
Abstract
Schizophrenia etiology is unknown, nevertheless imbalances occurring in an acute psychotic episode are important to its development, such as alterations in cellular energetic state, REDOX homeostasis and intracellular Ca2+ management, all of which are controlled primarily by mitochondria. However, mitochondrial function was always evaluated singularly, in the presence of specific respiratory substrates, without considering the plurality of the electron transport system. In this study, mitochondrial function was analyzed under conditions of isolated or multiple respiratory substrates using brain mitochondria isolated from MK-801-exposed mice. Results showed a high H2O2 production in the presence of pyruvate/malate, with no change in oxygen consumption. In the condition of multiple substrates, however, this effect is lost. The analysis of Ca2+ retention capacity revealed a significant change in the uptake kinetics of this ion by mitochondria in MK-801-exposed animals. Futhermore, when mitochondria were exposed to calcium, a total loss of oxidative phosphorylation and an impressive increase in H2O2 production were observed in the condition of multiple substrates. There was no alteration in the activity of the antioxidant enzymes analyzed. The data demonstrate for the first time, in an animal model of psychosis, two important aspects (1) mitochondria may compensate deficiencies in a single mitochondrial complex when they oxidize several substrates simultaneously, (2) Ca2+ handling is compromised in MK-801-exposed mice, resulting in a loss of phosphorylative capacity and an increase in H2O2 production. These data favor the hypothesis that disruption of key physiological roles of mitochondria may be a trigger in acute psychosis and, consequently, schizophrenia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Schizophrenia is a neuropsychiatric disorder that affects about 21 million people around the world (World Health Organization 2018) Its complex etiology includes genetic, environmental, psychological and social factors (Picchioni and Murray 2007). This is currently recognized as a neurodevelopmental disorder, including a prodromal phase characterized by attenuated symptoms, followed by an acute exacerbation of psychotic symptoms that usually lead to the chronic disturbance (McCutcheon et al. 2019). The first psychotic episode generally occurs in late adolescence and early adulthood, including mainly positive symptoms, such as delusions and hallucinations, but also negative symptoms, such as blunted affect and social withdrawal and cognitive deficits (Insel 2010; Lewis and Levitt 2002). Duration of untreated psychosis is a key factor for the prognostic (Harrigan et al. 2003), thus, understanding the changes occurring during the first psychotic episode may contribute to the understanding of schizophrenia development.
It is well established that there is an imbalance in dopamine and glutamate neurotransmitter systems related to the pathology (Insel 2010). This led to the emergence of neurochemical hypotheses to explain the signs and symptoms of schizophrenia. N-methyl-D-aspartate glutamate receptors (NMDAR) were first implicated in schizophrenia pathophysiology after the observation that NMDAR antagonists, such as phencyclidine (PCP) (Allen and Young 1978) and ketamine (Lahti et al. 1995), are able to induce delusional states, thought disorder as well as negative symptoms and cognitive deficits in healthy individuals, and exacerbate these symptoms in schizophrenia patients (Adler et al. 1998; Malhotra et al. 1997). In rodents, these antagonists (including MK-801) consistently induce behavioral and neurochemical endophenotypes analogous to acute psychosis manifestations, reproducing positive, negative and cognitive symptoms (Andine et al. 1999; Lodge and Mercier 2015; Manahan-Vaughan et al. 2008; Sams-Dodd 1996). For example, the impairment in sensory gating manifested by deficits in the prepulse inhibition of the startle reflex (PPI) is clearly observed in animals exposed to NMDAR antagonists (Jones et al. 2011; Mansbach 1991). Pharmacologically-induced hypofunction of NMDAR also leads to an acute increase in glutamate release and, consequently, disinhibition of brain regions such as the hippocampus and frontal cortex (Greene 2001), resembling the changes detected in schizophrenia patients by neuroimaging studies (Chiu et al. 2018; Theberge et al. 2002; Tregellas et al. 2014; Uhlhaas and Singer 2010). Moreover, acute challenges with NMDA antagonists induce metabolic changes similar to those described in first episode unmedicated schizophrenia patients (Soyka et al. 2005; Eyjolfsson et al. 2006). For these reasons, animals acutely exposed to NMDAR antagonists are used as models of NMDA hypofunction related to acute psychosis (Greene 2001; Howes et al. 2015; Jones et al. 2011). Such acute models are useful tools for the identification of initial changes related to NMDA receptors hypofunction that might occur during an acute psychotic episode and, consequently, contribute to the chronic state of the disorder.
The pathophysiology of psychotic episodes and schizophrenia is also correlated to changes in fundamental cellular processes which regulate the energetic and REDOX status. This is supported by several evidence found in patients brains: (i) reduction in glucose metabolism, related to several alterations in glycolytic enzymes [for review see (Martins-De-Souza et al. 2010)], impaired glucose tolerance and insulin resistance are already manifested in acutely psychotic first episode patients (Perry et al. 2016); (ii) decreased expression of the E1 subunit of the pyruvate dehydrogenase complex, which directly reflects in mitochondrial acetyl-CoA levels (Prabakaran et al. 2004); (iii) changes in expression and activity of the Krebs cycle enzymes (Bubber et al. 2011); (iv) detachment of hexokinase 1 from the external mitochondrial membrane, the main link between glucose metabolism and ATP production in brain (Regenold et al. 2012). In addition, several studies with patients or animal models show both an increase in the generation of reactive oxygen species (ROS) as well as a reduction in the efficiency of antioxidant systems, which leads to oxidative stress [for review see (Koga et al. 2016)]. Moreover, high baseline oxidative stress is present in first episode unmedicated patients and seems to be related to gray matter lost, cognitive outcomes and a worst disorder prognostic (Fraguas et al. 2017).
There is a lack of studies on mitochondrial function in acutely psychotic patients and the available data reflects activity of peripheral samples only. A decrease in mitochondrial DNA copy number was detected in blood samples of unmedicated patients (Hjelm et al. 2015; Li et al. 2015). However, many groups showed brain mitochondria changes in chronic schizophrenia (Rajasekaran et al. 2015). These changes are both quantitative and morphological, such as alterations in mitochondrial density and the structure of the cristae (Uranova et al. 2007). In addition, further evidence associates genetic polymorphisms in mitochondrial DNA to schizophrenia. Some of these polymorphisms result in structural and functional modifications in mitochondrial proteins such as OPA1 and components of the electron transport system (Ben-Shachar and Karry 2007; Bergman and Ben-Shachar 2016; Iwamoto et al. 2005; Marchbanks et al. 2003; Maurer et al. 2001; Prince et al. 1999; Rosenfeld et al. 2011). Taken together, these data point to mitochondrial imbalance as a hallmark in the pathophysiology of schizophrenia and understanding the early changes that might precede these chronicle alterations is a necessary to a further understanding of the disorder.
Mitochondrion is the organelle to which all cellular energy metabolism converges and the main source of ATP and ROS generation in brain cells. In addition, it plays a key role in synaptic plasticity and modulation of neuronal activity (Li et al. 2004) and changes in these processes may impact neural communication and development. Although mitochondrial complexes have been investigated due to its importance in the pathophysiology of psychosis (Bergman and Ben-Shachar 2016), they have always been evaluated in isolation, using enzymatic activity or oxygen consumption assays in the presence of specific substrates for each of them, without considering the plurality of the electron transport system and the mitochondrial ability to oxidize different substrates. Physiologically, mitochondria use several energetic substrates simultaneously and pathological conditions or stressors could modify this pattern, as previously shown in skeletal muscle (Martins et al. 2018). These changes may directly reflect on mitochondrial ATP synthesis and ROS generation, which are both altered in the disorder (Martins-de-Souza et al. 2011). In addition, brain mitochondria represent an important compartment for calcium storage and plays a central role in calcium homeostasis and modulation of neurotransmitter release (Jonas et al. 1999; Mnatsakanyan et al. 2017), which is also disrupted in schizophrenia (Lidow 2003; Lisek et al. 2016). Despite these facts, few studies have directly investigated mitochondrial calcium handling on experimental models of schizophrenia (Lisek et al. 2016; Bustamante et al. 2016).
Therefore, in this work, mitochondrial electron transport system, ROS generation and calcium uptake were evaluated in the presence of different respiratory substrates, used in isolation and together, in an animal model of NMDA hypofunction related to acute psychosis. Our results indicate that the disruption of NMDA activity may lead to disturbances in mitochondrial calcium handling in brain that follows a severe mitochondrial dysfunction under stress conditions which could be implicated in psychosis and, consequently, schizophrenia pathology.
Materials and methods
Animals
Adult male Swiss mice (PND60–90) were used. Animals were kept in groups of 4 or 5 in plastic cages with free access to filtered water and standard diet. Mice were kept under a 12 h light-dark cycle (lights off at 6:00 pm) with controlled temperature (23 ± 2 °C) and humidity (65%). All experimental protocols were developed according to the guidelines of the National Research Ethical Committee (published by National Heath Council-MS, 1998) and were approved by the local Ethical Committee (CEUA-UFRJ n. DFBCICB045).
MK-801 exposure
All the biochemical experiments were preceded by the pre-pulse inhibition of the startle reflex (PPI) test. Mice were transferred to the experimental room and remained for 1 h on-site for habituation. Subsequently, they were randomly divided into four groups: saline (NaCl 0.9%), MK-801 0.1 mg/kg, MK-801 0.3 mg/kg and MK-801 1.0 mg/kg. MK-801 hydrogen maleate (Sigma-Aldrich, Brazil) was dissolved in saline. All animals were injected intraperitoneally at 1 mL/100 g body weight and doses are expressed as free base. After injection, animals were kept undisturbed for 30 min and submitted to the PPI test.
Pre-pulse inhibition of startle reflex (PPI)
The experimental protocol was performed according to (Neves et al. 2013) with minor modifications. PPI test was performed in an isolated chamber containing a movement-sensitive platform and a loudspeaker to produce the acoustic stimulus (Panlab, Harvard Instruments, Barcelona, Spain). The experimental protocol consisted of 5 min habituation (background white noise 65 dB) followed by 5 exposures to the pulse (115 dB, 50 ms) for startle stabilization. Subsequently, animals were exposed to five different blocks of stimulus (10 times each, with 10 to 30 s intertrial interval): (1) control (background noise); (2) pulse (115 dB); (3) pulse preceded by pre-pulse of 72 dB; (4) pulse preceded by 80 dB pre-pulse; (5) pulse preceded by 90 dB pre-pulse. Interstimulus interval (prepulse-pulse) was fixed at 100 ms. Startle magnitude in each trial was digitalized and the percentual inhibition of the startle reflex for each pre-pulse intensity was calculated as: 100 - [(average startle from pre-pulse plus pulse trials / average startle from pulse alone trials) × 100].
Mitochondrial isolation
Soon after the PPI test, the animals were euthanized by decapitation and the forebrains were removed for mitochondrial isolation by digitonin according to the protocol described by Rosenthal et al. with small modifications (Rosenthal et al. 1987). To test if PPI alone would affect mitochondrial function, we compared mitochondrial respiration from animals that passed through PPI test or not. No difference was found in mitochondrial function between animals submitted or not to PPI test, thus no specific effect of PPI was detected (data not shown). Briefly, the forebrains were immersed in mitochondrial isolation buffer containing: 225 mM mannitol, 75 mM sucrose, 1 mM EGTA, 1 mM BSA, 10 mM HEPES pH 7.2 and kept on ice. This solution was repeatedly changed for excess blood removal. Subsequently, the brains were minced and softly homogenized 10x in glass potter with Teflon piston in isolation buffer. The produced homogenate was centrifuged at 1330 g for 3 min. After centrifugation, the pellet was discarded and the supernatant was separated and centrifuged again at 21200 g for 8 min. Thereafter, the resulting pellet was resuspended in 5 mL of isolation buffer and gently homogenized 3x in the glass potter in the presence of 0.1% (w/v) digitonin. This homogenate was centrifuged again at 21200 g for 8 min to remove the detergent. At the end of the process, pure mitochondria were obtained, since digitonin disrupts the cell membranes to promote an enriched mitochondrial preparation. The amount of mitochondrial protein was determined by the Lowry method using bovine serum albumin BSA as standard (Lowry et al. 1951).
High resolution respirometry
Measurements of mitochondrial oxygen consumption were performed with high-resolution oxygraphy (Oxygraph-2 k Oroboros Instruments, Innsbruck, Austria) at 37 °C with constant stirring. It was used 0.1 mg/mL of mitochondrial protein in MIR05 medium containing: 110 mM sucrose, 20 mM HEPES, 20 mM taurine, 60 mM K-lactobionate, 3 mM MgCl2, 10 mM KH2PO4, 0.5 mM EGTA, BSA 1 g/L, pH 7.1 (Gnaiger et al. 2000). Substrates and inhibitors of mitochondrial function were added sequentially, always after signal stabilization, using two different protocols. The following additions were made for experiments with isolated substrates: 2/2 mM Pyruvate/Malate or 5 mM Succinate; 1 mM ADP; 0.1 μg/mL Oligomycin; 2.5 μM Antimycin A. The following additions were made to the experiments in the presence of multiple substrates 2/2/10 mM Pyruvate/Malate/glutamate; 5 mM Succinate; 5 mM Glycerol Phosphate; 1 mM ADP; 0.1 μg/mL Oligomycin; 2.5 μM Antimycin A. Multiple substrate runs were performed in the absence or presence of 200 μM CaCl2. Data were collected through a specific software that shows, in real time, the concentration of oxygen and the specific oxygen flow, which means the negative time derivative of oxygen concentration (DatLab software 5.0, Oroboros Instruments, Innsbruck, Austria).
H2O2 production
The H2O2 generation was measured by two protocols. In the first part of the work (Fig. 2), when mitochondria were evaluated with isolated respiratory substrates, mitochondrial H2O2 release was measured by the fluorescence of Amplex Red probe using a spectrofluorometer (Varian Cary Eclipse; Agilent Technologies, Santa Clara, CA). Fluorescence was detected at an excitation wavelength of 563 nm (slit 5 nm) and an emission wavelength of 587 nm (slit 5 nm) in MIR05 medium in the presence of 0.1 mg/mL mitochondrial protein and 2 U/mL horseradish peroxidase. The titration sequence was identical to the respirometry experiments: 2/2 mM Pyruvate/Malate - or 5 mM Succinate; 1 mM ADP; 0.1 μg/mL Oligomycin; 2.5 μM Antimycin A. The rate of H2O2 production was determined using linear regression curve fitting with Origin 8.0.
In the second part of the work (Figs. 3, 6 and 7), the measurements of H2O2 production in the presence of multiple substrates were made simultaneously to the oxygen consumption measurements, through the coupling of an optical sensor to the oxygraph chambers (O2k-Fluo LED2-Module). This module, associated with the software (DatLab software 5.0, Oroboros Instruments, Innsbruck, Austria) is able to record the fluorescence emitted by the Amplex-Red-Peroxidase system. This protocol was performed in the absence and presence of 200 μM CaCl2. Equipment calibration was done by addition of a known concentration of H2O2 at the beginning of the run.
Enzymatic activities
Glutathione reductase (GR)
The activity of Glutathione reductase was measured in a reaction medium containing 0.1 M K+ phosphate buffer, pH 7 and 0.1 mM NADPH by following the consumption of NADPH, represented by the reduction of its light absorbance at 340 nm. The reaction was started by adding 1 mM of oxidized glutathione (GSSG) (adapted from (Flohe and Gunzler 1984)).
Glutathione peroxidase (GPx)
The activity of Glutathione peroxidase was measured indirectly through exogenous glutathione reductase activity coupling. The exogenous GR reduces the GSSG formed by the GPx coupled to the consumption of NADPH. The reaction medium contained 50 mM KH2PO4 pH 7, 1 mM NaN3, 0.2 mM NADPH, 1 U/mL exogenous GR, 1 mM GSH and the reaction was started with 0.25 mM H2O2. Reduction of light absorbance of NADPH was monitored at 340 nm (adapted from (Mills 1957)).
Thioredoxin reductase (TR)
The activity of Thioredoxin reductase was measured in medium containing 100 mM KH2PO4 pH 7, 2.5 mM DTNB, 10 mM EDTA. The reaction was started by adding 0.3 mM NADPH and the method is based on the ability of this enzyme to use 5,5 ¢ -dithiobis (2-nitrobenzoic acid) (DTNB) as the substrate to generate 5-thio-2-nitrobenzoic acid (TNB), which produces a strong yellow color that is measured at 412 nm (Holmgren and Bjornstedt 1995).
Superoxide dismutase (mnSOD and CuZnSOD isoforms)
The activity of mnSOD was monitored using superoxide radicals from the xanthine/xanthine oxidase system coupled to the reaction. The rate of cytochrome C reduction by the superoxide radical was monitored at 480 nm in medium containing 5–20 μg mitochondrial protein, 50 μM xanthine, 20 μM potassium cyanide (inhibitor of the CuZnSOD isoform), 20 μM cytochrome C and 2 U/mL xanthine oxidase (McCord and Fridovich 1969). The experiments were repeated in buffer without potassium cyanide to obtain the total SOD activity. The activity of the CuZnSOD isoform was determined by the difference between the total activity by the activity in the presence of potassium cyanide.
All enzymatic assays were performed on a Victor Perkin-Elmer plate reader at 37 °C with constant stirring.
Mitochondrial Ca2+ uptake
To evaluate the mitochondrial calcium retention capacity, mitochondria were submitted to CaCl2 titration, with additions of 100 μM in fixed 4 min intervals. The medium contained 10 mM HEPES, 2 mM MgCl2, 150 mM KCl, 2 mM KH2PO4, 1 mM ADP and 2 mM pyruvate and malate or 5 mM succinate and 0.1 μM of CaGreen, a probe that emits fluorescence when bound to Calcium (λex 506 nm and λem 532 nm) (Facundo et al. 2005; Rajdev and Reynolds 1993). Each CaCl2 addition resulted in fluorescence peak representing the binding of the ion to the fluorescent probe (Fig. 4a, b and 5a). Over time, a reduction in fluorescence is observed, indicating calcium detachment of the probe and its entry into the mitochondria. Titrations were performed until there was no more mitochondrial calcium uptake and the calculations were made after the subtraction of the baselines at the end of each calcium addition by the initial baseline. The assays were performed at 37 °C, with constant stirring in a spectrofluorometer (Varian Cary Eclipse; Agilent Technologies, Santa Clara, CA).
Statistics
Data were plotted on GraphPad Prism 6® software. PPI data was analyzed by a two-way repeated measure ANOVA, considering pre-pulse intensity as the first factor and treatment as the second, followed by Bonferroni post-test. For all other experiments, it was used One-way ANOVA with Bonferroni post-test for multiple comparisons. Differences were considered statistically significant when p < 0.05.
Results
Impairment of PPI by MK-801
PPI is the measure of startle reflex reduction induced by a pre-stimulus (Powell et al. 2009) and is considered an indicative of sensory-motor filter integrity. This filter allows the discrimination of external physiological or cognitive relevant from irrelevant stimulus and is extremely conserved among several species of mammals, including humans and rodents (Weiss and Feldon 2001). Pre-pulse inhibition is impaired in both acutely psychotic patients and animal models and reflects an important aspect of the disorder: the deficit in pre-attentional gating and information processing (Braff and Geyer 1990; Powell et al. 2009; Weiss and Feldon 2001).
Higher prepulse intensities induce a progressive startle inhibition, as shown in Fig. 1 (two-way RM ANOVA, Fintensity(2,94) = 43.98, p < 0.001). MK-801 treatment impaired PPI at all doses (two-way RM ANOVA, Ftreatment(3,94) = 23.65, p < 0.001, Bonferroni post-test p < 0.003) showing that NMDAR blockade had the expected effect. PPI disruption was slightly more prominent for the smaller doses of MK-801 (Bonferroni post-test p < 0.024 comparing 1.0 mg/kg effect with the other doses). There was no difference in startle amplitude between groups (ANOVA, F(3,47) = 1.73, p = 0.175). Taking together, these data validates the acute NMDAR hypofunction model in our experimental conditions.

NMDAR blockade by MK-801 impairs pre-pulse inhibition of the startle reflex. Mice were treated i.p. with saline or MK-801 30 min before test. Data are expressed as mean ± S.E.M. (n = 12–13). Data was analyzed by a two-way repeated measure ANOVA followed by Bonferroni post-test. ***p < 0.001 **p < 0.01 vs. saline group; #p < 0.05 vs. other MK-801 groups
Mitochondrial O2 and H2O2 fluxes with complex i, ii or multiple substrates
In order to evaluate the mitochondrial function in MK-801-treated animals we measured the fluxes of oxygen consumption and H2O2 generation in the presence of isolated substrates that feed the electron transport system via complex I (pyruvate/malate) or complex II (succinate). In Fig. 2a, d typical traces of the flux of oxygen consumption (solid black line) and H2O2 levels (solid gray line) are shown. As expected, both oxygen and H2O2 fluxes respond to sequential addition of mitochondrial substrates and inhibitors in accordance with the variation of the mitochondrial membrane potential (ΔΨM) variation (Korshunov et al. 1997). More importantly, H2O2 fluxes are higher with succinate than with pyruvate/malate (Fig. 2a, d, gray line). The O2 fluxes follow this same tendency for CII compared to CI -linked substrates (Fig. 2a, d, black lines). We detected a significant increase in H2O2 production in the MK-801 0.3 mg/kg group compared to controls in the presence of pyruvate/malate and ATP (Fig. 2c). However, there was no change in oxygen consumption (Fig. 2b). This parameter did not change in the presence of succinate alone (Fig. 2e, f).

Effect of MK-801 treatment on oxygen consumption and H2O2generation in isolated mitochondria using substrates linked to complexes I or II. a and d Representative experiments. The arrows represent the moments of the additions. PM - Pyruvate/Malate (2/2 mM); S - Succinate (5 mM); ADP (1 mM); O - Oligomycin (0.1 μg / mL); AA - Antimycin A (2.5 μM). The sequence of additions and substrates concentrations were identical in oxygen consumption and H2O2 generation experiments. b and e High resolution respirometry in the presence of pyruvate/malate (b) or succinate (e). c and f Production of H2O2 measured by fluorescence of Amplex Red in the presence of pyruvate/malate (c) or succinate (f). Data were analyzed by One-way ANOVA with Bonferroni post-test for multiple comparisons and are expressed as the mean ± standard error. ** p = 0.0093 compared to saline group. The numbers of individual experiments are in parentheses
Under physiological conditions, cells from the central nervous system (CNS) metabolize multiple substrates from NAD and FAD- enzymes simultaneously and converge the electrons to coenzyme Q (Q-junction) and finally to oxygen (Gnaiger 2014). We evaluated the O2 consumption and H2O2 production in the presence of pyruvate/malate/glutamate/succinate and glycerol 3-phosphate, simultaneously as a mimetic of in vivo system complexity. Figure 3a shows typical traces of O2 and H2O2 fluxes. There was no change between groups either in oxygen consumption (Fig. 3b) or H2O2 production (Fig. 3c) in the presence of multiple substrates. These results indicate that with the multiple substrate approach the observed effect of higher ROS generation via complex I (Fig. 2c, MK-801 0.3 mg/kg) is abolished.

Effect of MK-801 treatment on oxygen consumption and H2O2generation in isolated mitochondria in the presence of multiple substrates. a Representative experiment. The arrows represent the moments of the additions. PMG - Pyruvate/Malate/Glutamate (2/2/10 mM); S - Succinate (5 mM); GF - Glycerol Phosphate (5 mM); ADP (1 mM); O - Oligomycin (0.1 μg / mL); AA - Antimycin A (2.5 μM). b High resolution respirometry and (c) H2O2 production in the presence of multiple substrates. Data were analyzed by One-way ANOVA with Bonferroni post-test for multiple comparisons and are expressed as the mean ± standard error. MK-801 groups were not significantly different from saline group. The numbers of individual experiments are in parentheses
Effect of MK-801 in the antioxidant systems
In order to investigate whether the observed increase in H2O2 generation linked to complex I substrates was due to a reduction in enzymatic antioxidant capacity, the activities of important enzymes responsible for the clearance and control of ROS levels in neural cells were measured. As shown in Table 1, no changes were observed in the activities of the tested enzymes. This result suggests that the higher H2O2 levels in the presence of pyruvate/malate was due to an increase in H2O2 production instead of a decrease in the antioxidant enzymatic capacity.
Effects of MK-801 on mitochondrial Ca2+-uptake
In addition to oxygen consumption and H2O2 generation, calcium retention capacity is another important aspect of mitochondrial physiology, especially in the CNS where a precise balance in mitochondrial calcium has a key role in mechanisms of neuroplasticity, such as long-term potentiation (LTP) (Jonas et al. 1999). Intracellular calcium variations occur for a variety of reasons, including activation of NMDA receptors, and mitochondria play a key role in the uptake of this ion in order to control intracellular calcium levels. To evaluate this ability, the mitochondria were exposed to a titration of calcium until their maximum absorption capacity, in the presence of different substrates (Figs. 4 and 5). In Figs. 4a, d we show typical traces of mitochondrial Ca2+ uptake in the presence of pyruvate/malate or succinate, respectively. Although the maximum calcium retention capacity is the same for all MK-801 treated groups (Fig. 4c, f), the uptake kinetics is different in all drug treated groups relative to saline group (Fig. 4b, e, closed circles, dotted lines), starting from 400 μM in the presence of pyruvate/malate and from 300 μM succinate. In the presence of both pyruvate/malate or succinate, NMDA blockage led to a slowdown in calcium uptake into the mitochondria, observed in the graphs by a shift to the left of the titration curves, which implies a Ca2+ retention deficit (Fig. 4b, e). This displacement indicates a higher concentration of calcium outside the mitochondria and bound to the fluorescent probe CaGreen after the time set for titrations (increased residual baselines). This result suggests that the kinetic of calcium uptake is altered in the MK-801 treated groups.

Mitochondrial Ca2+uptake in MK-801 treated mice mitochondria in the presence of isolated substrates. a and d Representative experiments of calcium uptake curves in the presence of (a) pyruvate/malate or (d) succinate. Each peak represents the addition of 100 μM CaCl2. Additions were made every 4 min until maximal Ca2+ uptake. The baselines after each addition were used to calculate the ΔCa2+ fluorescence, after subtraction of the initial baseline. b and e Calcium uptake curves in the presence of (b) Pyruvate/Malate or (e) Succinate. c and f Maximal calcium absorption in the presence of (c) Pyruvate/Malate or (f) Succinate. Data were analyzed by One-way ANOVA with Bonferroni post-test for multiple comparisons and are expressed as the mean ± standard error. *** p < 0.0001 vs. Saline group. The numbers of individual experiments are in parentheses

Mitochondrial Ca2+uptake in MK-801 treated mice mitochondria in the presence in the multiple substrates. a Representative experiments of calcium uptake curves in the presence of pyruvate, malate, glutamate, succinate and glycerol phosphate as substrates. Each peak represents the addition of 100 μM CaCl2. Additions were made every 4 min until maximal Ca2+ uptake. The baselines after each addition were used to calculate the ΔCa2+ fluorescence, after subtraction of the initial baseline. b Calcium uptake curves in the presence of multiple substrates. c Maximal calcium absorption in the presence of multiple substrates. Data were analyzed by One-way ANOVA with Bonferroni post-test for multiple comparisons and are expressed as the mean ± standard error. ** p = 0.0014 vs. saline group. *** p = 0.0007 vs. saline group. The number of individual experiments is in parentheses
In the data presented in Fig. 5 the same experiment was conducted, but in the presence of multiple substrates feeding the electron transport system. Under this condition, the effect of NMDA blockade was exacerbated. Figure 5a shows a schematic representation of the Ca2+ titration. There is a significant shift to the left for all treated groups compared to the control group (Fig. 5b). Furthermore, the maximal Ca2+ retention capacity is reduced in the MK-801 treated groups in the presence of multiple substrates (Fig. 5c). Together, these results indicate that not only the kinetic of calcium uptake is affected by NMDA blockage but also the more physiological condition provided by the presence of multiple substrates exacerbates this effect.
Effect of calcium in O2 consumption and in H2O2 production
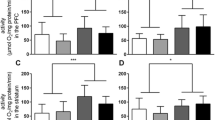
Since calcium handling is altered in the mitochondria of MK-801-exposed animals, we wondered whether, in the face of a stressful situation, such as an increase in calcium concentration, differences in oxygen consumption and H2O2 generation would be detected. To address this issue, we subjected the mitochondria to a condition of excess calcium (200 μM) as a stressor and performed the measurements in the presence of multiple substrates.
Figures 6 shows representative experiments of oxygen consumption (Fig. 6a) or H2O2 production (Fig. 6c). The stressful condition unmasked a drastic reduction in oxygen consumption by mitochondria from animals exposed to MK-801 in the highest doses (0.3 and 1.0 mg/kg) compared to saline and MK-801 0.1 mg/kg groups (Fig. 6b). The presence of calcium completely abolished oxidative phosphorylation in mitochondria from animals exposed to the highest doses of MK-801 (Fig. 6b). In addition, these doses of 0.3 and 1 mg/kg of MK-801 led to an impressive increase in H2O2 production in the presence of calcium (Fig. 6d). In this condition, the basal levels of H2O2 raised in the groups exposed to the highest doses of MK-801 in relation to the control group and this difference remains until the end of the experiment. It was also observed that the addition of ADP was not able to decrease H2O2 levels, in accordance with the loss of phosphorylating capacity (Fig. 6d). Together, these data show that treatment with MK-801 greatly alters oxygen uptake capacity and H2O2 generation in mitochondria exposed to high calcium concentrations.

Effect of calcium on oxygen consumption and H2O2generation in isolated brain mitochondria of treated mice in the presence of multiple substrates. a and c Representative experiments of oxygen consumption (a) and H2O2 generation (b) in the presence of multiple substrates. The arrows represent the moments of the additions. PMG - Pyruvate/Malate/Glutamate (2/2/10 mM); S - Succinate (5 mM); GF - Glycerol Phosphate (5 mM); ADP (1 mM); O - Oligomycin (0.1 μg / mL); AA - Antimycin A (2.5 μM). Experiments were already initiated in the presence of 200 μM CaCl2. b High resolution respirometry in the presence of 200 μM CaCl2 and multiple substrates. d Production of H2O2 in the presence of 200 μM CaCl2 and multiple substrates. Data were analyzed by One-way ANOVA with Bonferroni post-test for multiple comparisons and are expressed as the mean ± standard error. * p = 0.0018 vs saline and MK-801 0.1 mg/mL groups; ** p = 0.0002 vs saline and MK-801 0.1 mg/mL groups; *** p = 0.0001 vs saline and MK-801 0.1 mg/mL groups; # p = 0.0207 vs saline group; ## p = 0.0044 vs saline group. The numbers of individual experiments are in parentheses
Discussion
The present study shows for the first time that NMDAR blockade by acute MK-801 administration drastically alters calcium handling in brain mitochondria in the presence of multiple substrates, which results in reduction of oxygen consumption, loss of the capacity of ATP production and an increase in H2O2 generation.
The acute administration of MK-801 at different doses significantly impaired PPI response (Fig. 1), as expected from other studies (Andine et al. 1999; Varty and Higgins 1995). Importantly, this result relates the MK-801 doses used to impairments found in acutely psychotic patients, validating the model for the analysis of putative psychosis-related mitochondrial alterations in our experimental conditions. The relationship between changes in mitochondrial function and chronic schizophrenia is well established. Various studies described changes in mitochondrial DNA (Iwamoto et al. 2005; Marchbanks et al. 2003), in the expression and function of proteins related to mitochondrial structure (Rosenfeld et al. 2011) and ETS (Electron Transport System) (Ben-Shachar and Karry 2007; Bergman and Ben-Shachar 2016; Maurer et al. 2001; Prince et al. 1999) as well as functional losses of these complexes in patients (Bergman and Ben-Shachar 2016; Cavelier et al. 1995; Maurer et al. 2001; Prince et al. 1999). However, few studies have focused in unmedicated first episode patients or used animal models of acute NMDA hypofunction to thoroughly assess early acute changes in mitochondrial function that may be important for the disturbance. Most importantly, none of the previous studies have investigated the role of Ca2+ handling by brain mitochondria in a condition of multiple substrates. This situation mimics a physiologic stressful state of the mitochondria, which are exposed to large variations in calcium concentrations.
Acute NMDA blockade with MK-801 did not result in alteration in oxygen consumption when mitochondria were exposed to substrates of complex I or complex II in isolation (Fig. 2b, e). These data contrast with previous results published by Millo and Chari-Bitron (1973) where brain mitochondria from rats acutely treated with 10 mg/kg PCP showed a decrease in oxygen consumption in the presence of pyruvate/malate and in the presence of succinate, with a pronounced effect in complex I (Millo and Chari-Bitron 1973). Venancio et al. (2015) also showed a decrease in the activity of complex I with glutamate/malate after acute treatment with high doses of ketamine (Venancio et al. 2015). Differences between the experimental models, antagonists and doses used in each study may explain the divergence, since there is no study that relates the effects of these drugs on different species of rodents nor the equivalence among doses. In addition, it should be noted that the activity of the isolated complex I should be analyzed differently from its activity in an integrated and complex system.
In the same study, Venancio et al. (2015) also verified that mitochondria stimulated with glutamate/malate or with succinate alone showed a high rate of H2O2 production, which partially agrees with the present work, that showed higher H2O2 production in the presence of ADP only when mitochondria were exposed to substrates for complex I (Fig. 2c, f). In fact, several studies point to an increase in the production of ROS related to psychosis (for review, see (Koga et al. 2016)). Animal models based on repeated exposure to NMDA antagonists also show data consistent with our findings (de Oliveira et al. 2009, 2011; Faizi et al. 2014). For example, Faizi et al. (2014) observed in adult rats treated with 30 mg/kg ketamine for 5 consecutive days the collapse in mitochondrial membrane potential, increase in ROS production, mitochondrial swelling and cytochrome C release (Faizi et al. 2014). de Oliveira et al. (2011) treated adult rats with 25 mg/kg ketamine for one week and observed changes in the enzymatic activity of all mitochondrial complexes in different regions of the brain (de Oliveira et al. 2011). Zhou et al. (2012) observed changes in complexes I, II and III after seven days of treatment with 0.7 mg/kg MK-801(Zhou et al. 2012). Thus, the changes described in our work might represent an initial acute step that may lead to a more complex mitochondrial disfunction after a longer NMDA hypofunction. However, both changes in oxygen consumption as well as production of ROS seen in all these studies do not consider that, under physiological conditions, mitochondria feeds simultaneously NAD and FAD molecules with electrons derived from different metabolic sources. Moreover, the type and amount of ROS produced depends on the substrate used (Yin et al. 2014). In none of these animal models the oxygen consumption capacity or ROS production were measured in the presence of NAD and FAD electron donors.
In Fig. 3 we observed that, when substrates (NAD and FAD electron donors) other than pyruvate/malate (NAD electron donors) were introduced, the profile of higher H2O2 production via complex I was lost. In general, animal models related to psychosis and schizophrenia that evaluated mitochondrial function with isolated substrates found changes related to complex I. In addition, studies with tissue from schizophrenia patients show predominantly deficiencies in the same complex (Bergman and Ben-Shachar 2016). If there is a change in complex I, it is expected a decrease in O2 consumption and increase in ROS, especially if the experimental condition allows the entry of electrons into the system exclusively via complex I, as in most studies with isolated substrates. It is possible that physiologically, even if there is a defect in a certain mitochondrial complex, the simultaneous oxidation of several substrates may compensate for this deficiency, in order to reduce the impact of this defect on mitochondrial function. Thus, a compensatory effect could justify the results obtained with multiple substrates and reinforces the importance of a more detailed mitochondrial analysis.
As important as the evaluation of ROS production is the analysis of antioxidant systems, since the level of these species in the cells is a consequence of the balance between its production and purification. A huge range of studies shows a correlation between alteration in antioxidant enzyme systems and first episode psychosis or schizophrenia, as recently reviewed by Koga et al. (2016). Despite this, there is still considerable divergence among results for the same enzymes. For example, SOD has decreased activity in approximately half of the studies analyzed and increased activity in the other half, with a few showing no difference (Koga et al. 2016). Glutathione peroxidase presents reduced activity in most studies, but an increase in a few of them (Koga et al. 2016). A metanalysis including four studies in early onset unmedicated psychotic patients showed no differences in the activity of SOD, catalase and glutathione peroxidase when compared to blood samples of control individuals (Fraguas et al. 2017), in accordance with our findings. However, analysis of antioxidant enzymes activity also presents controversial results in other studies. While Venancio et al. (2015) showed an increase in the activity of SOD in acute high dose ketamine model, de Oliveira et al. (2009) evidenced a reduction in the activity of the enzyme in the same model with smaller doses. In the present study, no significant differences were observed in any of the antioxidant enzymes analyzed (Table 1), suggesting that, in our model, the most plausible explanation for the increase in ROS associated with acute psychosis is a higher production of ROS, and not a decrease in the clearance capacity.
Mitochondria have functions that go beyond the generation of ATP. In addition to act as a metabolic control center, it is associated with various cellular signaling pathways, which include not only the control of ROS generation but also the maintenance of intracellular calcium levels (Brini et al. 2014; Murphy 2009). For this reason, the analysis of mitochondrial function should consider oxygen consumption, ROS production and calcium handling. The analysis of calcium retention capacity in the presence of isolated substrates showed that, although total retention capacity was not altered (Fig. 4c, f) there was a drastic change in the kinetics of the uptake of this ion by mitochondria from animals treated with MK- 801 (Fig. 4b, e). When Ca2+ titration was performed in the presence of multiple substrates, a condition considered as more physiological, it was observed that both maximal Ca2+ retention capacity and the uptake kinetic were significantly modified by MK-801 treatment (Fig. 5). These data highlight the importance of the approach with multiple substrates when mitochondria function is analyzed since there are differences that can be abolished or revealed only in this scenario.
Although there is no similar data in the literature related to Ca2+ handling with acute treatment with MK-801 in mice, there are two recent studies with chronically treated animals with ketamine showing changes in calcium management. Bustamante et al. (2016) treated adult rats with ketamine 40 mg/kg for three consecutive days and observed a decrease in calcium concentration in the mitochondria from the hippocampus of these animals, as well as a reduction in the intracellular levels of Ca2+ induced by KCl (Bustamante et al. 2016). Lisek et al. (2016) injected 30 mg/kg ketamine in adult rats for five consecutive days and analyzed cells from various brain regions at the end of treatment, which led to a relevant increase in intracellular [Ca2+] in the cortex, cerebellum, hippocampus as well as altered expression of proteins related to the control of cellular calcium levels (Lisek et al. 2016). Although the experimental models are different and the acute and chronic treatments lead to distinct metabolic consequences, our results and previous evidences are consistent with the relevance of calcium management in psychosis.
The alterations in calcium handling (Figs. 4 and 5) raised a concern on the integrity of the mitochondria when it is exposed to high calcium concentrations. In the presence of calcium, we observed a complete loss of mitochondrial function in the groups treated with the highest doses of MK-801 in the presence of multiple substrates (Fig. 6). In addition, there was an impressive increase in H2O2 production in this condition (Fig. 6d). It is noteworthy that, in the presence of multiple substrates but without the calcium challenge, MK801-treated mitochondria presented exactly the same profile of oxygen consumption and H2O2 production as the control group (Fig. 3). Only under a stressful situation the difference emerges.
Ca2+ is an important second messenger that controls various metabolic pathways and is involved in fundamental processes, such as synaptic transmission, learning and memory (Brini et al. 2014; Friel 2000). The activation of voltage-gated channels, transmembrane receptors may lead to an increase of cytoplasmic calcium, which rapidly changes from nanomolar to micromolar concentrations (LaFerla 2002). The endoplasmic reticulum and the mitochondria are the main organelles involved in storage and release of intracellular calcium after the increase observed during depolarization, for example. In addition to Ca2+ uptake, mitochondria are also responsible for providing ATP to plasma membrane ATPases that transport Ca2+. The entrance of Ca2+ in mitochondria via Mitochondrial Calcium Uniporter (MCU) leads to an increase in the activity of several mitochondrial enzymes that will ultimately rise ATP generation (Brini et al. 2014). For this reason, mitochondria need to be able to tolerate large variations in Ca2+ concentration without losing the phosphorylative capacity or the control of ROS generation. However, as seen in Fig. 6, both processes become extremely affected by MK-801 acute treatment. The role of synaptic and non-synaptic mitochondria in neuroplasticity through appropriate calcium buffering may involve MCU (Kirichok et al. 2004; Penna et al. 2018), the interaction of endoplasmic reticulum with mitochondria (Park et al. 2017), and/or a fine regulation of permeability transition pore opening (Jonas et al. 1999; Mnatsakanyan et al. 2017). Our data give support for the proposal that alterations in one or more of these crucial steps may result in important cellular dysfunctions (Figs. 4, 5 and 6). However, at present, we do not know which of these targets are involved.
Schematic representations of control mitochondria (Fig. 7a) and MK-801-exposed mitochondria (Fig. 7b) highlight alterations in the mitochondrial function caused by high calcium concentrations in both cases. Data show that mitochondria derived from MK-801 treated animals present two negative characteristics concerning Ca2+ handling: (1) they have a reduced capacity of Ca2+ uptake and (2) they are extremely sensitive to incoming Ca2+, which abolishes oxidative phosphorylation and increases the generation of H2O2 in the condition of multiple substrates. On a real situation, where these mitochondria are in an active neuron, the result would be catastrophic, leading to a longer time for the clearance of Ca2+ from the cytosol, a drastic decrease in ATP production and an increase in H2O2 production. Altogether, this could impact several other cellular processes. In long term, the result of the impairment in Ca2+ management by mitochondria could lead to a pathological context with dysregulation of neural function.

Schematic representation of mitochondria fed by multiple substrates in the presence of calcium. In the scheme, mitochondrial complexes attached to the membrane are represented as indicated. OMM – Outer Mitochondrial Membrane; IMM – Inner Mitochondrial Membrane; CI – NADH Dehydrogenase; CII – Succinate Dehydrogenase; CIII – Ubiquinone Cytochrome C Oxidoreductase; CIV – Cytochrome C Oxidase; CV – ATP Synthase. Solid black lines represent the electron pathway through the ETS and the passing of protons through complex V resulting in ATP formation. Dotted black lines indicate the formation of superoxide as a result of the electron transport. Thick light blue arrows represent the proton pumping from the mitochondrial matrix to the intramembrane space through the indicated complexes. Red solid arrow indicates the entering of Ca2+ through Mitochondrial Calcium Uniporter (MCU). Mitochondria representations of saline (a) or MK-801 treated (b) groups show the consequences of calcium entrance in the matrix
Changes in critical cellular pathways, such as calcium signaling, are in focus and are proposed to be the main basis for pathologies with multiple potential targets (Sawa and Snyder 2002). Since early 80’s this proposal has been based on several studies that point to a dysregulation in intracellular calcium signaling in schizophrenia (Lidow 2003). Acute blockade of NMDAR is related to an increase in glutamate levels in key brain regions (Moghaddam et al. 1997). The excess of this neurotransmitter may activate all glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), kainic acid receptors (KAR) and metabotropic glutamate receptors (mGluR) that, ultimately, raises the intracellular calcium levels in these cells in a process that may lead to glutamate excitotoxicity (for review see (Kritis et al. 2015)). Thus, the deficit in mitochondrial calcium buffering could be explained by a previous rise in this ion due to an overflow of glutamate promoted by MK-801 treatment. Another possibility could be the rising in intracellular calcium concentration promoted by dopamine, since it is well established that NMDAR antagonists, such as MK-801, lead to dopamine release in the striatum and that dopamine receptor activation also result in increasing intracellular calcium concentrations (Breier et al. 1998; Hasbi et al. 2010; Whitton et al. 1992). It is important to highlight that, in the presence of calcium, mitochondria derived from treated groups are no longer able to generate ATP aerobically, resulting in a drastic energy imbalance. In this scenario, there is also a severe REDOX deregulation, caused by the significant increase in H2O2 production that could culminate in abnormal cellular signaling. Changes in glutamate signaling are related with the signs and symptoms of psychosis and the observed imbalance in energy production, REDOX equilibrium and calcium handling disruption might represent initial changes occurring in acute psychotic episodes that will possibly contribute to changes in neural circuits that can underline the emergence of the syndrome.
In summary, our data show that the analysis of mitochondrial function in the condition of isolated substrates should be carefully evaluated, since the mitochondria is able to oxidize several substrates simultaneously and to compensate for the deficiencies that may exist linked to a single mitochondrial complex. Moreover, the changes demonstrated in this study regarding the management of calcium in the presence of multiple substrates may have several implications in the context of acute psychosis and, possibly, schizophrenia and contribute to the hypothesis that the management of calcium may be a trigger for the pathophysiological changes seen in this complex pathology.
References
Adler CM, Goldberg TE, Malhotra AK, Pickar D, Breier A (1998) Effects of ketamine on thought disorder, working memory, and semantic memory in healthy volunteers. Biol Psychiatry 43(11):811–816
Allen RM, Young SJ (1978) Phencyclidine-induced psychosis. Am J Psychiatry 135(9):1081–1084. https://doi.org/10.1176/ajp.135.9.1081
Andine P, Widermark N, Axelsson R, Nyberg G, Olofsson U, Martensson E, Sandberg M (1999) Characterization of MK-801-induced behavior as a putative rat model of psychosis. J Pharmacol Exp Ther 290(3):1393–1408
Ben-Shachar D, Karry R (2007) Sp1 expression is disrupted in schizophrenia; a possible mechanism for the abnormal expression of mitochondrial complex I genes, NDUFV1 and NDUFV2. PLoS One 2(9):e817. https://doi.org/10.1371/journal.pone.0000817
Bergman O, Ben-Shachar D (2016) Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: possible interactions with cellular processes. Can J Psychiatr 61(8):457–469. https://doi.org/10.1177/0706743716648290
Braff DL, Geyer MA (1990) Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry 47(2):181–188
Breier A, Adler CM, Weisenfeld N, Su TP, Elman I, Picken L, Malhotra AK, Pickar D (1998) Effects of NMDA antagonism on striatal dopamine release in healthy subjects: application of a novel PET approach. Synapse 29(2):142–147. https://doi.org/10.1002/(SICI)1098-2396(199806)29:2<142::AID-SYN5>3.0.CO;2-7
Brini M, Cali T, Ottolini D, Carafoli E (2014) Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci 71(15):2787–2814. https://doi.org/10.1007/s00018-013-1550-7
Bubber P, Hartounian V, Gibson GE, Blass JP (2011) Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. Eur Neuropsychopharmacol 21(3):254–260. https://doi.org/10.1016/j.euroneuro.2010.10.007
Bustamante J, Czerniczyniec A, Lores-Arnaiz S (2016) Ketamine effect on intracellular and mitochondrial calcium mobilization. Biocell 40:11–14
Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, Oreland L, Gyllensten U (1995) Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics 29(1):217–224. https://doi.org/10.1006/geno.1995.1234
Chiu PW, Lui SSY, Hung KSY, Chan RCK, Chan Q, Sham PC, Cheung EFC, Mak HKF (2018) In vivo gamma-aminobutyric acid and glutamate levels in people with first-episode schizophrenia: a proton magnetic resonance spectroscopy study. Schizophr Res 193:295–303. https://doi.org/10.1016/j.schres.2017.07.021
de Oliveira L, Spiazzi CM, Bortolin T, Canever L, Petronilho F, Mina FG, Dal-Pizzol F, Quevedo J, Zugno AI (2009) Different sub-anesthetic doses of ketamine increase oxidative stress in the brain of rats. Prog Neuro-Psychopharmacol Biol Psychiatry 33(6):1003–1008. https://doi.org/10.1016/j.pnpbp.2009.05.010
de Oliveira L, Fraga DB, De Luca RD, Canever L, Ghedim FV, Matos MP, Streck EL, Quevedo J, Zugno AI (2011) Behavioral changes and mitochondrial dysfunction in a rat model of schizophrenia induced by ketamine. Metab Brain Dis 26(1):69–77. https://doi.org/10.1007/s11011-011-9234-1
Eyjolfsson EM, Brenner E, Kondziella D, Sonnewald U (2006) Repeated injection of MK801: an animal model of schizophrenia? Neurochem Int 48(6–7):541–546. https://doi.org/10.1016/j.neuint.2005.11.019
Facundo HT, de Paula JG, Kowaltowski AJ (2005) Mitochondrial ATP-sensitive K+ channels prevent oxidative stress, permeability transition and cell death. J Bioenerg Biomembr 37(2):75–82. https://doi.org/10.1007/s10863-005-4130-1
Faizi M, Salimi A, Rasoulzadeh M, Naserzadeh P, Pourahmad J (2014) Schizophrenia induces oxidative stress and cytochrome C release in isolated rat brain mitochondria: a possible pathway for induction of apoptosis and neurodegeneration. Iran J Pharm Res 13(Suppl):93–100
Flohe L, Gunzler WA (1984) Assays of glutathione peroxidase. Methods Enzymol 105:114–121
Fraguas D, Díaz-Caneja CM, Rodríguez-Quiroga A, Arango C (2017) Oxidative stress and inflammation in early onset first episode psychosis: a systematic review and meta-analysis. Int J Neuropsychopharmacol 20(6):435–444. https://doi.org/10.1093/ijnp/pyx015
Friel DD (2000) Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium 28(5–6):307–316. https://doi.org/10.1054/ceca.2000.0172
Gnaiger E (2014) Mitochondrial pathways and respiratory control - the blue book (Oroboros)
Gnaiger E, Kuznetsov AV, Schneeberger S, Seiler R, Brandacher G, Steurer W, Margreiter R (2000) Mitochondria in the cold. In Heldmaier G, Klingenspor M (ed) Life in the Cold. Heidelberg, Berlin, New York: Springer pp 431–442
Greene R (2001) Circuit analysis of NMDAR hypofunction in the hippocampus, in vitro, and psychosis of schizophrenia. Hippocampus 11(5):569–577. https://doi.org/10.1002/hipo.1072
Harrigan SM, McGorry PD, Krstev H (2003) Does treatment delay in first-episode psychosis really matter? Psychol Med 33(1):97–110. https://doi.org/10.1017/s003329170200675x
Hasbi A, O'Dowd BF, George SR (2010) Heteromerization of dopamine D2 receptors with dopamine D1 or D5 receptors generates intracellular calcium signaling by different mechanisms. Curr Opin Pharmacol 10(1):93–99. https://doi.org/10.1016/j.coph.2009.09.011
Hjelm BE, Rollins B, Mamdani F, Lauterborn JC, Kirov G, Lynch G, Gall CM, Sequeira A, Vawter MP (2015) Evidence of mitochondrial dysfunction within the complex genetic etiology of schizophrenia. Mol Neuropsychiatry 1(4):201–219. https://doi.org/10.1159/000441252
Holmgren A, Bjornstedt M (1995) Thioredoxin and thioredoxin reductase. Methods Enzymol 252:199–208
Howes O, McCutcheon R, Stone J (2015) Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol 29(2):97–115. https://doi.org/10.1177/0269881114563634
Insel TR (2010) Rethinking schizophrenia. Nature 468(7321):187–193. https://doi.org/10.1038/nature09552
Iwamoto K, Bundo M, Kato T (2005) Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet 14(2):241–253. https://doi.org/10.1093/hmg/ddi022
Jonas EA, Buchanan J, Kaczmarek LK (1999) Prolonged activation of mitochondrial conductances during synaptic transmission. Science 286(5443):1347–1350
Jones CA, Watson DJ, Fone KC (2011) Animal models of schizophrenia. Br J Pharmacol 164(4):1162–1194. https://doi.org/10.1111/j.1476-5381.2011.01386.x
Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427(6972):360–364. https://doi.org/10.1038/nature02246
Koga M, Serritella AV, Sawa A, Sedlak TW (2016) Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr Res 176(1):52–71. https://doi.org/10.1016/j.schres.2015.06.022
Korshunov SS, Skulachev VP, Starkov AA (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416(1):15–18
Kritis AA, Stamoula EG, Paniskaki KA, Vavilis TD (2015) Researching glutamate - induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci 9:91. https://doi.org/10.3389/fncel.2015.00091
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci 3(11):862–872. https://doi.org/10.1038/nrn960
Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995) Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13(1):9–19. https://doi.org/10.1016/0893-133X(94)00131-I
Lewis DA, Levitt P (2002) Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci 25:409–432. https://doi.org/10.1146/annurev.neuro.25.112701.142754
Li Z, Okamoto K, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119(6):873–887. https://doi.org/10.1016/j.cell.2004.11.003
Li Z, Hu M, Zong X, He Y, Wang D, Dai L, Dong M, Zhou J, Cao H, Lv L, Chen X, Tang J (2015) Association of telomere length and mitochondrial DNA copy number with risperidone treatment response in first-episode antipsychotic-naïve schizophrenia. Sci Rep 5:18553–18557. https://doi.org/10.1038/srep18553
Lidow MS (2003) Calcium signaling dysfunction in schizophrenia: a unifying approach. Brain Res Brain Res Rev 43(1):70–84
Lisek M, Boczek T, Ferenc B, Zylinska L (2016) Regional brain dysregulation of Ca(2+)-handling systems in ketamine-induced rat model of experimental psychosis. Cell Tissue Res 363(3):609–620. https://doi.org/10.1007/s00441-015-2332-3
Lodge D, Mercier MS (2015) Ketamine and phencyclidine: the good, the bad and the unexpected. Br J Pharmacol 172(17):4254–4276. https://doi.org/10.1111/bph.13222
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A (1997) Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17(3):141–150. https://doi.org/10.1016/S0893-133X(97)00036-5
Manahan-Vaughan D, von Haebler D, Winter C, Juckel G, Heinemann U (2008) A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus 18(2):125–134. https://doi.org/10.1002/hipo.20367
Mansbach RS (1991) Effects of NMDA receptor ligands on sensorimotor gating in the rat. Eur J Pharmacol 202(1):61–66
Marchbanks RM, Ryan M, Day IN, Owen M, McGuffin P, Whatley SA (2003) A mitochondrial DNA sequence variant associated with schizophrenia and oxidative stress. Schizophr Res 65(1):33–38
Martins EL, Ricardo JC, de-Souza-Ferreira E, Camacho-Pereira J, Ramos-Filho D, Galina A (2018) Rapid regulation of substrate use for oxidative phosphorylation during a single session of high intensity interval or aerobic exercises in different rat skeletal muscles. Comp Biochem Physiol B Biochem Mol Biol 217:40–50. https://doi.org/10.1016/j.cbpb.2017.11.013
Martins-De-Souza D, Dias-Neto E, Schmitt A, Falkai P, Gormanns P, Maccarrone G, Turck CW, Gattaz WF (2010) Proteome analysis of schizophrenia brain tissue. World J Biol Psychiatry 11(2):110–120. https://doi.org/10.3109/15622970903490626
Martins-de-Souza D, Harris LW, Guest PC, Bahn S (2011) The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid Redox Signal 15(7):2067–2079. https://doi.org/10.1089/ars.2010.3459
Maurer I, Zierz S, Moller H (2001) Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr Res 48(1):125–136
McCord JM, Fridovich I (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244(22):6049–6055
McCutcheon RA, Reis Marques T, Howes OD (2019) Schizophrenia - An Overview. JAMA Psychiatry 30:1–10. https://doi.org/10.1001/jamapsychiatry.2019.3360
Millo S, Chari-Bitron A (1973) Effect of phencyclidine on oxygen consumption of rat brain mitochondria in vitro and in vivo. Biochem Pharmacol 22(13):1661–1665
Mills GC (1957) Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem 229(1):189–197
Mnatsakanyan N, Beutner G, Porter GA, Alavian KN, Jonas EA (2017) Physiological roles of the mitochondrial permeability transition pore. J Bioenerg Biomembr 49(1):13–25. https://doi.org/10.1007/s10863-016-9652-1
Moghaddam B, Adams B, Verma A, Daly D (1997) Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17(8):2921–2927
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417(1):1–13. https://doi.org/10.1042/BJ20081386
Neves G, Antonio CB, Betti AH, Pranke MA, Fraga CA, Barreiro EJ, Noel F, Rates SM (2013) New insights into pharmacological profile of LASSBio-579, a multi-target N-phenylpiperazine derivative active on animal models of schizophrenia. Behav Brain Res 237:86–95. https://doi.org/10.1016/j.bbr.2012.09.016
Park SJ, Lee SB, Suh Y, Kim SJ, Lee N, Hong JH, Park C, Woo Y, Ishizuka K, Kim JH, Berggren PO, Sawa A, Park SK (2017) DISC1 modulates neuronal stress responses by gate-keeping ER-mitochondria Ca(2+) transfer through the MAM. Cell Rep 21(10):2748–2759. https://doi.org/10.1016/j.celrep.2017.11.043
Penna E, Espino J, De Stefani D, Rizzuto R (2018) The MCU complex in cell death. Cell Calcium 69:73–80. https://doi.org/10.1016/j.ceca.2017.08.008
Perry BI, McIntosh G, Weich S, Singh S, Rees K (2016) The association between first-episode psychosis and abnormal glycaemic control: systematic review and meta-analysis. Lancet Psychiatry 3(11):1049–1058. https://doi.org/10.1016/S2215-0366(16)30262-0
Picchioni MM, Murray RM (2007) Schizophrenia. BMJ 335(7610):91–95. https://doi.org/10.1136/bmj.39227.616447.BE
Powell SB, Zhou X, Geyer MA (2009) Prepulse inhibition and genetic mouse models of schizophrenia. Behav Brain Res 204(2):282–294. https://doi.org/10.1016/j.bbr.2009.04.021
Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S (2004) Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry 9(7):684–697, 643. https://doi.org/10.1038/sj.mp.4001511
Prince JA, Blennow K, Gottfries CG, Karlsson I, Oreland L (1999) Mitochondrial function is differentially altered in the basal ganglia of chronic schizophrenics. Neuropsychopharmacology 21(3):372–379. https://doi.org/10.1016/S0893-133X(99)00016-0
Rajasekaran A, Venkatasubramanian G, Berk M, Debnath M (2015) Mitochondrial dysfunction in schizophrenia: pathways, mechanisms and implications. Neurosci Biobehav Rev 48:10–21. https://doi.org/10.1016/j.neubiorev.2014.11.005
Rajdev S, Reynolds IJ (1993) Calcium green-5N, a novel fluorescent probe for monitoring high intracellular free Ca2+ concentrations associated with glutamate excitotoxicity in cultured rat brain neurons. Neurosci Lett 162(1–2):149–152
Regenold WT, Pratt M, Nekkalapu S, Shapiro PS, Kristian T, Fiskum G (2012) Mitochondrial detachment of hexokinase 1 in mood and psychotic disorders: implications for brain energy metabolism and neurotrophic signaling. J Psychiatr Res 46(1):95–104. https://doi.org/10.1016/j.jpsychires.2011.09.018
Rosenfeld M, Brenner-Lavie H, Ari SG, Kavushansky A, Ben-Shachar D (2011) Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol Psychiatry 69(10):980–988. https://doi.org/10.1016/j.biopsych.2011.01.010
Rosenthal RE, Hamud F, Fiskum G, Varghese PJ, Sharpe S (1987) Cerebral ischemia and reperfusion: prevention of brain mitochondrial injury by lidoflazine. J Cereb Blood Flow Metab 7(6):752–758. https://doi.org/10.1038/jcbfm.1987.130
Sams-Dodd F (1996) Phencyclidine-induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav Pharmacol 7(1):3–23
Sawa A, Snyder SH (2002) Schizophrenia: diverse approaches to a complex disease. Science 296(5568):692–695. https://doi.org/10.1126/science.1070532
Soyka M, Koch W, Moller HJ, Ruther T, Tatsch K (2005) Hypermetabolic pattern in frontal cortex and other brain regions in unmedicated schizophrenia patients. Results from a FDG-PET study. Eur Arch Psychiatry Clin Neurosci 255(5):308–312. https://doi.org/10.1007/s00406-005-0563-0
Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, Neufeld RW, Rogers J, Pavlosky W, Schaefer B, Densmore M, Al-Semaan Y, Williamson PC (2002) Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry 159(11):1944–1946. https://doi.org/10.1176/appi.ajp.159.11.1944
Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, Eichman LC, Lyons E, Freedman R (2014) Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry 171(5):549–556. https://doi.org/10.1176/appi.ajp.2013.13070981
Uhlhaas PJ, Singer W (2010) Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 11(2):100–113. https://doi.org/10.1038/nrn2774
Uranova N, Bonartsev P, Brusov O, Morozova M, Rachmanova V, Orlovskaya D (2007) The ultrastructure of lymphocytes in schizophrenia. World J Biol Psychiatry 8(1):30–37. https://doi.org/10.1080/15622970600960207
Varty GB, Higgins GA (1995) Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipsychotic drugs. Psychopharmacology 122(1):15–26
Venancio C, Felix L, Almeida V, Coutinho J, Antunes L, Peixoto F, Summavielle T (2015) Acute ketamine impairs mitochondrial function and promotes superoxide dismutase activity in the rat brain. Anesth Analg 120(2):320–328. https://doi.org/10.1213/ANE.0000000000000539
Weiss IC, Feldon J (2001) Environmental animal models for sensorimotor gating deficiencies in schizophrenia: a review. Psychopharmacology 156(2–3):305–326
Whitton PS, Biggs CS, Pearce BR, Fowler LJ (1992) Regional effects of MK-801 on dopamine and its metabolites studied by in vivo microdialysis. Neurosci Lett 142(1):5–8
World Health Organization (WHO) (2018) Mental Health: Schizophrenia. https://www.who.int/mental_health/management/schizophrenia/en/. Accessed 17 Jan 2019
Yin F, Boveris A, Cadenas E (2014) Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal 20(2):353–371. https://doi.org/10.1089/ars.2012.4774
Zhou K, Yang Y, Gao L, He G, Li W, Tang K, Ji B, Zhang M, Li Y, Yang J, Sun L, Zhang Z, Zhu H, He L, Wan C (2012) NMDA receptor hypofunction induces dysfunctions of energy metabolism and semaphorin signaling in rats: a synaptic proteome study. Schizophr Bull 38(3):579–591. https://doi.org/10.1093/schbul/sbq132
Acknowledgments
Antonio Galina had fellowships from the Brazilian National Concil for Research (CNPq 30712/2016-8) from 2015 to 2018 and from Cientista do Nosso Estado (CNE) of Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ E-26/203.004/2017). Mariana S. Silveira had a fellowship from the Brazilian National Concil for Research (CNPq 308910/2013-3) from 2013 to 2018. Gilda A. Neves received funding from Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ, PensaRio Grant number E-26/110.678/2012).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Monteiro, J., Assis-de-Lemos, G., de-Souza-Ferreira, E. et al. Energization by multiple substrates and calcium challenge reveal dysfunctions in brain mitochondria in a model related to acute psychosis. J Bioenerg Biomembr 52, 1–15 (2020). https://doi.org/10.1007/s10863-019-09816-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-019-09816-5