure profiles and b atom-resolved DOS profiles for BSTc, BSTt, BSTSc, and BSTSt
An analysis of the band structure and projected DOS are presented in Fig. 3a–b. For BSTc it was observed that the valence band (VB) is observed between 0 and − 4 eV. The conduction band (CB) was evaluated between 3.73 eV and 12 eV. The calculated indirect band gap energy was 3.73 eV, being an indirect electronic excitation between M and Γ points. On the other hand, for the BSTt structure the VB was taken between 0 and − 4 eV, while CB is between 3.76 and 12 eV. Regarding the bandgap region, an indirect electronic transition between M- Γ points was calculated around 3.76 eV. From BSTSc illustrated in Fig. 3, the VB was evaluated between 0 and − 4 eV. The CB is between 3.77 and 18 eV. For BSTSt, the VB was evaluated between 0 and − 4 eV and CB is between 3.77 and 18 eV, where a bandgap energy of 3.77 eV was calculated between M-G.
The introduction of Sn4+ dopants causes slight changes in the bandgap energy (3.77 eV) of BSTSc, originating from the intermediate electronic level due to the appearance of the localized states into the CB region from 4d Sn atomic orbitals. The dopant forms clusters that influence the short, medium and long range, the electronic density of the crystal, due to symmetry breaking. In this way, the results show the role of the dopant by introducing new properties to the semiconductor. This analysis is valid for the other dopants. Moreover, the detailed analysis of the upper panel of Fig. 3 indicates that Sn-doping mechanism affects the band distribution, mainly in the VB. Comparing the bands distribution for BST and BSTS models at different crystalline structures, it was noted that occupied electronic levels for BSTS are more degenerated in comparison with BST, which can be associated with the bonding character of Sn–O and Ti–O chemical bonds.
Besides, comparing the CB for both models of BST and BSTS enables us to interpretate the charge carriers properties (electrons) from the curvature of Conduction Band Minimum (CBM). Indeed, for BST models the CBM located at G point is almost linear up to X, showing a broad feature, while the CBM for BSTS models at the same point exhibit a parabolic-like distribution with a well-located minimum point. Based on the relation between the effective mass of charge carriers with the band curvature, we can argue that electron–hole recombination rate is distinct between BST and BSTS models. In this case, a broader band can induce a higher effective mass for the excited electrons reducing its mobility, while a well-defined parabolic band can be associated with a reduced effective mass and higher electron mobility. Therefore, the obtained results for band structure profiles of BST and BSTS indicate that Sn-doping induces a higher electron mobility, making BSTS a good candidate for electro-optical applications.
As regard the obtained bandgap values, experimental and theoretical values were compared, as shown in Table 2, evidencing a good agreement. Indeed, the B3LYP hybrid functional showed a very close representation of the experimental bandgap energy, showing deviation ranging from 2.19 to 13.5%. Here, it is important to point out that bandgap description is a challenging topic for quantum mechanical calculations due to the treatment of exchange–correlation effects. However, the obtained results confirm the predictive power of hybrid B3LYP functional for perovskite materials.
Additionally, Fig. 3b summarizes the analysis atomic contribution for both VB and CB, showing a well-defined pattern that is directly associated with the local clusters centered on Ba, Sr, Sn, Ti, and O atoms. The main contribution to the VB region is due to the 2p (px, py, pz) orbitals of the oxygen anions with a small content of Ba and Sr orbitals for both BST and BSTS models. In contrast, the CB was mostly based on empty valence (3dxz, 3dxy, 3dyz, 3dz2, 3dx2-y2) orbitals from Ti atom and a small content of Sn cations hybridized with oxygen atomic orbitals, revealing the role of [TiO6] and [SnO6] clusters. These results confirm the role of Sn-doping mechanism in the control of CBM distribution, which can be associated with the electron mobility along the electronic structure, as previously discussed.
Electronic density maps of the BSTc, BSTt, BSTSc, and BSTSt, were obtained from the optimized wavefunction, where the electronic density matrix was resolved as isolines that describe the density in an area, as an exhibit in Fig. 4(a-d). These electronic density maps were described along the Ba–O, Sr–O, Ti–O, and Sn–O bonds direction of the materials, which corresponds more specifically to the diagonal (110) plane for all models of cubic and tetragonal symmetries (Fig. 4a–d).

Electron density maps in the diagonal plane (110) for a BSTc, b BSTSc, c BSTt and d BSTSt crystals structures
Figure 4(a–d) illustrates the strongly covalent character in the interaction of the Ti and Sn atoms with the oxygen atoms on the analyzed (110) plane, which is represented by homogenous distributions of the contour lines. The observed behavior takes place because of the hybridization between the oxygen 2p atomic orbitals and the Ti (3d) and Sn (4d) atomic orbitals. The density distribution remained the same for Fig. 4(a–c) illustrating the inhomogeneous distribution of the contour lines that represent the ionic character of the Ba–O and Sr–O bonds.
Furthermore, the significant difference between the contour plots reported in Fig. 4(a–d) is associated with the charge corridor along the z-axis for BSTc, BSTt, BSTSc, and BSTSt models. In particular, for both cubic models (BSTc and BSTSc), the charge corridor exhibits a homogenous distribution for electron density along the Ti–O–Ti or Sn–O–Ti bond path. On the other hand, for BSTt and BSTSt, the intermetallic Ti–O–Ti showed a charge depletion area for a bridge-like oxygen atom, confirming the existence of spontaneous polarization along the [001] direction due to the off-centering atomic displacement for Ti atoms in tetragonal symmetry [3].
3.3 Vibrational properties
According to group theory, cubic perovskite with space group, \({O}_{h}^{1}\), Pm-3 m (221), has no Raman-active phonon modes. In contrast, tetragonal perovskite with space group,\({C}_{4v}^{1}\), P4mm (99) has the following phonon modes at the Γ point:
In the cubic phase (m-3 m), the zone-center optical phonons belong to 3F1u + F2u irreducible representations. Each of the Fu modes is triply degenerate, and all of them are of odd symmetry concerning the inversion, therefore, Raman inactive. The F1u modes are infrared active, while the F2u modes are silent. Upon transition to the tetragonal phase (4 mm), the F1u modes split into A1 and E modes, and the F2u phonon gives rise to B1 and E modes.
The A1 and E modes are both Raman and infrared active, while the B1 mode is only Raman active. The four optical E modes for the tetragonal phase are doubly degenerate and polarized along the x and y axes, and the three optical A1 modes are polarized along the z-axis. The E and A1 modes are split into longitudinal (LO) and transverse (TO) components as a result of long-range electrostatic forces associated with lattice ionicity [17, 61, 62]. Table 3 summarizes the experimental Raman active modes (cm−1) for tetragonal or pseudo-cubic perovskite BST.
Thus, tetragonal BST has the Raman active crystal symmetry of \({C}_{4v}^{1}\), showing distinguishable Raman peaks at room temperature, in contrast to the cubic BST of the Raman inactive \({O}_{h}^{1}\) symmetry, for which the peaks disappear. This means that it is possible to discern crystal phases according to the Raman spectrum.
Figure 5 shows the calculated Raman spectra of the BSTt and BSTSt at the B3LYP level of theory in the frequency range 100–900 cm−1. On the Raman spectrum of the tetragonal BST crystal structure, we observe seven active vibration modes observed at 121, 178, 280, 298, 546, 579, and 801 cm−1. It is well known that DFT calculation at the B3LYP method tends to overestimate the values of the vibrational frequencies; therefore, a scaling factor of 0.94 is used [66].

Theoretical Raman spectra calculated for BSTt and BSTSt structures
Analyzing Fig. 5, it was observed that theoretical results are in good agreement with experimental data reported by Chihaoui et al. and other papers presented in Table 3. The bands around 180, 280 and 579 cm−1 are assigned to the transverse optical (TO) mode of A1 symmetry, whereas the band around 298 and 801 cm−1, which are characteristic of the tetragonal phase, are attributed to the E (TO + LO) + B1 mode and A1(LO) + E(LO) [34]. The A1 longitudinal optic (LO) modes can be identified at 121 and 546 cm−1. Nine peaks ascribed to the BSTSt Raman spectrum are observed at 100–900 cm−1 and seven for BSTt. This behavior means that the substitution for Sr and Sn in the A and B sites of the perovskite structure introduces a significant disorder in the structure, favoring the relaxor character in the BSTS material.
3.4 Formation enthalpy and thermodynamic stability
The discussion involving the thermodynamic stability of perovskite polymorphs corresponds to an important topic due to the structural transformations associated with the symmetry order. In this context, the evaluation of Formation Enthalpy (ΔHf) from its forming binary oxides plays a fundamental role in the discussion of phase stability [67,68,69]. From a theoretical point of view, such a process can be interpreted as a function of the reaction involving the perovskite material (ABO3) and its component oxides (AO + BO2). In this case, the ΔHf can be calculated as the difference between the lattice energies, following the expression [70, 71]:
where Hi = ET + pV. Here, ET is the total energy the solid at P = 0, and the corresponding equilibrium structures were obtained by optimizing all of the geometric parameters. Additionally, the zero-point energy contribution was estimated from vibrational analyses for comparative purposes.
Herein, the overall phase stability was calculated for BSTc, BSTt, BSTSc, and BSTSt perovskites, respectively. Table 4 summarizes the obtained results for ΔHf as a function of the component oxides (BaO, SrO, TiO2 for BSTc and BSTt; BaO, SrO, TiO2, SnO2 for BSTSc and BSTSt, respectively).
At first glance, it was observed that all investigated reaction channels indicate that BSTc, BSTt, BSTSc, and BSTSt perovskites are stable in comparison with its component oxides, once negative values were founded. Moreover, the contribution of ZPE was calculated to be 0.022, 0.036, 0.038, and 0.041 eV for BSTc, BSTt, BSTSc, and BSTSt perovskites, respectively. However, the stability order remains the same, even with the ZPE correction.
Further, comparing the ΔHf for different polymorphs of BST (BSTc and BSTt), the calculated values indicate that the cubic phase is more stable in comparison with tetragonal symmetry. However, the values are quite similar, indicating the existence of a few differences between the crystalline structures. Similarly, the obtained values for tetragonal and cubic polymorphs of BSTSt indicate that the high-symmetric cubic lattice is more stable than tetragonal lattice, notwithstanding the polymorphs are very close in energy. Therefore, it was possible to observe that BST and BSTS perovskites are stable in comparison with their component oxides, being the cubic polymorph the low-energy arrangement expected in the complex phase diagram with the tetragonal phase.
3.5 Dielectric properties
Perovskite materials, such as BST, have been attracted an increased interest due to their inherent high dielectric constant [72,73,74]. In this context, the possibility to rationalize the dielectric properties through quantum–mechanical calculations correspond to an alternative tool to design new materials with potential technological applications. Herein, the static dielectric tensor (ε0) was evaluated by means of Couple-perturbed Kohn–Sham scheme (CPKS) combined with longitudinal-transverse optical (LO-TO) splitting, [75,76,77] as reported in Table 5.
Our theoretical results for both BSTt and BSTSt materials show that both oxides were described by two independent components of the static dielectric (ε0) tensor, where the \({{\varvec{\varepsilon}}}_{0}^{{\varvec{z}}{\varvec{z}}}\) is the dominant. Moreover, our purpose is to investigate the dielectric behavior of BSTSt materials in order to compare with BSTt. Therefore, it was observed that for BSTSt the calculated values of the static dielectric tensor are smaller than for BSTt for the dominant \({{\varvec{\varepsilon}}}_{0}^{{\varvec{z}}{\varvec{z}}}\) component. On the other hand, the \({{\varvec{\varepsilon}}}_{0}^{{\varvec{x}}{\varvec{x}}}\) component for BSTt becomes higher than for BSTSt. This effect may be related to a higher mobility of Sn atom, which induces a greater asymmetry in the electronic density of BO6 octahedral reducing the dielectric constant. In particular, the obtained results confirms the superior dielectric behavior of BSTt, but also shed a light on BSTSt perovskite materials as promising candidates for dielectric devices.
4 Conclusion
In summary, pristine cubic and tetragonal BST (BSTc and BSTt) and Sn-doped BST (BSTSc and BSTSt) were investigated from the Density Functional Theory calculations to gain an in-depth understanding of structural, electronic, vibrational and energetic properties. XRD patterns by VESTA software and DFT analysis confirmed the perovskite phases showing both Pm-3 m and P4mm space groups in pristine and Sn-doped BST structures. The building blocks of the BSTc, BSTt, BSTSc, and BSTSt crystals, i.e., a local coordination structure for both the Ba, Sr, Ti, and Sn atoms, were confirmed as deltahedral [Ba,SrO12], and octahedral [TiO6], and [SnO6] clusters. In the viewpoint of thermodynamic analysis, all models were calculated to be stable in comparison with their component oxides. The indirect bandgap energy was calculated as 3.73, 3.76, 3.77, and 3.77 eV for BSTc, BSTt, BSTSc, and BSTSt structures, respectively. Moreover, the detailed analysis of electronic structures for BST and BSTS models indicates that Sn-doping induces a higher electron mobility, being a potential candidate for electro-optical applications. Such results indicate that the change in geometry and insertion of the Sn atom generates an intrinsically order–disorder effect, which will promote a presence of intermediate levels, causing an increase in the gap energy. According to the charge density maps, the periodic models showed well-defined electron density distribution characterizing the symmetrical structure and confirmed the covalent bonds between transition Ti and Sn metals with oxygen atoms, while the Sr–O and Ba–O bonds were defined as ionic. Moreover, the vibrational analysis indicates the presence of singular disorders for BSTt and BSTSt associated with the doping process along with the A- and B-site of perovskite structure, resulting in a more pronounced relaxor character for BSTSt. For the dielectric applications, it was observed that dielectric tensor components for BSTSt were comparable with the calculated values for BSTt, suggesting a potential application in dielectric devices. These results were an essential tool for understanding the electronic and structural effects caused by a change in geometry and element doping on the perovskite BST structure.
References
Mohamed EA, Nabhan E, Ratep A, Hassan FM, Tahoon K (2020) Influence of BaTiO3 nanoparticles/clusters on the structural and dielectric properties of glasses-nanocomposites. Phys B Condens Matter 589:412220
Stanciu CA, Cernea M, Secu EC, Aldica G, Ganea P, Trusca R (2017) Lanthanum influence on the structure, dielectric properties and luminescence of BaTiO3 ceramics processed by spark plasma sintering technique. J Alloys Compd 706:538–545
Piskunov S, Eglitis RI (2015) First principles hybrid DFT calculations of BaTiO3/SrTiO3 (001) interface. Solid State Ion 274:29–33
Chao S, Dogan F (2011) BaTiO3–SrTiO3 layered dielectrics for energy storage. Mater Lett 65(6):978–981
Kang F, Zhang L, Huang B, Mao P, Wang Z, Sun Q, Wang J, Hu D (2020) Enhanced electromechanical properties of SrTiO3-BiFeO3-BaTiO3 ceramics via relaxor behavior and phase boundary design. J Eur Ceram Soc 40(4):1198–1204
Jongprateep O, Sato N (2019) Effects of sintering temperatures on microstructure and dielectric constant of Ba0.05SrxCa0.95-xTiO3 where (x = 0, 0.475 and 0.95). Mater Today-Proc 17:1898–1905
Fujisawa J-i, Eda T, Hanaya M (2017) Comparative study of conduction-band and valence-band edges of TiO2, SrTiO3, and BaTiO3 by ionization potential measurements. Chem Phys Lett 685:23–26
Gartnerova V, Pacherova O, Klinger M, Jelinek M, Jager A, Tyunina M (2017) Strain fluctuations in BaTiO3/SrTiO3 heterostructures. Mater Res Bull 89:180–184
Mbarki R, Haskins JB, Kinaci A, Cagin T (2014) Temperature dependence of flexoelectricity in BaTiO3 and SrTiO3 perovskite nanostructures. Phys Lett A 378(30):2181–2183
Assirey EAR (2019) Perovskite synthesis, properties and their related biochemical and industrial application. Saudi Pharm J 27(6):817–829
Kondo S, Yamada T, Tagantsev AK, Ma P, Leuthold J, Martelli P, Boffi P, Martinelli M, Yoshino M, Nagasaki T (2019) Large impact of strain on the electro-optic effect in (Ba, Sr)TiO3 thin films: experiment and theoretical comparison. Appl Phys Lett 115(9):092901
Gao L, Guan Z, Huang S, Liang K, Chen H, Zhang J (2019) Enhanced dielectric properties of barium strontium titanate thin films by doping modification. J Mater Sci Mater Electron 30(14):12821–12839
Xing Y, Liang H, Li X, Si L (2009) High-frequency dielectric properties of BSTO ceramic prepared with hydrothermal synthesized SrTiO3 and BaTiO3 powders. Particuology 7(5):414–418
Slimani Y, Unal B, Hannachi E, Selmi A, Almessiere MA, Nawaz M, Baykal A, Ercan I, Yildiz M (2019) Frequency and dc bias voltage dependent dielectric properties and electrical conductivity of BaTiO3SrTiO3/(SiO2)x nanocomposites. Ceram Int 45(9):11989–12000
Kim SW, Choi HI, Lee MH, Park JS, Kim DJ, Do D, Kim MH, Song TK, Kim WJ (2013) Electrical properties and phase of BaTiO3–SrTiO3 solid solution. Ceram Int 39:S487–S490
Yustanti E, Hafizah MAE, Manaf A (2016) Synthesis of strontium substituted barium titanate nanoparticles by mechanical alloying and high power ultrasonication destruction. AIP Conf Proc 1725(1):020102
Pasha UM, Zheng H, Thakur OP, Feteira A, Whittle KR, Sinclair DC, Reaney IM (2007) In situ Raman spectroscopy of A-site doped barium titanate. Appl Phys Lett 91(6):062908
Berbecaru C, Alexandru HV, Porosnicu C, Velea A, Ioachim A, Nedelcu L, Toacsan M (2008) Ceramic materials Ba(1–x)SrxTiO3 for electronics—synthesis and characterization. Thin Solid Films 516(22):8210–8214
Mohan CRK, Bajpai PK (2008) Effect of sintering optimization on the electrical properties of bulk BaxSr1−xTiO3 ceramics. Phys B Condens Matter 403(13):2173–2188
Shen Z-Y, Wang Y, Tang Y, Yu Y, Luo W-Q, Wang X, Li Y, Wang Z, Song F (2019) Glass modified barium strontium titanate ceramics for energy storage capacitor at elevated temperatures. J Materiomics 5(4):641–648
Hou B, Xu Y, Wu D, Sun Y (2006) Preparation and characterization of single-crystalline barium strontium titanate nanocubes via solvothermal method. Powder Technol 170(1):26–30
Irzaman PIR, Aminullah SH, Alatas H (2016) Development of ferroelectric solar cells of barium strontium titanate (BaxSr1-xTiO3) for subtituting conventional battery in LAPAN-IPB satellite (LISAT). Proc Environ Sci 33:607–614
Curecheriu LP, Mitoseriu L, Ianculescu A (2009) Nonlinear dielectric properties of Ba1−xSrxTiO3 ceramics. J Alloys Compd 482(1):1–4
Zuo XH, Deng XY, Chen Y, Ruan M, Li W, Liu B, Qu Y, Xu B (2010) A novel method for preparation of barium strontium titanate nanopowders. Mater Lett 64(10):1150–1153
Jung DS, Hong SK, Cho JS, Kang YC (2008) Morphologies and crystal structures of nano-sized Ba1−xSrxTiO3 primary particles prepared by flame spray pyrolysis. Mater Res Bull 43(7):1789–1799
Rashad MM, Turky AO, Kandil AT (2013) Optical and electrical properties of Ba1−xSrxTiO3 nanopowders at different Sr2+ ion content. J Mater Sci Mater Electron 24(9):3284–3291
Simões AZ, Moura F, Onofre TB, Ramirez MA, Varela JA, Longo E (2010) Microwave-hydrothermal synthesis of barium strontium titanate nanoparticles. J Alloys Compd 508(2):620–624
Alexandru HV, Berbecaru C, Ioachim A, Nedelcu L, Dutu A (2006) BST solid solutions, temperature evolution of the ferroelectric transitions. Appl Surf Sci 253(1):354–357
Veselinović L, Mitrić M, Mančić L, Vukomanović M, Hadžić B, Marković S, Uskoković D (2014) The effect of Sn for Ti substitution on the average and local crystal structure of BaTi1−xSnxO3 (0 ≤ x ≤ 0.20). J Appl Crystallogr 47(3):999–1007
Watanabe Y (2019) Accurate semiempirical analytical formulas for spontaneous polarization by crystallographic parameters of SrTiO3-BaTiO3 system by ab initio calculations. Comput Mater Sci 158:315–323
Atta NF, Ahmed Galal, El-Ads EH (2016) Perovskite nanomaterials—synthesis, characterization, and applications. In: Likun P, Zhu G (eds) Perovskite materials—synthesis, characterisation, properties, and applications. IntechOpen
Piskunov S, Eglitis RI (2016) Comparative ab initio calculations of SrTiO3/BaTiO3 and SrZrO3/PbZrO3 (001) heterostructures. Nucl Instrum Methods Phys Res B 374:20–23
Lee H-S, Mizoguchi T, Yamamoto T, Kang S-JL, Ikuhara Y (2007) First-principles calculation of defect energetics in cubic-BaTiO3 and a comparison with SrTiO3. Acta Mater 55(19):6535–6540
Zhang R-z, Hu X-y, Guo P, Wang C-l (2012) Thermoelectric transport coefficients of n-doped CaTiO3, SrTiO3 and BaTiO3: a theoretical study. Phys B Condens Matter 407(7):1114–1118
Rizwan M, Hajra ZI, Shakil M, Gillani SSA, Usman Z (2020) Electronic, structural and optical properties of BaTiO3 doped with lanthanum (La): insight from DFT calculation. Optik 211:164611
Dovesi R, Orlando R, Erba A, Zicovich-Wilson CM, Civalleri B, Casassa S, Maschio L, Ferrabone M, De La Pierre M, D’Arco P, Noël Y, Causà M, Rérat M, Kirtman B (2014) CRYSTAL14: a program for the ab initio investigation of crystalline solids. Int J Quantum Chem 114(19):1287–1317
Lee CT, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter 37:785–789
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Longo VM, Cavalcante LS, Erlo R, Mastelaro VR, de Figueiredo AT, Sambrano JR, de Lázaro S, Freitas AZ, Gomes L, Vieira ND, Varela JA, Longo E (2008) Strong violet–blue light photoluminescence emission at room temperature in SrZrO3: joint experimental and theoretical study. Acta Mater 56(10):2191–2202
Longo VM, Figueiredo ATd, Lázaro Sd, Gurgel MF, Costa MGS, Paiva-Santos CO, Varela JA, Longo E, Mastelaro VR, Vicente FSD, Hernandes AC, Franco RWA (2008) Structural conditions that leads to photoluminescence emission in SrTiO3: an experimental and theoretical approach. J Appl Phys 104(2):023515
Longo VM, Cavalcante LS, Costa MGS, Moreira ML, de Figueiredo AT, Andrés J, Varela JA, Longo E (2009) First principles calculations on the origin of violet-blue and green light photoluminescence emission in SrZrO3 and SrTiO3 perovskites. Theor Chem Acc 124(5):385
Moreira E, Henriques JM, Azevedo DL, Caetano EWS, Freire VN, Albuquerque EL (2011) Structural, optoelectronic, infrared and Raman spectra of orthorhombic SrSnO3 from DFT calculations. J Solid State Chem 184(4):921–928
Oliveira MC, Gracia L, de Assis M, Rosa ILV, do Carmo Gurgel MF, Longo E, Andrés J (2017) Mechanism of photoluminescence in intrinsically disordered CaZrO3 crystals: first principles modeling of the excited electronic states. J Alloys Compd 722:981–995
Oliveira MC, Ribeiro RAP, Gracia L, de Lazaro SR, de Assis M, Oliva M, Rosa ILV, Gurgel MFdC, Longo E, Andrés J (2018) Experimental and theoretical study of the energetic, morphological, and photoluminescence properties of CaZrO3:Eu3+. CrystEngComm 20(37):5519
Tranquilin RL, Lovisa LX, Almeida CRR, Paskocimas CA, Li MS, Oliveira MC, Gracia L, Andres J, Longo E, Motta FV, Bomio MRD (2019) Understanding the white-emitting CaMoO4 Co-Doped Eu3+, Tb3+, and Tm3+ phosphor through experiment and computation. J Phys Chem C 123(30):18536–18550
Oliveira MC, Andrés J, Gracia L, de Oliveira MSMP, Mercury JMR, Longo E, Nogueira IC (2019) Geometry, electronic structure, morphology, and photoluminescence emissions of BaW1-xMoxO4 (x = 0, 0.25, 0.50, 0.75, and 1) solid solutions: theory and experiment in concert. Appl Surf Sci 463:907–917
Eglitis R, Kruchinin SP (2020) Ab initio calculations of ABO3perovskite (001), (011) and (111) nano-surfaces, interfaces and defects. Mod Phys Lett B 34(19n20):2040057
Eglitis RI (2019) Ab initio calculations of CaZrO3, BaZrO3, PbTiO3 and SrTiO3 (001), (011) and (111) surfaces as well as their (001) interfaces. Integr Ferroelectr 196(1):7–15
Eglitis R, Purans J, Popov AI, Jia R (2019) Systematic trends in YAlO3, SrTiO3, BaTiO3, BaZrO3 (001) and (111) surface ab initio calculations. Int J Mod Phys B 33(32):1950390
Maul J, Santos IMG, Sambrano JR, Erba A (2016) Thermal properties of the orthorhombic CaSnO3 perovskite under pressure from ab initio quasi-harmonic calculations. Theor Chem Acc 135(2):36
Cavalcante LS, Batista NC, Badapanda T, Costa MGS, Li MS, Avansi W, Mastelaro VR, Longo E, Espinosa JWM, Gurgel MFC (2013) Local electronic structure, optical bandgap and photoluminescence (PL) properties of Ba(Zr0.75Ti0.25)O3 powders. Mat Sci Semicon Proc 16(3):1035–1045
Momma K, Izumi F (2011) VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 44(6):1272–1276
Kokalj A (1999) XCrySDen-a new program for displaying crystalline structures and electron densities. J Mol Graph Model 17(3–4):176–216
Monkhorst HJ, Pack JD (1976) Special points for Brillouin-zone integrations. Phys Rev B 13(12):5188
Shannon R (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst Sec A 32(5):751–767
Souza IA, Cavalcante LS, Sczancoski JC, Moura F, Paiva-Santos CO, Varela JA, Simões AZ, Longo E (2009) Structural and dielectric properties of Ba0.5Sr0.5(SnxTi1−x)O3 ceramics obtained by the soft chemical method. J Alloys Compd 477(1):877–882
Chihaoui S, Seveyrat L, Perrin V, Kallel I, Lebrun L, Khemakhem H (2017) Structural evolution and electrical characteristics of Sn-doped Ba0.8Sr0.2TiO3 ceramics. Ceram Int 43(1, Part A):427–432
Joseph J, Vimala TM, Raju J, Murthy VRK (1999) Structural investigations on the (Ba, Sr)(Zr, Ti)O3 system. J Phys D Appl Phys 32(9):1049–1057
Turky AO, Rashad MM, Bechelany M (2016) Tailoring optical and dielectric properties of Ba0.5Sr0.5TiO3 powders synthesized using citrate precursor route. Mater Des 90:54–59
Souza IA, Simões AZ, Longo E, Varela JA, Pizani PS (2006) Photoluminescence at room temperature in disordered Ba0.50Sr0.50(Ti0.80Sn0.20)O3 thin films. Appl Phys Lett 88(21):211911
Kuo S-Y, Liao W-Y, Hsieh W-F (2001) Structural ordering transition and repulsion of the giant LO-TO splitting in polycrystalline BaxSr1-xTiO3. Phys Rev B 64(22):224103
Tenne DA, Soukiassian A, Xi XX, Choosuwan H, Guo R, Bhalla AS (2004) Lattice dynamics in BaxSr1-xTiO3 single crystals: a Raman study. Phys Rev B 70(17):174302
Souza IA, Gurgel MFC, Santos LPS, Góes MS, Cava S, Cilense M, Rosa ILV, Paiva-Santos CO, Longo E (2006) Theoretical and experimental study of disordered Ba0.45Sr0.55TiO3 photoluminescence at room temperature. Chem Phys 322(3):343–348
Ganjir R, Bajpai PK (2019) Influence of Co doping on the structural, dielectric and Raman properties of Ba0.75Sr0.25Ti1−xCoxO3. J Electron Mater 48(1):634–641
Bajpai PK, Mohan CRK, Ganjir R, Kumar R, Kumar A, Katiyar RS (2018) Swift heavy ion induced material modifications in Ba1-xSrxTiO3 ceramics as probed by temperature-dependent Raman spectroscopy. J Raman Spectrosc 49(2):324–335
Foresman J, Frisch A (2015) Exploring chemistry with electronic structure methods, 3rd edn. Gaussian Inc, Pittsburgh
Hautier G, Ong SP, Jain A, Moore CJ, Ceder G (2012) Accuracy of density functional theory in predicting formation energies of ternary oxides from binary oxides and its implication on phase stability. Phys Rev B 85(15):155208
Emery AA, Wolverton C (2017) High-throughput DFT calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci Data 4(1):170153
Kojitani H, Navrotsky A, Akaogi M (2001) Calorimetric study of perovskite solid solutions in the CaSiO3–CaGeO3 system. Phys Chem Miner 28(6):413–420
Beltrán A, Gracia L, Andrés J (2019) Polymorphs of ZnV2O6 under pressure: a first-principle investigation. J Phys Chem C 123(5):3239–3253
Beltrán A, Gracia L, Andrés J, Longo E (2017) First-principles study on polymorphs of AgVO3: assessing to structural stabilities and pressure-induced transitions. J Phys Chem C 121(49):27624–27642
Nagaraj B, Sawhney T, Perusse S, Aggarwal S, Ramesh R, Kaushik VS, Zafar S, Jones RE, Lee JH, Balu V, Lee J (1999) (Ba, Sr)TiO3 thin films with conducting perovskite electrodes for dynamic random access memory applications. Appl Phys Lett 74(21):3194–3196
Kuroiwa T, Tsunemine Y, Horikawa T, Makita T, Tanimura J, Mikami N, Sato K (1994) Dielectric properties of (BaxSr1-x)TiO3 thin films prepared by RF sputtering for dynamic random access memory application. Jpn. J Appl Phys 33(Part 1, No. 9B):5187–5191
Serraiocco J, Acikel B, Hansen P, Taylor T, Xu H, Speck JS, York RA (2002) Tunable passive integrated circuits using BST thin films. Integr Ferroelectr 49(1):161–170
Ferrero M, Rérat M, Orlando R, Dovesi R (2008) The calculation of static polarizabilities of 1–3D periodic compounds the implementation in the crystal code. J Comput Chem 29(9):1450–1459
Ferrero M, Rérat M, Orlando R, Dovesi R (2008) Coupled perturbed Hartree-Fock for periodic systems: the role of symmetry and related computational aspects. J Chem Phys 128(1):014110
Ferrero M, Rérat M, Kirtman B, Dovesi R (2008) Calculation of first and second static hyperpolarizabilities of one- to three-dimensional periodic compounds. Implementation in the CRYSTAL code. J Chem Phys 129(24):244110
Acknowledgements
The authors thank the following Brazilian research financing institutions for financial support: National Council for Scientific and Technological Development (CNPQ), Coordination for the Improvement of Higher Education Personnel (CAPES), the Goiás Research Foundation – FAPEG, the São Paulo Research Foundation—FAPESP (2013/07296-2), Graduate Program in Chemistry of Federal University of Catalão, Graduate Program in Materials Science and Engineering (PPGCEM-UFRN) and Federal University of São Carlos. M. C. Oliveira acknowledges the financial support from PNPD/CAPES (88887.319041/2019-00). The authors acknowledge the National Laboratory for Scientific Computing (LNCC) and High-Performance Computing Center (NACAD) of the Federal University of Rio de Janeiro (COPPE-UFRJ) for providing the computational resources of Lobo Carneiro supercomputer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mesquita, W.D., de Jesus, S.R., Oliveira, M.C. et al. Barium strontium titanate-based perovskite materials from DFT perspective: assessing the structural, electronic, vibrational, dielectric and energetic properties. Theor Chem Acc 140, 27 (2021). https://doi.org/10.1007/s00214-021-02723-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-021-02723-2