
Abstract
Flavone apigenin has an anti-inflammatory effect. We assessed whether apigenin may reduce the inflammatory mediator production, which is regulated by the Toll-like receptor-4-dependent activation of the Akt, mTOR, and NF-κB pathways, and activation of JNK and p38-MAPK in HEK001 keratinocytes and primary keratinocytes. Apigenin, the Akt inhibitor, Bay 11-7085, and N-acetylcysteine inhibited the lipopolysaccharide-stimulated production of cytokines IL-1β and IL-6 and chemokines CCL17 and CCL27; the expression of cyclooxygenase-2; the increase in the levels of Toll-like receptor-4, phosphorylated Akt, and mTOR; the activation of NF-κB; the activation of the JNK and p38-MAPK; and the production of reactive oxygen/nitrogen species in keratinocytes. Inhibitors of the c-JNK (SP600125) and p38-MAPK (SB203580) reduced lipopolysaccharide-induced production of inflammatory mediators and activation of the JNK and p38-MAPK in keratinocytes. These results show that apigenin may inhibit the lipopolysaccharide-caused inflammatory mediator production in keratinocytes by reducing the Toll-like receptor-4-dependent activation of Akt, mTOR, and NF-κB pathways, and activation of JNK and p38-MAPK. The suppressive effect of apigenin may be achieved by the inhibition of reactive oxygen/nitrogen species production. Additionally, apigenin appears to reduce the Akt, mTOR, and NF-κB pathway- and the JNK and p38-MAPK-mediated inflammatory skin diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Keratinocytes respond to microbial products, such as lipopolysaccharide, through the expression of Toll-like receptors (TLRs), thereby evoking immune and inflammatory responses (Baker 2006; Begon et al. 2007; Jin et al. 2015). TLR-4, one of the Toll-like receptors, plays a critical role in recognizing microbial lipopolysaccharide (Takeda and Akira 2005; Morris et al. 2015). Lipopolysaccharide binding to keratinocyte CD14 and subsequent activation of TLR-4 results in the nuclear translocation of nuclear factor (NF)-κB, leading to the production of cytokines and chemokines (Song et al. 2002; Morris et al. 2015). Lipopolysaccharide activates the phosphatidylinositol (PI) 3-kinase/Akt/mammalian target of rapamycin (mTOR) pathways, which is followed by activation of NF-κB (Thomson et al. 2009; Zhong et al. 2012; Ma et al. 2015). Further, lipopolysaccharide exhibits a proinflammatory effect on various tissues through activation of the c-Jun N-terminal kinases (JNKs) and p38-mitogen-activated protein kinases (MAPK) (Bi et al. 2016; Kim et al. 2016; Wu et al. 2016; Zhu et al. 2016).
The flavone apigenin has been shown to exhibit an anti-inflammatory effect and modulate an immune response (Lim et al. 2013; Wang et al. 2014; Patil et al. 2016). Apigenin reduces lipopolysaccharide-stimulated proinflammatory cytokine production by reducing cyclooxygenase (COX) and NF-κB activation (Lim et al. 2013; Wang et al. 2014). Apigenin inhibits the lipopolysaccharide-induced expression of inducible nitric oxide synthase, COX-2, and expression of proinflammatory cytokines (Patil et al. 2016) in human lung epithelial cells—A549 cells. Apigenin reduces lipopolysaccharide-stimulated expression of proinflammatory cytokines and COX-2 mRNA expression by inhibiting NF-κB in placenta and fetal membranes (Lim et al. 2013). Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-κB (Nicholas et al. 2007). Apigenin-7-glycoside not only inhibits the lipopolysaccharide-enhanced inflammatory activity in lung but also exhibits anti-inflammatory effect through inhibition of the MAPK and NF-κB pathways. Apigenin reduces lipopolysaccharide-stimulated IL-1β mRNA expression in both normoxia and hypoxia in human periodontal ligament cells (Pumklin et al. 2016). Apigenin reduces lipopolysaccharide-induced expression of IL-6, IL-8, and COX-2 mRNA in IPEC-J2 nontransformed intestinal epithelial cells (Farkas et al. 2015).
In contrast, apigenin suppresses the lipopolysaccharide-stimulated production of chemokines through the extracellular signal-regulated kinase (ERK)/MAPK pathway without inhibition of the NF-κB activation (Huang et al. 2010). Apigenin inhibits the di-(2-ethylhexyl)phthalate-stimulated expression of inflammatory mediators by activation of JNKs but not by activation of NF-κB and ERK/MAPK pathways (Wang et al. 2012).
Apigenin is demonstrated to have an anti-inflammatory effect. However, whether apigenin reduces lipopolysaccharide-stimulated production of inflammatory mediators in keratinocytes remains uncertain. Furthermore, the apigenin effect on the lipopolysaccharide-caused activation of the Akt, mTOR, and NF-κB signaling pathways and the JNK and p38-MAPK in keratinocytes has not been studied. In this respect, we assessed the apigenin effect on lipopolysaccharide-stimulated inflammatory mediator production in HEK001 keratinocytes and primary keratinocytes.
Materials and methods
Materials
Apigenin, Bay 11-7085 (an inhibitor of NF-κB activation and phosphorylation of IκB-α, (2E)-3-[[4-(1,1-dimethylethyl)phenyl]sulfonyl]-2-propenenitrile), the Akt inhibitor (type II, SH-5)), rapamycin (a mTOR inhibitor), SB203580 (a selective inhibitor of p38-MAPK), and horseradish peroxidase-conjugated anti-mouse IgG were purchased from EMD-Calbiochem Co. (La Jolla, CA, USA). Enzyme-linked immunosorbent assay (ELISA) kits for human CXCL1/IL1β, human IL6, human thymus and activation-regulated chemokine (TARC/CCL17), human cutaneous T cell-attracting chemokine (CTACK/CCL27), human cyclooxygenase (COX)-2, and human/mouse/rat phosphorylated Akt (Pan) were obtained from R&D Systems, Inc. (Minneapolis, MN, USA). Antibodies (Toll-like receptor (TLR)-4, NF-κB p65, NF-κB p50, phosphorylated IκB-α, Akt1, phosphorylated Akt1, mTOR, JNK, phosphorylated JNK, p38, phosphorylated p38, and β-actin) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The TransAM™ NF-κB p65 to DNA binding assay kit was purchased from Active Motif® (Carlsbad, CA, USA). Lipopolysaccharide (from Escherichia coli), SP600125 (a selective inhibitor of c-JNK), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO), and N G-methyl-L-arginine acetate salt (L-NMMA) were obtained from Sigma-Aldrich Inc. (St. Louis, MO, USA).
Keratinocyte culture
Human keratinocytes (HEK001, tissue: skin; morphology: epithelial; cell type: human papillomavirus 16 E6/E7 transformed) were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in keratinocyte-serum free media supplemented with bovine pituitary extract and recombinant epidermal growth factor (GIBCO®, Invitrogen Co., Grand Island, NY, USA).
Normal human keratinocytes were provided from the Department of Urology, Chung-Ang University Hospital (Seoul, South Korea). Keratinocytes were prepared from neonatal foreskin discarded after circumcision (Kim et al. 2005) in accordance with ethical guidelines. Neonatal foreskin was chopped and split overnight in a sucrose-trypsin solution (0.1% sucrose, 0.25% trypsin, and 1 mM EDTA) at 4 °C. Keratinocyte suspension was cultured in EpiLife® medium supplemented with growth factor (Cascade Biologics™, Portland, OR, USA).
Cell numbers used in the ELISA were based on manufacturers’ protocols and adjusted to measure the values under suitable experimental conditions.
Immunoassays for IL-1β, IL-6, COX-2, CCL17, and CCL27
Keratinocytes (1 × 106 cells/ml of medium for the cytokine and COX assay, and 5 × 106 cells/ml of medium for the chemokine assay) were grown in 24-well plate and treated with 1 μg/ml lipopolysaccharide for 24 h. After centrifugation at 412g for 10 min, the amounts of IL-1β, IL-6, COX-2, CCL17, and CCL27 in the culture supernatants were analyzed using ELISA kits, according to the manufacturer’s instructions. Absorbance of mixture was measured at 450 nm using a microplate reader (Magellan, Tecan, Salzburg, Austria).
Preparation of cytosolic and nuclear extracts
HEK001 keratinocytes (5 × 106 cells/ml of medium) were treated with lipopolysaccharide for 30 min at 37 °C. The levels of TLR-4 in keratinocytes were measured after a 24-h incubation. Keratinocyte cytosolic and nuclear extracts were prepared, as in previously described methods (Schreiber et al. 1989). Keratinocytes collected by centrifugation at 412g for 10 min were washed twice with phosphate buffered saline (PBS). Keratinocytes were suspended in 100 μl lysis buffer (10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 10 mM HEPES-KOH, pH 7.8) and were made to swell on ice for 10 min. Next, 6 μl of a 10% Nonidet NP-40 solution (final concentration of approximately 0.6%) was added to lysis buffer, and the mixtures were vigorously vortexed for 10 s. The homogenates were centrifuged at 12,000g for 10 min at 4 °C. The supernatants were stored as cytoplasmic extracts and kept at − 70 °C. The nuclear pellets were resuspended in 75 μl of an ice-cold hypertonic solution containing 5% glycerol and 0.4 M NaCl. The tubes were located on ice for 30 min and then centrifuged at 12,000 g for 15 min at 4 °C. The supernatants as the nuclear extracts were stored at − 70 °C. Protein concentrations were measured, according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot assays for the levels of signaling proteins
The cytosolic and nuclear extracts for Western blot were treated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and then boiled. Samples (45 μg protein/well) were loaded onto each lane of a 10–12% SDS-polyacrylamide gel, electrophoresis was done, and the proteins were transferred onto polyvinylidene difluoride membranes (GE Healthcare Chalfont St. Giles, Buckinghamshire, UK). The membranes were blocked for 2 h in TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20). The membranes were labeled with the appropriate antibody (TLR-4, NF-κB p65, NF-κB p50, phosphorylated IκB-α, Akt1, phosphorylated Akt, mTOR, JNK, phosphorylated JNK, p38, phosphorylated p38, and β-actin) overnight at 4 °C with gentle agitation. After 4 washes in TBST (TBS containing 0.1% Tween 20), the membranes were treated with horseradish peroxidase-conjugated anti-mouse IgG. The membranes were then treated with SuperSignal® West Pico chemiluminescence substrate, and the protein bands which have enhanced chemiluminescence were visualized using an image analyzer (ImageQuant™ LAS4000, GE Healthcare Bio-Sciences AB, Bjӧrkgatan 30, 751 84 Uppsala, Sweden).
The densities of the protein bands were determined using TINA 2.10g software licensed for Seoul National University (SNU and SNUMD, Seoul, South Korea) and expressed as a fold increase compared to the control density.
Assay for the DNA binding activity of NF-κB p65
The biding of NF-κB p65 to DNA was measured, according to the TransAM™ NF-κB kit user’s manual. Keratinocytes (2 × 106 cells/ml of medium) were treated with lipopolysaccharide for 30 min. Nuclear extracts were prepared, according to the procedure described in the user’s manual, and added to a 96-well plate to which oligonucleotides containing an NF-κB p65 consensus binding site (5′-GGGACTTTCC-3′) were immobilized. The amount of nuclear extract in the sample was determined to be 5 μg/20 μl. The DNA-binding active NF-κB p65 was treated with a primary antibody specific for NF-κB p65 and then with anti-rabbit horseradish peroxidase-conjugated IgG. Next, the color developer and stop solutions were sequentially added to the well plates. The absorbance of mixture was measured at 450 nm with a reference wavelength of 655 nm in a microplate reader.
Enzyme-linked immunosorbent assay for phosphorylated Akt
Keratinocytes (1 × 106 cells/ml of medium) were treated with lipopolysaccharide for 4 h. Keratinocytes were collected by centrifugation at 412g for 10 min, washed twice with PBS, and suspended in the lysis buffer provided from R&D Systems for whole cell lysates. The homogenates were centrifuged at 2000g for 5 min and the supernatants were used for the ELISA. The amount of protein in the sample was determined to be 60 μg/100 μl. The amounts of phosphorylated Akt were measured, according to the manufacturer’s instructions for the immunoassays. The supernatants were treated with the antibodies for the phosphorylated forms of the kinases, next with the biotinylated detection antibodies, and finally with streptavidin-horseradish-peroxidase. The absorbance was measured at 405 nm.
Measurement of intracellular reactive oxygen species production
The dye 2′,7′-dichlorofluorescin-diacetate (DCFH2-DA), which is oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF) by hydroperoxides, was used to determine relative levels of cellular peroxides (Fu et al. 1998). Keratinocytes (1 × 106 cells/ml of medium in 24-well plate) were treated with lipopolysaccharide 24 h at 37 °C. The cells were washed, suspended in keratinocyte-serum free media, treated with 50 μM dye for 30 min at 37 °C, and washed with PBS. The cell suspensions were centrifuged at 412g for 10 min and the medium was removed. The cells were lysed with 100 μl of dimethyl sulfoxide, and the fluorescence was measured at an excitation wavelength 485 nm and an emission wavelength 530 nm using a fluorescence microplate reader (SPECTRAFluor, Tecan, Salzburg, Austria).
Measurement of nitrite/nitrate production
The nitric oxide liberated from keratinocytes was assayed by measuring nitric oxide metabolites, nitrite, and nitrate (NO x ). Keratinocytes (1 × 105 cells/400 μl in 24-well plate) were treated with lipopolysaccharide for 24 h at 37 °C. The nitrate in the mixture was reduced to nitrite by being treated with 500 mU/ml nitrate reductase, 160 μM NADPH, and 4 μM flavin adenine dinucleotide at room temperature for 2 h. The mixture was treated with an equal amount of Griess reagent (Sigma-Aldrich Inc.). The absorbance of mixture was measured at 540 nm, and the amount of nitrite was assayed using sodium nitrite as the standard. The results were expressed as total nitrite equivalents (NO x ).
Measurement of cell viability
Cell viability was measured using an MTT reduction assay, which is based on the conversion of MTT to formazan crystals by mitochondrial dehydrogenases (Mosmann 1983). HEK001 keratinocytes (3 × 104 cells/200 μl) were treated with apigenin for 24 h at 37 °C. The mixture (200 μl) was treated with 10 μl of a 10 mg/ml MTT solution for 2 h at 37 °C. After centrifugation at 412g for 10 min, the mixture was removed and 100 μl of dimethyl sulfoxide was added to well plates to dissolve the formazan. The absorbance of mixture was measured at 570 nm using a microplate reader (Magellan, Tecan, Salzburg, Austria). Cell viability was expressed as a percentage of the value in untreated control cells.
Statistical analysis
The data are expressed as the mean ± SEM. The data from these experiments were analyzed using a randomized block design analysis of variance (ANOVA) with Tukey’s post hoc test. A probability value of p < 0.05 was considered statistically significant.
Results
Apigenin suppressed the production of the inflammatory mediators
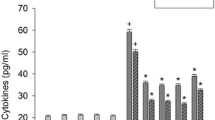
The apigenin effect on the cytokine production in keratinocytes treated with lipopolysaccharide was examined. Apigenin reduced the lipopolysaccharide-stimulated cytokine IL-1β and IL-6 production in HEK001 keratinocytes (Fig. 1a). It had a maximal inhibition of approximately 68–74% at 10 μM; beyond this concentration, the effect somewhat declined. Apigenin alone less than 15 μM did not significantly induce cytokine production in cells not treated with lipopolysaccharide. Apigenin at 15 μM alone increased the levels of cytokines, which was not statistically significant, compared to control values. Apigenin also inhibited the lipopolysaccharide-caused increase in the levels of an inducible enzyme COX-2, which plays a critical role in inflammation (Fig. 1c). We examined whether the lipopolysaccharide-stimulated inflammatory mediator production was regulated by the NF-κB, Akt, and mTOR pathways. Bay 11-7085 (2.5 μM), 0.5 μM Akt inhibitor, and 0.5 μM mTOR inhibitor rapamycin reduced the lipopolysaccharide-caused increase in the levels of IL-1β, IL-6, and COX-2 (Fig. 1b, c). We examined the combined effect of Bay 11-7085, Akt inhibitor, and rapamycin on the production of cytokines and COX. The inhibitors alone reduced lipopolysaccharide-stimulated production of cytokines and COX by 50–62, 50–64, and 53–63%, respectively, in HEK001 keratinocytes and primary keratinocytes. However, the combination reduced lipopolysaccharide-stimulated production of cytokines and COX by 88–90%. We examined whether lipopolysaccharide-stimulated cytokine production was mediated by reactive oxygen species. One millimolar thiol antioxidant N-acetylcysteine reduced the lipopolysaccharide-caused increase in the levels of inflammatory mediators (Fig. 1b, c). Signaling inhibitors and N-acetylcysteine alone did not induce the production of inflammatory mediators in HEK001 keratinocytes.

Apigenin suppressed the production of cytokines and increases in the levels of COX-2. In a, HEK001 keratinocytes were pretreated with 1–15 μM apigenin for 20 min and then treated with lipopolysaccharide (LPS) in combination with apigenin for 24 h. In b–d, HEK001 keratinocytes were pretreated with 2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, 0.5 μM rapamycin, 1 mM N-acetylcysteine (NAC), 20 μM SP600125, or 10 μM SB203580 for 20 min and then treated with lipopolysaccharide (LPS) in combination with compounds for 24 h. The amounts of IL-1β and IL-6 and the levels of COX-2 were measured using ELISA kits. Data indicate the mean ± SEM, n = 6. + p < 0.05 compared to the control; *p < 0.05 compared to lipopolysaccharide alone. BAR indicates combination of Bay 11-7085, Akt inhibitor, and rapamycin, and Ji-p38i indicates combination of JNK inhibitor and p38 inhibitor
We examined whether the lipopolysaccharide-stimulated inflammatory mediator production was mediated by activation of the JNK and p38-MAPK. Twenty micromolars JNK inhibitor SP600125 and 10 μM p38-MAPK inhibitor SB203580 reduced the IL-1β production (Fig. 1d). Alone, the inhibitors did not affect IL-1β production. We examined the combined effect of JNK inhibitor and p38 inhibitor on IL-1β production. The inhibitors alone reduced lipopolysaccharide-stimulated IL-1β production by 32–33 and 37–38%, respectively, in HEK001 keratinocytes and primary keratinocytes. However, the combination reduced lipopolysaccharide-stimulated IL-1β production by 61–63%.
In primary keratinocytes treated with lipopolysaccharide, the apigenin effect on the IL-1β production was investigated. Apigenin reduced the lipopolysaccharide-stimulated IL-1β production (Fig. 2a). In primary keratinocytes, apigenin had a maximal inhibition at 10 μM; beyond this concentration, the effect declined. We examined whether the lipopolysaccharide-stimulated cytokine production was mediated by the NF-κB, Akt, and mTOR signaling pathways and by reactive oxygen species. Bay 11-7085 (2.5 μM), 0.5 μM Akt inhibitor, 0.5 μM rapamycin, and 1 mM N-acetylcysteine reduced the lipopolysaccharide-stimulated IL-1β production (Fig. 2b). Alone, the inhibitors and N-acetylcysteine did not affect the IL-1β levels. Lastly, we examined the involvement of JNK and p38 in IL-1β production. SP600125 (20 μM) and 10 μM SB203580 reduced the IL-1β production (Fig. 2b). Alone, the inhibitors did not induce the IL-1β production.

Apigenin, signaling inhibitors, and N-acetylcysteine suppressed IL-1β production in primary keratinocytes. In a, primary keratinocytes were pretreated with 1–15 μM apigenin for 20 min and then treated with lipopolysaccharide (LPS) in combination with apigenin for 24 h. In b, primary keratinocytes were pretreated with 2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, 0.5 μM rapamycin, 1 mM N-acetylcysteine (NAC), 20 μM SP600125, or 10 μM SB203580 for 20 min and then treated with lipopolysaccharide (LPS) in combination with compounds for 24 h. The amounts of IL-1β were measured using ELISA kit. Data indicate the mean ± SEM, n = 6. + p < 0.05 compared to the control; *p < 0.05 compared to lipopolysaccharide alone. BAR indicates combination of Bay 11-7085, Akt inhibitor, and rapamycin, and Ji-p38i indicates combination of JNK inhibitor and p38 inhibitor
The apigenin effect on the lipopolysaccharide-stimulated chemokine production in keratinocytes was examined. Apigenin reduced the lipopolysaccharide-stimulated production of chemokines CCL27 and CCL17 in HEK001 keratinocytes (Fig. 3a). It had a maximal inhibition at 10 μM; beyond this concentration, the effect somewhat declined. Apigenin at 15 μM alone increased the levels of chemokines, which was not statistically significant, compared to control values. We examined whether the lipopolysaccharide-stimulated chemokine production was regulated by the NF-κB, Akt, and mTOR pathways and by reactive oxygen species. Bay 11-7085 (2.5 μM), 0.5 μM Akt inhibitor, 0.5 μM rapamycin, and 1 mM N-acetylcysteine reduced the lipopolysaccharide-stimulated production of CCL17 and CCL27 (Fig. 3b). Alone, the inhibitors did not alter the chemokine production. Lastly, we examined the involvement of JNK and p38 in CCL17 production. SP600125 (20 μM) and 10 μM SB203580 reduced the CCL17 production (Fig. 3c). Alone, the inhibitors did not affect CCL17 production. We examined the combined effect of Bay 11-7085, Akt inhibitor, and rapamycin on chemokine production. The inhibitors alone reduced lipopolysaccharide-stimulated chemokine production by 56–57, 51–52, and 61–62%, respectively, in HEK001 keratinocytes. However, the combination reduced lipopolysaccharide-stimulated chemokine production by 89–90%. Next, we examined the combined effect of JNK inhibitor and p38 inhibitor on CCL17 production. The inhibitors alone reduced lipopolysaccharide-stimulated IL-1β production by 37 and 41%, respectively, in HEK001 keratinocytes. However, the combination reduced lipopolysaccharide-stimulated IL-1β production by 68%.

Apigenin suppressed the chemokine production. In a, HEK001 keratinocytes were pretreated with 1–15 μM apigenin for 20 min and then treated with 1 μg/ml lipopolysaccharide (LPS) in combination with apigenin for 24 h. In b and c, HEK001 keratinocytes were pretreated with 2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, 0.5 μM rapamycin, 1 mM N-acetylcysteine (NAC), 20 μM SP600125, or 10 μM SB203580 for 20 min and then treated with lipopolysaccharide (LPS) in combination with compounds for 24 h. The amounts of CCL17 and CCL27 were measured using ELISA kits. Data indicate the mean ± SEM, n = 6. + p < 0.05 compared to the control; *p < 0.05 compared to lipopolysaccharide alone. BAR indicates combination of Bay 11-7085, Akt inhibitor, and rapamycin, and Ji-p38i indicates combination of JNK inhibitor and p38 inhibitor
Apigenin suppressed the increase in TLR-4 levels and activation of NF-κB
We examined the apigenin effect on the activation of the signaling pathway at the concentration 7.5 μM, which did not affect cell viability. Lipopolysaccharide increased the TLR-4 levels, which was inhibited by the addition of 5–10 μM apigenin and 1 mM N-acetylcysteine (Fig. 4a). Next, we investigated whether the apigenin effect on the lipopolysaccharide-stimulated inflammatory mediator production in keratinocytes was achieved by reducing NF-κB activation. Lipopolysaccharide caused an increase in the levels of NF-κB p65, NF-κB p50, and phosphorylated IκB in HEK001 keratinocytes, which was inhibited by the addition of 7.5 μM apigenin, 2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, and 0.5 μM rapamycin (Fig. 4b).

Apigenin suppressed the increases in the TLR-4 levels and activation of NF-κB. HEK001 keratinocytes were pretreated with 5–10 μM apigenin, 2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, 0.5 μM rapamycin, or 1 mM N-acetylcysteine (NAC) for 20 min and treated with 1 μg/ml lipopolysaccharide in combination with compounds for 30 min (for NF-κB assay) or 24 h (for TLR-4 assay). In a and b, the levels of TLR-4, NF-κB p65, NF-κB p50, and phosphorylated IκB-α were detected by Western blotting with specific antibodies. Data indicate representative of three to four independent experiments. The densities of protein bands were determined using TINA 2.10g software, and data indicate a fold increase from the control density. In c, the NF-κB p65 to DNA binding activity was measured using an assay kit. Data indicate the mean ± SEM, n = 5. + p < 0.05 compared to the control; *p < 0.05 compared lipopolysaccharide alone. BAR indicates combination of Bay 11-7085, Akt inhibitor, and rapamycin, and Ji-p38i indicates combination of JNK inhibitor and p38 inhibitor
The suppressive effect of apigenin on the lipopolysaccharide-caused NF-κB activation was confirmed by measuring the binding of NF-κB p65 to DNA. Nonstimulated cells showed a small increase in the NF-κB p65 to DNA binding. Lipopolysaccharide increased the NF-κB p65-DNA binding activity. Apigenin, Bay 11-7085, the Akt inhibitor, rapamycin, and N-acetylcysteine inhibited the lipopolysaccharide-caused increase in the NF-κB p65 to DNA binding activity (Fig. 4c). Alone, the compounds did not increase the binding of NF-κB p65 to DNA.
We examined the combined effect of Bay 11-7085, Akt inhibitor, and rapamycin on the NF-κB p65 to DNA binding activity. The inhibitors alone reduced lipopolysaccharide-caused increase in the NF-κB p65 to DNA binding activity by 51, 77, and 62%, respectively, in HEK001 keratinocytes. However, the combination reduced lipopolysaccharide-caused increase in the NF-κB p65 to DNA binding activity by 91%. Next, we examined the combined effect of the JNK inhibitor and p38 inhibitor on the NF-κB p65 to DNA binding activity. The inhibitors alone reduced lipopolysaccharide-caused increase in the NF-κB p65 to DNA binding activity by 32 and 37%, respectively, in HEK001 keratinocytes. However, the combination reduced lipopolysaccharide-caused increase in the NF-κB p65 to DNA binding activity by 57%.
Apigenin suppressed the activation of JNK and p38-MAPK
Lipopolysaccharide-caused activation of the JNK and p38-MAPK was investigated by monitoring Western blot finding. Lipopolysaccharide increased the levels of phosphorylated JNK and phosphorylated p38 in HEK001 keratinocytes (Fig. 5a). Apigenin, 2.5 μM Bay 11-7085, 20 μM SP600125, and 10 μM SB203580 reduced the lipopolysaccharide-caused phosphorylation of the JNK and p38 in HEK001 keratinocytes (Fig. 5a). Next, we examined whether the lipopolysaccharide-caused increase in the binding of NF-κB p65 to DNA was regulated by JNK and p38. Twenty micromolars SP600125 and 10 μM SB203580 reduced the lipopolysaccharide-caused increase in the NF-κB p65 to DNA binding in HEK001 keratinocytes (Fig. 5b). Alone, the inhibitors did not affect the NF-κB p65-DNA binding activity.

Apigenin suppressed the activation of JNK and p38. In a, HEK001 keratinocytes were pretreated with 20 μM SP600125 or 10 μM SB203580 for 20 min and then treated with 1 μg/ml lipopolysaccharide in combination with SP600125 or SB203580 for 4 h. The levels of JNK, phosphorylated JNK, p38, and phosphorylated p38 were detected by Western blotting with specific antibodies. The densities of protein bands were determined using TINA 2.10g software, and data indicate a fold increase from the control density. Data indicate representative of three to four different experiments. In b, the NF-κB p65 to DNA binding activity was measured using an assay kit. Data indicate the mean ± SEM, n = 6. + p < 0.05 compared to the control; *p < 0.05 compared to lipopolysaccharide alone
Apigenin suppressed Akt activation and increase in mTOR levels
We examined whether the lipopolysaccharide-stimulated inflammatory mediator production was regulated by the Akt/mTOR pathway. To define the inhibitory effect of apigenin, we examined the apigenin effect on the changes in the levels of phosphorylated Akt at a 4-h exposure to lipopolysaccharide. Apigenin, the Akt inhibitor, and N-acetylcysteine inhibited the lipopolysaccharide-caused activation of Akt, i.e., increase in the phosphorylated Akt1 levels, while lipopolysaccharide did not affect the levels of Akt1 (upper part in Fig. 6a). We further examined whether the apigenin reduced Akt phosphorylation. Apigenin, the Akt inhibitor, and N-acetylcysteine inhibited the lipopolysaccharide-caused phosphorylation of Akt (lower part in Fig. 6a). Alone, apigenin, the Akt inhibitor, and N-acetylcysteine did not cause phosphorylation of Akt.

Apigenin suppressed changes in the levels of phosphorylated Akt and mTOR. In a, keratinocytes were treated with 1 μg/ml lipopolysaccharide in combination with compounds (7.5 μM apigenin, 0.5 μM Akt inhibitor, and 1 mM N-acetylcysteine (NAC)) for 4 h. Next, the levels of phosphorylated Akt1 and the phosphorylation of Akt were detected by Western blotting and ELISA kit. In Western blotting, the densities of protein bands were determined using TINA 2.10g software, and data represent a fold increase from the control density. Data indicate representative of three to four independent experiments. Data represent the mean ± SEM, n = 6. + p < 0.05 compared to the control; *p < 0.05 compared to lipopolysaccharide alone. In b, HEK001 keratinocytes were treated with 1 μg/ml lipopolysaccharide in combination with compounds (7.5 μM apigenin 0.5 μM Akt inhibitor, 0.5 μM rapamycin, and 1 mM N-acetylcysteine (NAC)) for 4 h, and the levels of mTOR were detected by Western blotting and data indicate representative of three to four independent experiments
We examined whether the lipopolysaccharide-stimulated inflammatory mediator production was regulated by mTOR signaling. HEK001 keratinocytes treated with lipopolysaccharide exhibited an increase in the levels of mTOR. Apigenin, the Akt inhibitor, rapamycin, and N-acetylcysteine inhibited the increase in the levels of mTOR (Fig. 6b).
Apigenin reduced the production of reactive oxygen species and nitric oxide
The production of reactive oxygen species within keratinocytes treated with lipopolysaccharide cells was measured by measuring a conversion of DCFH2-DA to fluorescent DCF. HEK001 keratinocytes treated with lipopolysaccharide showed an increase in DCF fluorescence. Apigenin and the oxidant scavengers (N-acetylcysteine and 40 μM trolox (a cell-permeable, water-soluble analog of vitamin E with quenching effect on free radicals) inhibited the lipopolysaccharide-caused increase in DCF fluorescence (Fig. 7a). Alone, the oxidant scavengers did not induce changes in DCF fluorescence.

Apigenin suppressed the production of reactive oxygen species and nitric oxide. In a, HEK001 keratinocytes were treated with 1 μg/ml lipopolysaccharide in combination with compounds (7.5 μM apigenin, 1 mM N-acetylcysteine (NAC), and 40 μM trolox) for 24 h. The increase in DCF fluorescence, which indicates production of reactive oxygen species production, was measured and data indicate arbitrary units (a.u.) of fluorescence. In b, HEK001 keratinocytes were treated with 1 μg/ml lipopolysaccharide in combination with compounds (7.5 μM apigenin, 1 mM N-acetylcysteine (NAC), 50 μM rutin (nitric oxide synthase inhibitor), 50 μM carboxy-PTIO, and 500 μM L-NMMA (nonselective inhibitor of all nitric oxide synthase isoforms) for 24 h. The amounts of NO x , the nitric oxide metabolites nitrite and nitrate, were measured and data indicate mean ± SEM, n = 6. + p < 0.05 compared to control; *p < 0.05 compared to lipopolysaccharide alone
The production of nitric oxide in keratinocytes treated with lipopolysaccharide was examined. HEK001 keratinocytes treated with lipopolysaccharide for 24 h liberated 4.50 ± 0.10 μM NO x , the nitric oxide metabolites nitrite and nitrate (mean ± SEM, n = 6). Apigenin, N-acetylcysteine, 50 μM rutin (nitric oxide synthase inhibitor), 50 μM carboxy-PTIO (nitric oxide scavenger), and 500 μM L-NMMA (nitric oxide synthase inhibitor) inhibited the lipopolysaccharide-caused NO x production (Fig. 7b). The compounds alone did not induce changes in the NO x levels.
Apigenin (up to 10 μM) did not cause cell death
To assess whether the inhibitory effect of apigenin on the stimulated keratinocyte responses can be affected by changes in cell viability, we examined the cytotoxicity of apigenin using the MTT assay, which provides rapid and precise data for cellular growth and survival. When the keratinocytes were treated with apigenin for 24 h, approximately 5 and 9% of cell death at 10 and 15 μM were detected, respectively (Fig. 8).

Apigenin (up to 10 μM) did not cause cell death. Keratinocytes were treated with 1–15 μM of apigenin for 24 h and then cell viability was measured using the MTT reduction assay. Data indicate mean ± SEM, n = 6. + p < 0.05 compared to the control (percentage of control)
Discussion
Apigenin has been shown to exhibit anti-inflammatory and regulatory effect on immune responses. However, the apigenin effect on the lipopolysaccharide-stimulated inflammatory mediator production in keratinocytes has not been studied. Apigenin inhibited the lipopolysaccharide-stimulated production of cytokines IL-1β and IL-6 and chemokines CCL17 and CCL27 and the increase in the levels of an inducible inflammatory enzyme COX-2 in HEK001 keratinocytes and primary keratinocytes. Proinflammatory cytokines and chemokines are involved in skin inflammation and immune reaction (Schaerli and Moser 2005; Pastore et al. 2006). COX-2 upregulated during inflammatory states produces the prostanoids responsible for the generation of pain and inflammation (Vane and Botting 1998). These results indicate that apigenin may reduce the inflammatory and immune reactions in skin by reducing the production of cytokines and chemokines and the expression of COX-2.
Lipopolysaccharide stimulates the production of proinflammatory mediators through the TLR-4-mediated NF-κB activation (Song et al. 2002; Lee et al. 2010; Ge et al. 2012). The phosphorylation and proteolytic degradation of IκB and, subsequently, the translocation of NF-κB dimers to the nucleus trigger the activation of target genes responsible for the synthesis of cytokines and chemokines (Ghosh and Hayden 2008; Napetschnig and Wu 2013). In the present study, lipopolysaccharide increased the levels of the TLR-4, the levels of phosphorylated IκB and NF-κB p65/50, and the binding of NF-κB p65 to DNA in HEK001 keratinocytes. These results show that lipopolysaccharide stimulates the production of inflammatory mediators by causing the TLR-4-dependent NF-κB pathway activation, i.e., the nuclear translocation and binding of activated NF-κB dimers to DNA at specific sites. Connectively, apigenin may inhibit the lipopolysaccharide-stimulated inflammatory mediator production by reducing the TLR-4-mediated NF-κB activation.
The PI3K, Akt, and mTOR pathways regulate NF-κB activation (Lee et al. 2008; Thomson et al. 2009; Zhong et al. 2012). Lipopolysaccharide has been shown to cause NF-κB activation by activating the PI3K/Akt/mTOR pathways (Thomson et al. 2009; Zhong et al. 2012; Ma et al. 2015). We examined whether the lipopolysaccharide-caused NF-κB activation-mediated inflammatory mediator production was regulated by the Akt and mTOR signaling in keratinocytes. Apigenin, the Akt inhibitor, and rapamycin inhibited the lipopolysaccharide-caused increases in the levels of phosphorylated Akt and mTOR, activation of NF-κB, and production of inflammatory mediators. These results suggest that apigenin may inhibit the lipopolysaccharide-stimulated inflammatory mediator production by reducing the activation of the Akt, mTOR, and NF-κB pathways.
Reactive oxygen and nitrogen species, including nitric oxide, have been shown to be engaged in the physiological regulation of cellular functions and also in pathologic conditions such as chronic inflammatory diseases (Gloire et al. 2006; Sharma et al. 2007; Sun and Zhang 2007; Siomek 2012). Lipopolysaccharide stimulates the production of reactive oxygen and nitrogen species (Hsu and Wen 2002; Lee et al. 2003; Sharma et al. 2007). The thiol antioxidant N-acetylcysteine inhibits lipopolysaccharide-stimulated cytokine production by reducing reactive oxygen species production (Hsu and Wen 2002). In the present study, N-acetylcysteine inhibited the lipopolysaccharide-caused increases in the levels and activity of TLR-4, phosphorylated Akt, and mTOR; NF-κB activation; and production of reactive oxygen/nitrogen species. It has been shown that reactive oxygen species activate NF-κB through PI3K/Akt signaling pathways (Korbecki et al. 2013). Thus, the present study suggests that the lipopolysaccharide-caused activation of the Akt, mTOR, and NF-κB pathways may be achieved by the production of reactive oxygen species and nitrogen species. Apigenin attenuates the lipopolysaccharide-stimulated release of 8-isoprostane, a marker of oxidative stress (Lim et al. 2013). Unlike this report, apigenin at 25 μM increases the proinflammatory cytokine-caused production of peroxides and nitric oxide (Crespo et al. 2008). Thus, it is unclear whether the inhibitory effect of apigenin on the NF-κB-mediated inflammatory responses is associated with inhibition of reactive oxygen species production. In the present study, apigenin and the oxidant scavengers inhibited the lipopolysaccharide-caused activation of Akt, mTOR, and NF-κB pathways and the production of reactive oxygen species and nitric oxide. These results suggest that apigenin may inhibit the Akt, mTOR, and NF-κB activation-stimulated inflammatory mediator production by reducing the reactive oxygen species and nitric oxide production.
In the present study, the combination of Bay 11-7085, Akt inhibitor, and rapamycin did not completely inhibit the lipopolysaccharide-caused production of cytokines, COX, and chemokines and the binding of NF-κB p65 to DNA. These findings suggest that in addition to the Akt, mTOR, and NF-κB pathways, other regulatory processes appear to be involved in the lipopolysaccharide-caused inflammatory production.
We examined whether the apigenin effect on the inflammatory mediator production may be attributed to cytotoxic effect. Apigenin caused cell death by approximately 3 and 5% at 7.5 and 10 μM in HEK001 keratinocytes, respectively. Apigenin caused cell death by approximately 9% at 15 μM in keratinocytes. Thus, apigenin (up to 10 μM) may reduce the production of inflammatory mediators, production of reactive oxygen species, and activation of signaling pathways without influence on cell viability. However, the apigenin effect at higher concentrations appears to be affected by cell viability changes.
The MAPK subfamilies JNK and p38-MAPK play a critical role in the maintenance of inflammation and innate immunity (Kim and Choi 2015; Kim et al. 2016; Liu et al. 2016). Lipopolysaccharide stimulates the expression of inducible nitric oxide synthase (iNOS), COX-2, and proinflammatory cytokines in microglial cells through the activation of NF-κB and JNK/p38-MAPKs/Akt pathways (Kim et al. 2016; Liu et al. 2016). Lipopolysaccharide induces the production of proinflammatory mediators and expressions of iNOS and COX-2 in RAW264.7 cell lines through activation of JNK and p38-MAPK (Park et al. 2016). Lipopolysaccharide induces the cytokine expression in RAW264.7 cells by activating the NF-κB, p38-MAPK, JNK, and ERK (Wu et al. 2016).
In this respect, we examined whether apigenin can inhibit lipopolysaccharide-stimulated inflammatory mediator production by affecting JNK and p38-MAPK activation. HEK001 keratinocytes treated with lipopolysaccharide showed an increase in the cytosolic levels of phosphorylated JNK and phosphorylated p38. Apigenin, N-acetylcysteine, the JNK inhibitor (SP600125), and the p38-MAPK inhibitor (SB203580) reduced the lipopolysaccharide-stimulated production of the cytokines and chemokines and the increase in the cytosolic and nuclear levels of NF-κB p65, NF-κB p50, phosphorylated IκB, phosphorylated JNK, and phosphorylated p38. These results suggest that apigenin may reduce the lipopolysaccharide-stimulated inflammatory mediator production in keratinocytes by reducing JNK and p38-MAPK activation. Further, the inhibitory effect of N-acetylcysteine on JNK and p38-MAPK activation indicates that the lipopolysaccharide-caused activation of JNK and p38-MAPK appears to be achieved by reactive oxygen species production in keratinocytes. Thus, the suppressive effect of apigenin on JNK and p38-MAPK activation may be achieved by the inhibition of reactive oxygen/nitrogen species production.
In the present study, the combination of JNK inhibitor and p38 inhibitor did not completely inhibit lipopolysaccharide-caused production of IL-1β and CCL17 and the binding of NF-κB p65 to DNA. These finding suggests that along with the JNK and p38-MAPK signaling, other regulatory processes appear to be involved in the lipopolysaccharide-caused inflammatory production.
Apigenin has been shown to suppress lipopolysaccharide-stimulated IL-1β mRNA expression in human periodontal ligament cells (Pumklin et al. 2016). Apigenin attenuates lipopolysaccharide-induced expression of cytokine, chemokine, and COX-2 mRNA in IPEC-J2 nontransformed intestinal epithelial cells (Farkas et al. 2015). These reports suggest that apigenin may interfere with lipopolysaccharide-caused inflammatory production at a very early step in the lipopolysaccharide-acting pathway.
Overall, the results show that apigenin may reduce the lipopolysaccharide-caused proinflammatory mediator production in keratinocytes by reducing the TLR-4-dependent activation of the Akt, mTOR, and NF-κB pathways, and activation of the JNK and p38-MAPK. The suppressive effect of apigenin on the activation of signaling transduction pathways and production of inflammatory mediators appears to be achieved by the inhibition of reactive oxygen/nitrogen species production. Additionally, apigenin appears to reduce the Akt, mTOR, and NF-κB pathway- and the JNK and p38-MAPK-mediated inflammatory skin diseases.
References
Baker BS (2006) The role of microorganisms in atopic dermatitis. Clin Exp Immunol 144(1):1–9. https://doi.org/10.1111/j.1365-2249.2005.02980.x
Begon E, Michel L, Flageul B, Beaudoil I, Jean-Louis F, Bachelez H, Dubertret L, Musette P (2007) Expression, subcellular localization and cytokinic modulation of Toll-like receptors (TLRs) in normal human keratinocytes: TLR2 up-regulation in psoriatic skin. Eur J Dermatol 17(6):497–506. https://doi.org/10.1684/ejd.2007.0264
Bi C, Jiang Y, Fu T, Hao Y, Zhu X, Lu Y (2016) Naringin inhibits lipopolysaccharide-induced damage in human umbilical vein endothelial cells via attenuation of inflammation, apoptosis and MAPK pathways. Cytotechnology 68(4):1473–1487. https://doi.org/10.1007/s10616-015-9908-3
Crespo I, García-Mediavilla MV, Almar M, González P, Tuñón MJ, Sánchez-Campos S, González-Gallego J (2008) Differential effects of dietary flavonoids on reactive oxygen and nitrogen species generation and changes in antioxidant enzyme expression induced by proinflammatory cytokines in Chang liver cells. Food Chem Toxicol 46(5):1555–1569. https://doi.org/10.1016/j.fct.2007.12.014
Farkas O, Palócz O, Pászti-Gere E, Gálfi P (2015) Polymethoxyflavone apigenin-trimethylether suppresses LPS-induced inflammatory response in nontransformed porcine intestinal cell line IPEC-J2. Oxidative Med Cell Longev 2015:1–10. https://doi.org/10.1155/2015/673847
Fu W, Luo H, Parthasarathy S, Mattson MP (1998) Catecholamines potentiate amyloid β-peptide neurotoxicity: involvement of oxidative stress, mitochondrial dysfunction, and perturbed calcium homeostasis. Neurobiol Dis 5(4):229–243. https://doi.org/10.1006/nbdi.1998.0192
Ge Y, Xu Y, Sun W, Man Z, Zhu L, Xia X, Zhao L, Zhao Y, Wang X (2012) The molecular mechanisms of the effect of dexamethasone and cyclosporin A on TLR4/NF-κB signaling pathway activation in oral lichen planus. Gene 508(2):157–164. https://doi.org/10.1016/j.gene.2012.07.045
Ghosh S, Hayden MS (2008) New regulators of NF-κB in inflammation. Nat Rev Immunol 8(11):837–848. https://doi.org/10.1038/nri2423
Gloire G, Legrand-Poels S, Piette J (2006) NF-κB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72(11):1493–1505. https://doi.org/10.1016/j.bcp.2006.04.011
Hsu HY, Wen MH (2002) Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem 277(25):22131–22139. https://doi.org/10.1074/jbc.M111883200
Huang CH, Kuo PL, Hsu YL, Chang TT, Tseng HI, Chu YT, Kuo CH, Chen HN, Hung CH (2010) The natural flavonoid apigenin suppresses Th1- and Th2-related chemokine production by human monocyte THP-1 cells through mitogen-activated protein kinase pathways. J Med Food 13(2):391–398. https://doi.org/10.1089/jmf.2009.1229
Jin SE, Lim HS, Kim Y, Seo CS, Yoo SR, Shin HK, Jeong SJ (2015) Traditional herbal formula banhasasim-tang exerts anti-inflammatory effects in RAW 264.7 macrophages and HaCaT keratinocytes. Evid Based Complement Alternat Med 2015:728380
Kim EK, Choi EJ (2015) Compromised MAPK signaling in human diseases: an update. Arch Toxicol 89(6):867–882. https://doi.org/10.1007/s00204-015-1472-2
Kim JE, Kim BJ, Jeong MS, Seo SJ, Kim MN, Hong CK, Ro BI (2005) Expression and modulation of LL-37 in normal human keratinocytes, HaCaT cells and inflammatory skin diseases. J Korean Med Sci 20(4):649–654. https://doi.org/10.3346/jkms.2005.20.4.649
Kim AR, Lee B, Joung EJ, Gwon WG, Utsuki T, Kim NG, Kim HR (2016) 6,6′-Bieckol suppresses inflammatory responses by down-regulating nuclear factor-κB activation via Akt, JNK, and p38 MAPK in LPS-stimulated microglial cells. Immunopharmacol Immunotoxicol 38(3):244–252. https://doi.org/10.3109/08923973.2016.1173060
Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D (2013) The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol 64(4):409–421
Lee AK, Sung SH, Kim YC, Kim SG (2003) Inhibition of lipopolysaccharide-inducible nitric oxide synthase, TNF-α and COX-2 expression by sauchinone effects on I-κBα phosphorylation, C/EBP and AP-1 activation. Br J Pharmacol 139(1):11–20. https://doi.org/10.1038/sj.bjp.0705231
Lee SA, Park SH, Kim BC (2008) Raloxifene, a selective estrogen receptor modulator, inhibits lipopolysaccharide-induced nitric oxide production by inhibiting the phosphatidylinositol 3-kinase/Akt/NF-κB pathway in RAW264.7 macrophage cells. Mol Cells 26(1):48–52
Lee CS, Jang ER, Kim YJ, Lee MS, Seo SJ, Lee MW (2010) Hirsutenone inhibits lipopolysaccharide-activated NF-κB-induced inflammatory mediator production by suppressing Toll-like receptor 4 and ERK activation. Int Immunopharmacol 10(4):520–525. https://doi.org/10.1016/j.intimp.2010.01.015
Lim R, Barker G, Wall CA, Lappas M (2013) Dietary phytophenols curcumin, naringenin and apigenin reduce infection-induced inflammatory and contractile pathways in human placenta, foetal membranes and myometrium. Mol Hum Reprod 19(7):451–462. https://doi.org/10.1093/molehr/gat015
Liu SY, Xu P, Luo XL, Hu JF, Liu XH (2016) (7R,8S)-dehydrodiconiferyl alcohol suppresses lipopolysaccharide-induced inflammatory responses in BV2 microglia by inhibiting MAPK signaling. Neurochem Res 23:541–549
Ma C, Zhu L, Wang J, He H, Chang X, Gao J, Shumin W, Yan T (2015) Anti-inflammatory effects of water extract of Taraxacum mongolicum hand.-Mazz on lipopolysaccharide-induced inflammation in acute lung injury by suppressing PI3K/Akt/mTOR signaling pathway. J Ethnopharmacol 168:349–355. https://doi.org/10.1016/j.jep.2015.03.068
Morris MC, Gilliam EA, Li L (2015) Innate immune programing by endotoxin and its pathological consequences. Front Immunol 5:680
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1–2):55–63. https://doi.org/10.1016/0022-1759(83)90303-4
Napetschnig J, Wu H (2013) Molecular basis of NF-κB signaling. Annu Rev Biophys 42(1):443–468. https://doi.org/10.1146/annurev-biophys-083012-130338
Nicholas C, Batra S, Vargo MA, Voss OH, Gavrilin MA, Wewers MD, Guttridge DC, Grotewold E, Doseff AI (2007) Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-kappaB through the suppression of p65 phosphorylation. J Immunol 179(10):7121–7127. https://doi.org/10.4049/jimmunol.179.10.7121
Park JY, Kim SI, Lee HJ, Kim SS, Kwon YS, Chun W (2016) Isorhamnetin-3-O-glucuronide suppresses JNK and p38 activation and increases heme-oxygenase-1 in lipopolysaccharide-challenged RAW264.7 cells. Drug Dev Res 77(3):143–151. https://doi.org/10.1002/ddr.21301
Pastore S, Mascia F, Girolomoni G (2006) The contribution of keratinocytes to the pathogenesis of atopic dermatitis. Eur J Dermatol 16:125–113
Patil RH, Babu RL, Naveen Kumar M, Kiran Kumar KM, Hegde SM, Nagesh R, Ramesh GT, Sharma SC (2016) Anti-inflammatory effect of apigenin on LPS-induced pro-inflammatory mediators and AP-1 factors in human lung epithelial cells. Inflammation 39(1):138–147. https://doi.org/10.1007/s10753-015-0232-z
Pumklin J, Bhalang K, Pavasant P (2016) Hypoxia enhances the effect of lipopolysaccharide-stimulated IL-1β expression in human periodontal ligament cells. Odontology 104(3):338–346. https://doi.org/10.1007/s10266-015-0223-4
Schaerli P, Moser B (2005) Chemokines: control of primary and memory T-cell traffic. Immunol Res 31(1):57–74. https://doi.org/10.1385/IR:31:1:57
Schreiber E, Matthias P, Müller MM, Schaffner W (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 17(15):6419. https://doi.org/10.1093/nar/17.15.6419
Sharma JN, Al-Omran A, Parvathy SS (2007) Role of nitric oxide in inflammatory diseases. Inflammopharmacology 6:252–259
Siomek A (2012) NF-κB signaling pathway and free radical impact. Acta Biochim Pol 59(3):323–331
Song PI, Park YM, Abraham T, Harten B, Zivony A, Neparidze N, Armstrong CA, Ansel JC (2002) Human keratinocytes express functional CD14 and toll-like receptor 4. J Investig Dermatol 119(2):424–432. https://doi.org/10.1046/j.1523-1747.2002.01847.x
Sun XF, Zhang H (2007) NFκB and NFκBI polymorphisms in relation to susceptibility of tumour and other diseases. Histol Histopathol 22(12):1387–1398. https://doi.org/10.14670/HH-22.1387
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17(1):1–14. https://doi.org/10.1093/intimm/dxh186
Thomson AW, Turnquist HR, Raimondi G (2009) Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 9(5):324–337. https://doi.org/10.1038/nri2546
Vane JR, Botting RM (1998) Anti-inflammatory drugs and their mechanism of action. Inflamm Res 47:S78–S87
Wang J, Liao Y, Fan J, Ye T, Sun X, Dong S (2012) Apigenin inhibits the expression of IL-6, IL-8, and ICAM-1 in DEHP-stimulated human umbilical vein endothelial cells and in vivo. Inflammation 35(4):1466–1476. https://doi.org/10.1007/s10753-012-9460-7
Wang J, Liu YT, Xiao L, Zhu L, Wang Q, Yan T (2014) Anti-inflammatory effects of apigenin in lipopolysaccharide-induced inflammatory in acute lung injury by suppressing COX-2 and NF-κB pathway. Inflammation 37(6):2085–2090. https://doi.org/10.1007/s10753-014-9942-x
Wu H, Zhao G, Jiang K, Chen X, Zhu Z, Qiu C, Li C, Deng G (2016) Plantamajoside ameliorates lipopolysaccharide-induced acute lung injury via suppressing NF-κB and MAPK activation. Int Immunopharmacol 35:315–322. https://doi.org/10.1016/j.intimp.2016.04.013
Zhong LM, Zong Y, Sun L, Guo JZ, Zhang W, He Y, Song R, Wang WM, Xiao CJ, Lu D (2012) Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS One 7(2):e32195. https://doi.org/10.1371/journal.pone.0032195
Zhu W, Li J, Liu Y, Xie K, Wang L, Fang J (2016) Mesencephalic astrocyte-derived neurotrophic factor attenuates inflammatory responses in lipopolysaccharide-induced neural stem cells by regulating NF-κB and phosphorylation of p38-MAPKs pathways. Immunopharmacol Immunotoxicol 38(3):205–213. https://doi.org/10.3109/08923973.2016.1168433
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kim, A., Lee, C.S. Apigenin reduces the Toll-like receptor-4-dependent activation of NF-κB by suppressing the Akt, mTOR, JNK, and p38-MAPK. Naunyn-Schmiedeberg's Arch Pharmacol 391, 271–283 (2018). https://doi.org/10.1007/s00210-017-1454-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-017-1454-4