
Abstract
The evaluation of drug-induced mitochondrial impairment may be important in drug development as well as in the comprehension of molecular mechanisms of the therapeutic and adverse effects of drugs. The primary aim of this study was to investigate the effects of four drugs for treatment of depression (bupropion, fluoxetine, amitriptyline, and imipramine) and five drugs for bipolar disorder treatment (lithium, valproate, valpromide, lamotrigine, and carbamazepine) on cell energy metabolism. The in vitro effects of the selected psychopharmaca were measured in isolated pig brain mitochondria; the activities of citrate synthase (CS) and electron transport chain (ETC) complexes (I, II + III, and IV) and mitochondrial respiration rates linked to complex I and complex II were measured. Complex I was significantly inhibited by lithium, carbamazepine, fluoxetine, amitriptyline, and imipramine. The activity of complex IV was decreased after exposure to carbamazepine. The activities of complex II + III and CS were not affected by any tested drug. Complex I-linked respiration was significantly inhibited by bupropion, fluoxetine, amitriptyline, imipramine, valpromide, carbamazepine, and lamotrigine. Significant inhibition of complex II-linked respiration was observed after mitochondria were exposed to amitriptyline, fluoxetine, and carbamazepine. Our outcomes confirm the need to investigate the effects of drugs on both the total respiration rate and the activities of individual enzymes of the ETC to reveal the risk of adverse effects as well as to understand the molecular mechanisms leading to drug-induced changes in the respiratory rate. Our approach can be further replicated to study the mechanisms of action of newly developed drugs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dysfunctions of mitochondria and impairments of energy metabolism have been implicated in the pathophysiology of bipolar affective disorder (BAD) (Cikánková et al. 2017; Kato 2006). The mitochondrial hypothesis of BAD is extensive and includes a range of features, such as reduced cell energy metabolism characterized by decreased ATP synthesis, a shift from oxidative phosphorylation (OXPHOS) to glycolysis (Frey et al. 2007; Stork and Renshaw 2005), increased expression of proapoptotic genes (Benes et al. 2006), mutations or polymorphisms of mitochondrial genes (Kato et al. 2001; Munakata et al. 2004), ubiquitin-proteasome system dysregulation (Bousman et al. 2010), increased oxidative stress (Valvassori et al. 2018), a reduction in antioxidant capacity (decreased glutathione peroxidase and superoxide dismutase) (Konradi et al. 2004), structural mitochondrial abnormalities (Cataldo et al. 2010), and calcium signaling abnormalities (Kato 2008).
Pharmacotherapy of BAD is based on the administration of drugs with mood-stabilizing effects, mainly lithium, valproate, lamotrigine, and carbamazepine. These drugs extent various pleotropic effects; carbamazepine, valproate, and lamotrigine are classified as neuron inhibitors with pleotropic effects (NIPEs). Recent studies have also provided data about the positive effects of chronic treatment with mood-stabilizing drugs (MSs) on mitochondrial functions (Finsterer and Mahjoub 2012).
Lithium is a metal alkali ion and is typically used for the treatment of manic episodes of BAD. Lithium also augments antidepressant (AD) treatment in severe depression; the coadministration of lithium during AD therapy may affect neurotransmission through 5-HT1A and 5-HT1B receptors (Haddjeri et al. 2000; Redrobe and Bourin 1999). Lithium has multiple mechanisms of action and a neuroprotective property in terms of preventing apoptosis. Inhibition of inositol monophosphatase participates in the action of lithium in treating BAD. There is evidence that lithium inhibits the activity of glycogen synthase kinase-3 (GSK-3), a regulatory enzyme involved in the gene expression of proteins responsible for apoptosis, cellular resilience, and synaptic plasticity (Zarate et al. 2006). Lithium increases mitochondrial calcium concentrations and thus desensitizes mitochondria to depolarization and prevents the swelling of organelles and the release of cytochrome c from mitochondria. A high level of calcium ions is related to neurotoxicity and apoptosis through the induction of depolarization, which causes OXPHOS inhibition and the activation of mitochondrial apoptogenic proteins (Cebers et al. 1997).
Valproate also demonstrates a significant antimanic effect and is often administered in the case of lithium ineffectiveness or intolerance (Janicak 1993). Valproate medications very often contain a combination of valproic acid, sodium salts of valproate, and/or valpromide-containing preparations (a carboxamide derivative of valproic acid) (Macritchie et al. 2003). Valproate as well as lithium reduces rotenone-induced toxicity, cytochrome c release, and caspase-3 activation to prevent apoptosis (Lai et al. 2006). However, valproate also has deleterious effects on mitochondria, such as inhibiting mitochondrial β-oxidation and thus ATP production through a deficiency in lipid metabolism cofactors (Aires et al. 2010; Silva et al. 2008). The effects of valproate have been shown in experiments in isolated rat mitochondria; valproate causes oxidative stress by diverse ways, such as through the significant inhibition of enzymatic complex II activity and mitochondrial permeability transition pore (MPTP) and ion channel opening, which results in mitochondrial swelling and the release of cytochrome c (Jafarian et al. 2013).
Carbamazepine, like valproate, is an antiepileptic used in the treatment of BAD, most often in cases of intolerance for or ineffectiveness of lithium and valproate (Post and Weiss 2011). Carbamazepine also decreases the respiratory control rate, ATP synthesis, and the membrane potential and causes the impairment of calcium metabolism in mitochondria. This suggests that carbamazepine causes the uncoupling effect of mitochondrial respiration and induces hepatic mitochondrial toxicity (Santos et al. 2008). Eight months of carbamazepine treatment significantly decreases the production of ATP from all substrates (complexes I–IV) in lymphocyte homogenates from children (Berger et al. 2010). Carbamazepine gives rise to a neuroprotective effect in rotenone-induced striatal neuronal dysfunction in rats resulting from the inhibition of mitochondrial complex I (Costa et al. 2008).
Lamotrigine, a third-generation anticonvulsant, acts as an inhibitor of glutamatergic neurotransmission and is used in the treatment of depressive episodes of BAD (Walden et al. 2000). There is evidence that lamotrigine has a neuroprotective effect. Lamotrigine attenuates the MPTP and the inhibitory effect of rotenone on complex I and thus prevents the induction of apoptosis (Kim et al. 2007; Lagrue et al. 2007). The effect of lamotrigine on the OXPHOS process was investigated in a study of peripheral white blood cells in children. It has been shown that 2 years of lamotrigine treatment significantly increases ATP production in lymphocytes, in presence of pyruvate + malate or succinate (Berger et al. 2010).
ADs are widely used in psychiatry, not only for the treatment of BAD (Ghaemi et al. 2003). Their mechanism of action is diverse; ADs not only act as monoamine reuptake inhibitors or antagonists/agonists of neurotransmitter receptors but also have been shown to have neurotrophic effects and thus modulate neuronal plasticity (Castrén 2004). Imipramine and amitriptyline are classified as nonselective monoamine reuptake inhibitors (NSMRIs) and represent the first generation of drugs in a treatment of depression. They inhibit the reuptake of norepinephrine, dopamine, and serotonin from the synaptic cleft. However, their clinical usage is complicated by frequent adverse effects such as QT prolongation, anticholinergic effects (dry mouth, constipation, tachycardia, etc.), due to their antagonist effects on histamine receptors (sedation, and increased appetite followed by weight gain), and antiadrenergic effects (postural hypotension and subsequent tachycardia). NSMRIs exhibit some characteristic of uncouplers of oxidative phosphorylation; i.e., they inhibit NADH oxidation and ATP synthesis (Weinbach et al. 1986).
Selective serotonin reuptake inhibitors (SSRIs) are currently the first-choice drugs for depression therapy, can be administered over the long term, and can prevent the relapse of disease. The side effects of SSRIs include nausea, vomiting, fatigue, diarrhea, and sexual disorders. Overall, their tolerance is considered very good (Hyttel 1994). Fluoxetine, an SSRI tested in our study, significantly inhibits ETC mitochondrial enzymes (complexes I, II + III, and IV) and impairs mitochondrial functions (Abdel-Razaq et al. 2011).
Bupropion is a norepinephrine-dopamine reuptake inhibitor (NDRI). It affects dopamine and norepinephrine signaling and it is also a noncompetitive antagonist of nicotinic acetylcholine receptors and bupropion is usually well tolerated, and the most common side effects are cephalgia, dryness of the mucous membranes, dizziness, and nausea. A common side effect is insomnia, which is often transient. Rarely, weight loss, glucose deficiency, and hypertension have been observed (Ascher et al. 1995). Bupropion contributes to mitochondrial collapse and cytotoxicity through oxidative stress-induced production of reactive oxygen species and lipoperoxidation; it also causes the depletion of intracellular glutathione (Ahmadian et al. 2017). Bupropion induces mitochondrial cytochrome c release, activates caspase-9, caspase-8, and caspase-3, and consequently causes apoptotic cell death in SH-SY5Y cells (Jang et al. 2011).
The mechanisms of action and adverse effects of selected drugs used for treatment of depression and mood disorders are well known, but their effects on mitochondria and cell energy metabolism remain to be fully elucidated. Mitochondrial changes may be related to psychiatric medications. Drug-induced effects manifest as a global downregulation of mitochondrial genes (genes encoding respiratory chain components) (Iwamoto et al. 2005). In contrast, long-term treatment may have neuroprotective effects via increasing the level of N-acetyl-aspartate (NAA). Decreased NAA is likely related to the pathogenesis of BAD. It has been suggested that the NAA level can serve as a biomarker of mitochondrial metabolism or a predictor of treatment response or of disease progression (Strakowski et al. 2005). Valproate, lithium, and lamotrigine treatment increase NAA levels in patients with BAD (Soeiro-de-Souza et al. 2012; Zanetti et al. 2015).
Some side effects of the drugs used for treatment of depression and/or mood disorders may be the consequence of impairments at the level of mitochondrial respiration (QTc prolongation and metabolic syndrome). It can be supposed that QTc interval prolongation and cardiac anomalies are related to mitochondrial myopathies. Mitochondrial myopathy caused by mitochondrial defects at the level of ETC enzyme complexes may manifest by changes in ECG based on the ionic imbalance (alteration in mitochondrial metabolism of calcium) (Anan et al. 1995; Karanikis et al. 2005; Liu et al. 2014; Maron et al. 2006; Ogasahara et al. 1989). Higher prevalence of metabolic syndrome in BAD patients could be related to mitochondrial malfunction (inhibition of enzyme activities, mitochondrial respiration decrease, and alteration in mitochondrial lipid metabolism) caused by BAD treatment or the BAD pathology (Taylor and MacQueen 2006). Valproate alters negatively lipid spectrum (Silva et al. 2008). Treatment of depression (primarily NSMRIs, e.g., imipramine) is associated with higher incident of diabetes mellitus type 2 (Deuschle 2013; van Reedt Dortland et al. 2010).
We investigated the in vitro effects of bupropion, fluoxetine, amitriptyline, imipramine, lithium, valproate, valpromide, lamotrigine, and carbamazepine (Tables 1 and 2) on the activities of mitochondrial enzymes (citrate synthase (CS) and ETC complexes I, II + III, and IV) and on the rate of oxygen consumption by mitochondria (depicted in Fig. 1). We hypothesize that the effects of some drugs for treatment of depression and BAD interfere with synaptic activity and cell energy metabolism. To verify this hypothesis, we studied the effects of the abovementioned drugs on the oxidative phosphorylation (OXPHOS) system and the activities of ETC complexes.

Scheme of mitochondrial functions and enzyme activities, which were tested in our measurements; examined parameters are depicted in black color. ADP adenosine diphosphate, ATP adenosine triphosphate, IMM inner mitochondrial membrane, NAD+ nicotinamide adenine dinucleotide (oxidized form), NADH nicotinamide adenine dinucleotide (reduced form), Pi phosphate, Q coenzyme Q10 (ubiquinone), QH2 reduced form of coenzyme Q10 (ubiquinol), TCA cycle tricarboxylic acid cycle
Methods and materials
Media and chemicals
To isolate and preserve mitochondria, we used buffered sucrose (0.32 mol/L sucrose and 4 mmol/L HEPES, pH 7.4) as a medium. In addition, Krebs-Henseleit (KH) buffer consisted of 118 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4·7H2O, 25 mmol/L NaHCO3, and 11.1 mmol/L glucose, pH 7.4. Mitochondrial respiration was measured using MiR05 without albumin (110 mmol/L sucrose, 60 mmol/L K+-lactobionate, 20 mmol/L taurine, 3 mmol/L MgCl2·6H2O, 10 mmol/L KH2PO4, 0.5 mmol/L EGTA, and 20 mmol/L HEPES, pH adjusted to 7.1 with KOH) as a medium (Pesta and Gnaiger 2012). Other media were utilized for specific enzyme assays.
Isolation of brain mitochondria
Mitochondria were isolated from pig brain gray matter and purified on a sucrose gradient as described previously (Fisar et al. 2010; Pinna et al. 2003). The mitochondrial respiration rate was measured using freshly purified mitochondria, and the activities of individual enzymes were measured using frozen mitochondria (stored at − 70 °C). The protein concentrations were determined by the Lowry method.
Activities of citrate synthase and complexes of the electron transport chain
Suspensions of purified mitochondria in hypotonic buffer (25 mmol/L potassium phosphate and 5 mmol/L MgCl2, pH 7.2) were ultrasonicated 3 times to stimulate the enzymes to the highest level of activity. Samples were exposed to the examined drugs for 30 min at a temperature of 30 °C, which was maintained throughout the entire experiment, and the drugs were applied at a concentration of 50 μmol/L. We compared each measurement to that obtained from a control set of solvents instead of drug. The total volume of the samples was 1 mL. Depending on the method applied, the final concentration of protein varied. The temperature during the measurement was 30 °C. We employed a Uvicon XL spectrophotometer (SECOMAM, Alès, France) to examine the activities of CS and ETC complexes.
Complex I (NADH dehydrogenase (ubiquinone), EC 1.6.5.3)
The activity of mitochondrial complex I was examined based on the rotenone sensitivity rate of NADH oxidation at 340 nm using a spectrophotometer. The oxidoreductive reaction was started by infusing coenzyme Q10 (final concentration of 33 μmol/L) and NADH solution and measured for 3 min (Folbergrova et al. 2010; Hroudova and Fisar 2010). The final protein concentration was higher compared to the other methods (1 mg/mL) to improve the signal and subsequently the quality of the measurement.
Complex II + III (succinate cytochrome c oxidoreductase, EC 1.3.5.1; EC 1.10.2.2)
The activity of complex II + III was measured based on the antimycin A sensitivity rate of cytochrome c reduction (Trounce et al. 1996). The reduction of cytochrome c was examined using a spectrophotometer at 550 nm for 1 min. The substrate was based on succinate. The final protein concentration was 500 μg/mL.
Complex IV (cytochrome c oxidase, EC 1.9.3.1)
Cytochrome c oxidase (COX) activity was measured by monitoring the decrease in absorbance during the oxidation of reduced cytochrome c at 550 nm for 3 min (Rustin et al. 1994). The final protein concentration was 2.2 μg/mL and the stock solutions were prepared fresh each day.
Citrate synthase (EC 2.3.3.1)
The spectrophotometric measurement of CS activity was based on the observed color change of DNTB (5,5′-dithiobis-(2-nitrobenzoic) acid). The reaction was initiated by adding the substrate oxaloacetate (0.5 mmol/L) and measured at 412 nm for 3 min (Srere 1969). The final protein concentration was 20 μg/mL and the stock solutions were being prepared fresh for each run of the experiment.
Mitochondrial respiration
We employed an oxygraph (Oxygraph-2k, Oroboros Instruments, Innsbruck, Austria) with Clark-type electrodes to measure the mitochondrial oxygen consumption rate. The protocol design was inspired by a previous study (Hroudova and Fisar 2012; Pesta and Gnaiger 2012). The oxygraph chambers were filled with MiR05 respiration medium (oxygen solubility factor: 0.92) to a final volume of 2 mL. The final concentration of mitochondrial protein was 0.05–0.20 mg/mL. We applied specific mitochondrial substrates, namely malate and pyruvate, to examine the respiratory rate of mitochondria during electron transport through complex I. Other substrates, namely succinate and rotenone, were utilized to measure the respiration of complex II. We were able to run two parallel experiments on the machine. The final concentrations of the drugs were between 5 and 750 μmol/L. To inhibit complex I and complex III, we applied rotenone and antimycin A, respectively.
Data analysis and statistics
The activities of mitochondrial enzymes were measured as the slope of time dependence of absorbance using LabPower Junior software (SECOMAM). Relative changes in enzyme activities induced by the drugs were determined assuming that the control sample activity was equal to 100%. High-resolution respirometry data were recorded and analyzed using DatLab 4.3 software. Respiration rates were expressed as mass-specific oxygen flux (pmol O2 consumed per second/mg of protein in the sample).
The inhibition of the respiration rate was analyzed using four-parameter logistic regression with SigmaPlot software (Systat Software. Inc., Richmond, USA) to establish the half maximal inhibitory concentration (IC50), the Hill slope, and residual activity at a high drug concentration.
The results were analyzed by the data analysis software system STATISTICA (TIBCO Software Inc., Palo Alto, USA). One-sample t tests for single means were used to determine whether enzyme activity in samples treated with drug was significantly different from that of the control. The data are expressed as the mean ± standard deviation or mean ± standard error.
Results
Activities of CS and respiratory chain complexes
The activities of ETC complexes were measured after 30 min of incubation with five drugs with mood-stabilizing effects (lithium, valproate, valpromide, lamotrigine, and carbamazepine) and four ADs (bupropion, fluoxetine, amitriptyline, and imipramine) at a concentration of 50 μmol/L.
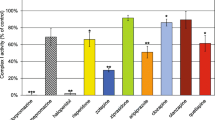
Complex I was the most sensitive to drug-induced inhibitory effects (Fig. 2). Significant inhibitory effects were observed after exposure to lithium, carbamazepine, fluoxetine, amitriptyline, and imipramine. Amitriptyline, fluoxetine, carbamazepine, and imipramine exerted the most significant inhibitory effects. Conversely, valproate, valpromide, and lamotrigine had no significant effect on complex I activity.

The effects of selected drugs used for treatment of depression and mood disorders on respiratory chain complex I activity in brain mitochondria. The samples were incubated with 50 μmol/L drugs at 30 °C for 30 min and enzyme activities were measured spectrophotometrically as described in the “Methods and materials” section. The relative activities are displayed (100% = control sample without the drug). The values are the means ± standard deviation of at least three independent measurements. Comparisons between controls and samples treated with drug were performed using the Wilcoxon matched pairs test (*p < 0.05, **p < 0.01)
None of the tested drugs significantly affected complex II + III activity (Fig. 3). The activity of complex IV was significantly inhibited by carbamazepine only (Fig. 4).

The effects of selected drugs used for treatment of depression and mood disorders on respiratory chain complex II + III activity in brain mitochondria. The samples were incubated with 50 μmol/L drugs at 30 °C for 30 min and enzyme activities were measured spectrophotometrically as described in the “Methods and materials” section. The relative activities are displayed (100% = control sample without the drug). The values are the means ± standard deviation of at least three independent measurements. Comparisons between controls and samples treated with drug were performed using the Wilcoxon matched pairs test (*p < 0.05, **p < 0.01)

The effects of selected drugs used for treatment of depression and mood disorders on respiratory chain complex IV activity in brain mitochondria. The samples were incubated with 50 μmol/L drugs at 30 °C for 30 min and enzyme activities were measured spectrophotometrically as described in the “Methods and materials” section. The relative activities are displayed (100% = control sample without the drug). The values are the means ± standard deviation of at least three independent measurements. Comparisons between controls and samples treated with drug were performed using the Wilcoxon matched pairs test (*p < 0.05, **p < 0.01)
CS activity was not affected by any of the tested drugs (Fig. 5).

The effects of selected drugs used for treatment of depression and mood disorders on citrate synthase (CS) activity in brain mitochondria. The samples were incubated with 50 μmol/L drugs at 30 °C for 30 min and enzyme activities were measured spectrophotometrically as described in the “Methods and materials” section. The relative activities are displayed (100% = control sample without the drug). The values are the means ± standard deviation of at least three independent measurements. Comparisons between controls and samples treated with drug were performed using the Wilcoxon matched pairs test (*p < 0.05, **p < 0.01)
Mitochondrial respiration
The inhibitory effects of the tested ADs and MSs on mitochondrial respiratory rate are shown in Figs. 6 and 7, and the parameters characterizing the potency of inhibition are summarized in Tables 3 and 4. Complex I-linked respiration was strongly inhibited by fluoxetine, amitriptyline, and imipramine, with IC50 = 65.5 ± 1.9, 226.1 ± 4.0, and 473.1 ± 9.7 μM, respectively. These drugs were full inhibitors of mitochondrial complex I-linked respiration. Fluoxetine fully inhibited complex I-linked respiration in concentrations at about 250 μmol/L. Amitriptyline and imipramine were also found to be full inhibitors at higher concentrations (> 750 μmol/L). Other examined drugs (bupropion, valpromide, carbamazepine, and lamotrigine) were revealed to be partial inhibitors of complex I-linked respiration (at > 100 μmol/L) (Fig. 6). We did not find any inhibitory effects of lithium and valproate on complex I-linked respiration, as described previously (Hroudova and Fisar 2012).

Drug-induced inhibition of the respiratory rate in pig brain mitochondria incubated with substrates for electron supply through complex I. Changes in the respiratory rate induced by selected drugs used for treatment of depression and mood disorders were measured. Dose-response curves are displayed as plots of the relative respiratory rate against the drug concentration. The samples were continuously stirred and incubated at 37 °C; titration with drugs was achieved in 2–3-min intervals. Following the subtraction of residual oxygen consumption from the respiratory rates at different drug/medium concentrations, differences among drug-titrated and medium-titrated samples were calculated, and relative drug-induced changes in respiratory rate were determined with the presumption that the relative respiratory rate was equal to 1 at zero drug concentration. The half maximal inhibitory concentration (IC50) and Hill slope were calculated using nonlinear regression analysis software. The values are the means from at least 3 independent measurements. The lines represent the best fitted curves using the four-parameter logistic function

Drug-induced inhibition of the respiratory rate in pig brain mitochondria incubated with substrates for electron supply through complex II. Changes in the respiratory rate induced by selected drugs used for treatment of depression and mood disorders were measured. Dose-response curves are displayed as plots of the relative respiratory rate against the drug concentration. The samples were continuously stirred and incubated at 37 °C; titration with drugs was achieved in 2–3-min intervals. Following the subtraction of residual oxygen consumption from the respiratory rates at different drug/medium concentrations, differences among drug-titrated and medium-titrated samples were calculated, and relative drug-induced changes in respiratory rate were determined with the presumption that the relative respiratory rate was equal to 1 at zero drug concentration. The half maximal inhibitory concentration (IC50) and Hill slope were calculated using nonlinear regression analysis software. The values are the means from at least 3 independent measurements. The lines represent the best fitted curves using the four-parameter logistic function
Significant inhibition of complex II-linked respiration was observed after exposure to amitriptyline (IC50 = 582.4 ± 96.6 μM), which can be described as a full inhibitor. Fluoxetine, carbamazepine, and lamotrigine partially inhibited complex II-linked respiration (Fig. 7). We did not find any inhibitory effects of bupropion, lithium, and valproate on complex II-linked respiration (Hroudova and Fisar 2012).
Correlation between relative complex I activity at a final drug concentration of 50 μmol/L (Fig. 2) and IC50 values determined for drug-induced inhibition of complex I-linked respiration (Table 3) in isolated pig brain mitochondria was found significant for ADs (r = 0.989, p = 0.011, N = 4), but not for drugs with mood-stabilizing effects (r = − 0.987, p = 0.102, N = 3) (Fig. 8).

Correlation between relative complex I activity at a final drug concentration of 50 μmol/L (Fig. 2) and IC50 values determined for drug-induced inhibition of complex I-linked respiration (Table 3) in isolated pig brain mitochondria. 95% confidence intervals are displayed for each point. The line shows the simple linear regression for antidepressants (with correlation coefficient r = 0.989, p = 0.011, N = 4), and the dotted line represents 95% confidence band
Discussion
The in vitro effects of tested drugs with pharmacologically different mechanisms of action were examined on mitochondrial energy metabolism. The mitochondrial respiratory rate and enzymatic activities (the activities of CS and ETC complexes) were studied with the aim of supporting the hypothesis that drug-induced mitochondrial changes can affect cell energy metabolism and may be related to the adverse effects of drugs.
Of the examined enzymes, complex I was most affected by the drugs (Fig. 2). Complex I is the key to OXPHOS control; its impaired function can have consequences on the energy metabolism of cells and overall neuronal activity. Significant inhibition was observed after exposure to lithium, carbamazepine, fluoxetine, amitriptyline, and imipramine. Lithium, valproate, amitriptyline, and imipramine were also studied in our earlier in vitro experiment (Hroudova and Fisar 2010), and we partially confirmed these results, except for those for valproate. Valproate was previously found to inhibit complex I; however, this study did not show that it decreased complex I activity. Lithium did not have a statistically significant effect during the previous study, unlike in this study. Reasons for the conflicting results of these studies may be the use of different drug concentrations (the earlier study used 5 mmol/L, and the current study used 50 μmol/L) and the use of the crude synaptosomal fraction in the earlier study (Hroudova and Fisar 2010) compared to the use of purified mitochondria in the current study. In another in vivo study, lithium increased the activity of complex I in depressive episodes of BAD, which correlated with plasma lithium levels. As such, mitochondrial respiratory chain enzyme activities remain unchanged during depressive episodes (de Sousa et al. 2014). The exposure of the postmortem human cortex to lithium at clinically relevant concentrations (up to 10 mM) increases the activity of complexes I + III and II + III (Maurer et al. 2009). Our measurements reflect the direct effects of acute drug exposure on isolated mitochondria, which may be the cause of the different outcomes of the abovementioned studies. In a study on mitochondria isolated from rat hearts, treatment with norfluoxetine (the active metabolite of fluoxetine) at concentrations of 20–50 μmol/L for 18 h significantly decreased the activities of mitochondrial complexes (I, II + III, and IV) and reduced integrated mitochondrial function (the membrane potential and mitochondrial state-3 respiration) at concentrations above 10 μmol/L (Abdel-Razaq et al. 2011). Carbamazepine significantly decreased the activity of complex I in our measurements. Our findings of spectrophotometric measurements of complex I activity match the data from mitochondrial respiration measurements (Fig. 8). These results are in line with the data available in the abovementioned study (Santos et al. 2008) and indicate that antidepressant-induced reduction of complex I-linked respiration is largely due to impaired complex I function. Contrary, mood stabilizer-induced decrease in complex I-linked respiration seems to be not related to complex I inhibition except for carbamazepine, which effects are similar as with ADs. The low inhibitory effect of carbamazepine on complex I-linked respiration complements experiments in which carbamazepine exhibits a neuroprotective effect against rotenone-induced neuronal dysfunction (Costa et al. 2008). Except for the abovementioned drugs (amitriptyline, imipramine, fluoxetine, and carbamazepine), the tested drugs (bupropion, valpromide, carbamazepine, and lamotrigine) partially inhibited complex I-linked respiration at high concentration.
Based on spectrophotometric measurements of complex II + III activity, we did not observe any significant drug-induced changes (Fig. 3). In the previous experiment, we observed a significant inhibition of complex II by amitriptyline and imipramine. In this previous study, we used 10 times higher concentrations of ADs and we measured the activity of enzymatic complex II individually (Hroudova and Fisar 2010), whereas in the present study, we studied the activity of complexes II and III together, which may have been the cause of the inconsistent results. Mitochondrial respiratory rates supported by complex II were inhibited by amitriptyline, fluoxetine, carbamazepine, and lamotrigine. Mitochondrial complex II-linked respiratory rates were inhibited by amitriptyline, fluoxetine, carbamazepine, and lamotrigine, which, in the case of fluoxetine and amitriptyline, correlates with previous studies and other available data (de Oliveira 2016; Hroudova and Fisar 2012). Imipramine-induced increase of complex II-linked respiration cannot be explained by our measurements.
Carbamazepine was the only tested drug that significantly decreased the activity of complex IV (Fig. 7). The results fit with an in vivo study on lymphocyte homogenates from children, in which carbamazepine inhibited all mitochondrial complexes, including complex IV (Berger et al. 2010). Our findings for valproate do not fit other available data, which suggest that valproate decreases the activity of complex IV (Hroudova and Fisar 2010; Ponchaut et al. 1991; Ponchaut et al. 1992). Reasons for this may be the different drug concentrations used, the experimental biological model, and the experimental conditions. Fluoxetine slightly inhibited enzymatic complex IV, but the results were not statistically significant. In the previously mentioned study, fluoxetine showed the most noticeable inhibitory effects on complex IV (Abdel-Razaq et al. 2011). Acute and chronic exposure to fluoxetine (24 h vs. 28 days) have different impacts. The acute administration of fluoxetine induces the activities of respiratory chain enzymes, with the exception of complex I. However, after 28 days of fluoxetine exposure, the inhibition of complex IV in the hippocampus is observed (Agostinho et al. 2011b).
CS activity was not significantly influenced by the tested drugs (Fig. 5). CS is an important mitochondrial enzyme that is responsible for regulatory function in the energy metabolism of the cell. It is located in the mitochondrial matrix, and as a part of the Krebs cycle, it links it to the respiratory chain (Srere 1969). Differences in the effects of the drugs on mitochondrial enzyme activity, which depend on acute or chronic administration, are described in the fluoxetine study; CS activity was increased in rat brain regions (the striatum, prefrontal cortex, and hippocampus) after acute administration of fluoxetine (25 mg/kg i.p. for 24 h), but chronic administration for 28 days did not affect the activity of CS in these rat brain regions (Agostinho et al. 2011a).
We noticed a possible relationship between complex I inhibition and prolongation of the QTc interval. Our results showed that lithium, carbamazepine, fluoxetine, amitriptyline, and imipramine inhibit complex I activity (Fig. 2). For these drugs, there are available data of changes in ECG measurements; according to case reports, NSMRIs (amitriptyline, imipramine) induced torsade de pointes. Potassium channel blockade could explain the ability of NSMRIs to prolong the QTc interval and provoke torsade de pointes (Vieweg and Wood 2004). Fluoxetine did not show any ECG conductance disturbances in registration trials but some case reports with abnormal QTc interval or torsade de pointes after administration of fluoxetine have been published (Lherm et al. 2000; Varriale 2001). Further research is required to elucidate the connectivity between QTc prolongation and alterations in ETC enzyme activities.
There is evidence that mitochondrial dysfunction is related to pathophysiology of insulin resistance (Kim et al. 2008). Decreased activity of ETC complexes could link to insulin resistance; some ADs and MSs contribute to the development of metabolic syndrome. This hypothesis should be also further investigated.
In contrast to long-term therapy in both diseases, all our measurements simulate a single acute drug administration and do not reflect the effect of drugs after long-term administration. The drugs indicated for BAD (lithium, valproate, valpromide, lamotrigine, and carbamazepine) were measured in concentrations corresponding to therapeutic drug concentrations in serum (tens to hundreds μmol/L). However, drugs for treatment of depression (bupropion, amitriptyline, imipramine, and fluoxetine) were measured at a concentration of 2 to 3 orders of magnitude higher than the therapeutic serum concentration. No significant correlation between the therapeutic concentration of tested drugs drug-induced inhibition of mitochondrial enzymes supports the hypothesis that the direct mitochondrial effects of drugs are related to their adverse rather than therapeutic effects.
Mitochondria isolated from pig brains were used in our experiments. The pig seems to be an uncommon species for pharmacological studies, but pig brain mitochondria are more like human mitochondria than rodent mitochondria are (Hroudova and Fisar 2010).
Effects of drugs used for treatment of schizophrenia and acute mania (mGPCR antagonists) were described in our previous studies (Cikankova et al. 2019; Hroudova and Fisar 2012).
Conclusions
From our data, tested drugs influenced both ETC mitochondrial enzyme activity and mitochondrial respiration. The activity of these enzymes can be investigated as a predictor of treatment response to or the adverse effects of examined drugs. The inhibitory effects of the tested drugs for treatment of depression and BAD on mitochondrial functions may confirm to the adverse effects of drugs. Considerably, more research is needed to determine the molecular mechanism by which ADs and MSs affect mitochondrial enzymes. Further clinical research must specify whether mitochondrial effects are causally connected to clinically manifested effects, such as QTc interval prolongation or metabolic syndrome. Due to the complex processes associated with OXPHOS activity, it is necessary to measure the effect of test substances not only on individual ETC complexes but also on the total respiration rate of the mitochondria. The experimental methods can be applied in toxicity trials of newly developed drugs.
Abbreviations
- AD:
-
Antidepressant
- BAD:
-
Bipolar affective disorder
- COX:
-
Cytochrome c oxidase
- CS:
-
Citrate synthase
- ETC:
-
Electron transport chain
- MPTP:
-
Mitochondrial permeability transition pore
- MS:
-
Mood-stabilizing drug
- NAA:
-
N-Acetyl-aspartate
- NIPEs:
-
Neuron inhibitor with pleotropic effects
- NSMRI:
-
Nonselective monoamine reuptake inhibitor
- OXPHOS:
-
Oxidative phosphorylation
- SSRI:
-
Selective serotonin reuptake inhibitor
References
Abdel-Razaq W, Kendall DA, Bates TE (2011) The effects of antidepressants on mitochondrial function in a model cell system and isolated mitochondria. Neurochem Res 36:327–338. https://doi.org/10.1007/s11064-010-0331-z
Agostinho FR et al (2011a) Treatment with olanzapine, fluoxetine and olanzapine/fluoxetine alters citrate synthase activity in rat brain. Neurosci Lett 487:278–281. https://doi.org/10.1016/j.neulet.2010.10.037
Agostinho FR et al (2011b) Olanzapine plus fluoxetine treatment alters mitochondrial respiratory chain activity in the rat brain. Acta Neuropsychiatr 23:282–291. https://doi.org/10.1111/j.1601-5215.2011.00569.x
Ahmadian E, Babaei H, Mohajjel Nayebi A, Eftekhari A, Eghbal MA (2017) Mechanistic approach for toxic effects of bupropion in primary rat hepatocytes. Drug Res (Stuttg) 67:217–222. https://doi.org/10.1055/s-0042-123034
Aires CC et al (2010) Inhibition of hepatic carnitine palmitoyl-transferase I (CPT IA) by valproyl-CoA as a possible mechanism of valproate-induced steatosis. Biochem Pharmacol 79:792–799. https://doi.org/10.1016/j.bcp.2009.10.011
Anan R et al (1995) Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation 91:955–961
Ascher JA, Cole JO, Colin JN, Feighner JP, Ferris RM, Fibiger HC, Golden RN, Martin P, Potter WZ, Richelson E (1995) Bupropion: a review of its mechanism of antidepressant activity. J Clin Psychiatry 56:395–401
Benes FM, Matzilevich D, Burke RE, Walsh J (2006) The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry 11:241–251. https://doi.org/10.1038/sj.mp.4001758
Berger I, Segal I, Shmueli D, Saada A (2010) The effect of antiepileptic drugs on mitochondrial activity: a pilot study. J Child Neurol 25:541–545. https://doi.org/10.1177/0883073809352888
Bousman CA et al (2010) Preliminary evidence of ubiquitin proteasome system dysregulation in schizophrenia and bipolar disorder: convergent pathway analysis findings from two independent samples. Am J Med Genet B Neuropsychiatr Genet 153b:494–502. https://doi.org/10.1002/ajmg.b.31006
Castrén E (2004) Neurotrophic effects of antidepressant drugs. Curr Opin Pharmacol 4:58–64. https://doi.org/10.1016/j.coph.2003.10.004
Cataldo AM et al (2010) Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol 177:575–585. https://doi.org/10.2353/ajpath.2010.081068
Cebers G, Zhivotovsky B, Ankarcrona M, Liljequist S (1997) AMPA neurotoxicity in cultured cerebellar granule neurons: mode of cell death. Brain Res Bull 43:393–403
Cikankova T, Sigitova E, Zverova M, Fisar Z, Raboch J, Hroudova J (2017) Mitochondrial dysfunctions in bipolar disorder: effect of the disease and pharmacotherapy. CNS Neurol Disord Drug Targets 16:176–186. https://doi.org/10.2174/1871527315666161213110518
Cikankova T, Fisar Z, Bakhouche Y, Luptak M, Hroudova J (2019) In vitro effects of antipsychotics on mitochondrial respiration. Naunyn Schmiedeberg's Arch Pharmacol. https://doi.org/10.1007/s00210-019-01665-8
Costa C et al (2008) Electrophysiology and pharmacology of striatal neuronal dysfunction induced by mitochondrial complex I inhibition. J Neurosci 28:8040–8052. https://doi.org/10.1523/jneurosci.1947-08.2008
de Oliveira MR (2016) Fluoxetine and the mitochondria: a review of the toxicological aspects. Toxicol Lett 258:185–191. https://doi.org/10.1016/j.toxlet.2016.07.001
de Sousa RT et al (2014) Lithium increases leukocyte mitochondrial complex I activity in bipolar disorder during depressive episodes. Psychopharmacology. https://doi.org/10.1007/s00213-014-3655-6
Deuschle M (2013) Effects of antidepressants on glucose metabolism and diabetes mellitus type 2 in adults. Curr Opin Psychiatry 26:60–65. https://doi.org/10.1097/YCO.0b013e32835a4206
Finsterer J, Mahjoub SZ (2012) Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol 8:71–79. https://doi.org/10.1517/17425255.2012.644535
Fisar Z, Hroudova J, Raboch J (2010) Inhibition of monoamine oxidase activity by antidepressants and mood stabilizers. Neuro Endocrinol Lett 31:645–656
Fisar Z, Hroudova J, Singh N, Maceckova D, Koprivova A (2017) Protocols for high-resolution respirometry experiments to test the activity of electron transfer system of pig brain mitochondria. Indian Journal of Biochemistry & Biophysics 54:258–272
Folbergrova J, Jesina P, Haugvicova R, Lisy V, Houstek J (2010) Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem Int 56:394–403. https://doi.org/10.1016/j.neuint.2009.11.011
Frey BN et al (2007) Abnormal cellular energy and phospholipid metabolism in the left dorsolateral prefrontal cortex of medication-free individuals with bipolar disorder: an in vivo 1H MRS study. Bipolar Disord 9(Suppl 1):119–127. https://doi.org/10.1111/j.1399-5618.2007.00454.x
Ghaemi SN, Hsu DJ, Soldani F, Goodwin FK (2003) Antidepressants in bipolar disorder: the case for caution. Bipolar Disord 5:421–433
Haddjeri N, Szabo ST, de Montigny C, Blier P (2000) Increased tonic activation of rat forebrain 5-HT(1A) receptors by lithium addition to antidepressant treatments. Neuropsychopharmacology 22:346–356. https://doi.org/10.1016/s0893-133x(99)00138-4
Hroudova J, Fisar Z (2010) Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuro Endocrinol Lett 31:336–342
Hroudova J, Fisar Z (2012) In vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol Lett 213:345–352. https://doi.org/10.1016/j.toxlet.2012.07.017
Hyttel J (1994) Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int Clin Psychopharmacol 9(Suppl 1):19–26
Iwamoto K, Bundo M, Kato T (2005) Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet 14:241–253. https://doi.org/10.1093/hmg/ddi022
Jafarian I, Eskandari MR, Mashayekhi V, Ahadpour M, Hosseini MJ (2013) Toxicity of valproic acid in isolated rat liver mitochondria. Toxicol Mech Methods 23:617–623. https://doi.org/10.3109/15376516.2013.821567
Jang EH, Park CS, Kang JH (2011) Bupropion, an atypical antidepressant, induces endoplasmic reticulum stress and caspase-dependent cytotoxicity in SH-SY5Y cells. Toxicology 285:1–7. https://doi.org/10.1016/j.tox.2011.02.006
Janicak PG (1993) The relevance of clinical pharmacokinetics and therapeutic drug monitoring: anticonvulsant mood stabilizers and antipsychotics. J Clin Psychiatry 54(Suppl):35–41 discussion 55-36
Karanikis P, Korantzopoulos P, Kountouris E, Dimitroula V, Patsouras D, Pappa E, Siogas K (2005) Kearns-Sayre syndrome associated with trifascicular block and QT prolongation. Int J Cardiol 101:147–150. https://doi.org/10.1016/j.ijcard.2004.01.027
Kato T (2006) The role of mitochondrial dysfunction in bipolar disorder. Drug News Perspect 19:597–602. https://doi.org/10.1358/dnp.2006.19.10.1068006
Kato T (2008) Role of mitochondrial DNA in calcium signaling abnormality in bipolar disorder. Cell Calcium 44:92–102. https://doi.org/10.1016/j.ceca.2007.11.005
Kato T, Kunugi H, Nanko S, Kato N (2001) Mitochondrial DNA polymorphisms in bipolar disorder. J Affect Disord 62:151–164
Kim YJ, Ko HH, Han ES, Lee CS (2007) Lamotrigine inhibition of rotenone- or 1-methyl-4-phenylpyridinium-induced mitochondrial damage and cell death. Brain Res Bull 71:633–640. https://doi.org/10.1016/j.brainresbull.2006.12.006
Kim JA, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102:401–414. https://doi.org/10.1161/circresaha.107.165472
Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S (2004) Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry 61:300–308. https://doi.org/10.1001/archpsyc.61.3.300
Lagrue E, Chalon S, Bodard S, Saliba E, Gressens P, Castelnau P (2007) Lamotrigine is neuroprotective in the energy deficiency model of MPTP intoxicated mice. Pediatr Res 62:14–19. https://doi.org/10.1203/PDR.0b013e31806790d7
Lai JS, Zhao C, Warsh JJ, Li PP (2006) Cytoprotection by lithium and valproate varies between cell types and cellular stresses. Eur J Pharmacol 539:18–26. https://doi.org/10.1016/j.ejphar.2006.03.076
Lherm T, Lottin F, Larbi D, Bray M, Legall C, Caen D (2000) Torsade de pointes after poisoning with fluoxetine alone. Presse Med 29:306–307
Liu T et al (2014) Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ Res 115:44–54. https://doi.org/10.1161/circresaha.115.303062
Macritchie K, Geddes JR, Scott J, Haslam D, de Lima M, Goodwin G (2003) Valproate for acute mood episodes in bipolar disorder. Cochrane Database Syst Rev:CD004052. https://doi.org/10.1002/14651858.CD004052
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart Association., Council on Clinical Cardiology, Heart Failure and Transplantation Committee., Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups., Council on Epidemiology and Prevention (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113:1807–1816. https://doi.org/10.1161/circulationaha.106.174287
Maurer IC, Schippel P, Volz HP (2009) Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord 11:515–522. https://doi.org/10.1111/j.1399-5618.2009.00729.x
Munakata K et al (2004) Mitochondrial DNA 3644T-->C mutation associated with bipolar disorder. Genomics 84:1041–1050. https://doi.org/10.1016/j.ygeno.2004.08.015
Ogasahara S, Engel AG, Frens D, Mack D (1989) Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci U S A 86:2379–2382
Pesta D, Gnaiger E (2012) High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol (Clifton, NJ) 810:25–58. https://doi.org/10.1007/978-1-61779-382-0_3
Pinna G, Broedel O, Eravci M, Stoltenburg-Didinger G, Plueckhan H, Fuxius S, Meinhold H, Baumgartner A (2003) Thyroid hormones in the rat amygdala as common targets for antidepressant drugs, mood stabilizers, and sleep deprivation. Biol Psychiatry 54:1049–1059
Ponchaut S, Draye JP, Van Hoof F, Veitch K (1991) Loss of hepatic cytochrome aa3 during chronic valproate treatment: dissociation of proton pumping and electron transport in complex IV. Biochem Soc Trans 19:253S
Ponchaut S, van Hoof F, Veitch K (1992) In vitro effects of valproate and valproate metabolites on mitochondrial oxidations. Relevance of CoA sequestration to the observed inhibitions. Biochem Pharmacol 43:2435–2442
Post RM, Weiss SR (2011) Tolerance to the prophylactic effects of carbamazepine and related mood stabilizers in the treatment of bipolar disorders. CNS Neurosci Ther 17:649–660. https://doi.org/10.1111/j.1755-5949.2010.00215.x
Redrobe JP, Bourin M (1999) Evidence of the activity of lithium on 5-HT1B receptors in the mouse forced swimming test: comparison with carbamazepine and sodium valproate. Psychopharmacology 141:370–377
Rustin P, Chretien D, Bourgeron T, Gerard B, Rotig A, Saudubray JM, Munnich A (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 228:35–51
Santos NA, Medina WS, Martins NM, Mingatto FE, Curti C, Santos AC (2008) Aromatic antiepileptic drugs and mitochondrial toxicity: effects on mitochondria isolated from rat liver. Toxicol in Vitro 22:1143–1152. https://doi.org/10.1016/j.tiv.2008.03.004
Silva MF et al (2008) Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: a review. J Inherit Metab Dis 31:205–216. https://doi.org/10.1007/s10545-008-0841-x
Soeiro-de-Souza MG, Dias VV, Figueira ML, Forlenza OV, Gattaz WF, Zarate CA Jr, Machado-Vieira R (2012) Translating neurotrophic and cellular plasticity: from pathophysiology to improved therapeutics for bipolar disorder. Acta Psychiatr Scand 126:332–341. https://doi.org/10.1111/j.1600-0447.2012.01889.x
Srere (1969) Citrate synthase: [EC 4.1.3.7 citrate oxaloacetate-lyase (CoA acetylating)]. Methods Enzymol 13:3–11
Stork C, Renshaw PF (2005) Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry 10:900–919. https://doi.org/10.1038/sj.mp.4001711
Strakowski SM, Delbello MP, Adler CM (2005) The functional neuroanatomy of bipolar disorder: a review of neuroimaging findings. Mol Psychiatry 10:105–116. https://doi.org/10.1038/sj.mp.4001585
Taylor V, MacQueen G (2006) Associations between bipolar disorder and metabolic syndrome: a review. J Clin Psychiatry 67:1034–1041. https://doi.org/10.4088/jcp.v67n0704
Trounce IA, Kim YL, Jun AS, Wallace DC (1996) Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 264:484–509
Valvassori SS et al (2018) Increased oxidative stress in the mitochondria isolated from lymphocytes of bipolar disorder patients during depressive episodes. Psychiatry Res 264:192–201. https://doi.org/10.1016/j.psychres.2018.03.089
van Reedt Dortland AK, Giltay EJ, van Veen T, Zitman FG, Penninx BW (2010) Metabolic syndrome abnormalities are associated with severity of anxiety and depression and with tricyclic antidepressant use. Acta Psychiatr Scand 122:30–39. https://doi.org/10.1111/j.1600-0447.2010.01565.x
Varriale P (2001) Fluoxetine (Prozac) as a cause of QT prolongation. Arch Intern Med 161:612
Vieweg WV, Wood MA (2004) Tricyclic antidepressants, QT interval prolongation, and torsade de pointes. Psychosomatics 45:371–377. https://doi.org/10.1176/appi.psy.45.5.371
Walden J, Schaerer L, Schloesser S, Grunze H (2000) An open longitudinal study of patients with bipolar rapid cycling treated with lithium or lamotrigine for mood stabilization. Bipolar Disord 2:336–339
Weinbach EC, Costa JL, Nelson BD, Claggett CE, Hundal T, Bradley D, Morris SJ (1986) Effects of tricyclic antidepressant drugs on energy-linked reactions in mitochondria. Biochem Pharmacol 35:1445–1451
Zanetti MV, Otaduy MC, de Sousa RT, Gattaz WF, Busatto GF, Leite CC, Machado-Vieira R (2015) Bimodal effect of lithium plasma levels on hippocampal glutamate concentrations in bipolar II depression: a pilot study. Int J Neuropsychopharmacol:18. https://doi.org/10.1093/ijnp/pyu058
Zarate CA Jr, Singh J, Manji HK (2006) Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry 59:1006–1020. https://doi.org/10.1016/j.biopsych.2005.10.021
Acknowledgments
The authors thank Zdeněk Hanuš for his assistance.
Funding
This study is supported by a grant GA ČR 17-07585Y from the Grant Agency, Czech Republic, and by a grant 17-05292S from the Czech Science Foundation, Czech Republic.
Author information
Authors and Affiliations
Contributions
JH and ZF conceived and designed the research. TC, ZF, and JH conducted experiments, analyzed data, and wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cikánková, T., Fišar, Z. & Hroudová, J. In vitro effects of antidepressants and mood-stabilizing drugs on cell energy metabolism. Naunyn-Schmiedeberg's Arch Pharmacol 393, 797–811 (2020). https://doi.org/10.1007/s00210-019-01791-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-019-01791-3