
Abstract
Ilex Rotunda Thunb has been shown to have anti-inflammatory and antioxidant effects. In human keratinocytes, we investigated the effect of rotundarpene (4-caffeoyl-3-methyl-but-2-ene-1,4-diol) on the TNF-α-stimulated production of inflammatory mediators in relation to the Akt, mTOR, and NF-κB pathways, and the JNK and p38-MAPK. Rotundarpene, Akt inhibitor, Bay 11-7085, rapamycin, and N-acetylcysteine inhibited the TNF-α-stimulated production of cytokines and chemokines, increase in the levels of p-Akt and mTOR, activation of NF-κB, and production of reactive oxygen species in keratinocytes. TNF-α treatment induced phosphorylation of the JNK and p38-MAPK. Inhibitors of the c-JNK (SP600125) and p38-MAPK (SB203580) reduced the TNF-α-induced production of inflammatory mediators, binding of NF-κB to DNA, and activation of the JNK and p38-MAPK in keratinocytes. The results show that rotundarpene may reduce the TNF-α-stimulated inflammatory mediator production by suppressing the reactive oxygen species-dependent activation of the Akt, mTOR, and NF-κB pathways, and activation of the JNK and p38-MAPK in human keratinocytes. Additionally, rotundarpene appears to attenuate the Akt, mTOR, and NF-κB pathways and the JNK and p38-MAPK-mediated inflammatory skin diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tumor necrosis factor (TNF)-α is an inflammatory cytokine that is released by a variety of cell types, including immune effector cells and tissue-specific cells [1]. TNF-α is involved in the inflammation and immune response in inflammatory skin diseases [2, 3]. TNF-α activates the PI3K/Akt (protein kinase B) pathway [4–6] and regulates its downstream targets, mammalian target of rapamycin (mTOR), and FoxO3a [7]. The PI3K/Akt and mTOR signaling pathways regulate the activation of transcription factors, including activator protein-1 and nuclear factor (NF)-κB [8, 9]. NF-κB regulates genes responsible for the innate and adaptive immune responses as well as inflammation [10]. TNF-α binding to cell surface receptors induces activation of mitogen-activated protein (MAP) kinases, extracellular signal-regulated kinases (ERKs), the c-Jun NH2-terminal kinases (JNKs), and the p38-mitogen-activated protein kinase (p38-MAPK) [11, 12]. These MAP kinase pathways induce a secondary response by increasing the expression of inflammatory cytokines that contribute to the biological activity of TNFα [11]. TNF-α induces activation of JNK and p38-MAPK in HaCaT keratinocytes [13, 14].
The bark of Ilex rotunda Thunb is used for the treatment of colds, tonsillitis, sore throat, acute gastroenteritis, and dysentery in China [15, 16]. The main bioactive constituents of I. rotunda are triterpenoid saponins, phenolic acid compounds, and flavonoids. Trilex consisted of Ilex latifolia, Ilex asprella, and I. rotunda inhibits acetic acid-induced inflammatory exudation in mice abdominal cavity [15]. Syringin isolated from I. rotunda reduces carrageenan-induced paw edema in rats [17]. Hemiterpene glycosides from the leaves of I. rotunda Thunb exhibit an antioxidative activity by scavenging 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical and superoxide radicals [18]. Caffeoylquinic acid derivatives, such as 3,5-dicaffeoylquinic acid and 4,5-dicaffeoylquinic acid, are isolated from the barks of I. rotunda Thunb [18]. Caffeoyl derivatives isolated from Laggera alata inhibit acute and chronic inflammation in mice and rats [19]. Caffeoyl derivatives have a scavenging action on the DPPH radical and attenuate hydrogen peroxide-induced cell death [20, 21]. They inhibit the expression of inducible nitric oxide synthase and cyclooxygenase-2 and the production of nitric oxide in RAW264.7 macrophages and HaCat cells treated with lipopolysaccharide [21].
Ilex Rotunda Thunb has been shown to have anti-inflammatory and antioxidant effects. Nevertheless, whether the effect of rotundarpene (4-caffeoyl-3-methyl-but-2-ene-1,4-diol) on the TNF-α-induced activation of NF-κB is mediated by its effect on the Akt and mTOR pathways and the JNK and p38-MAPK has not been studied. We examined the effect of rotundarpene on TNF-α-induced inflammatory mediator production in relation to activation of the Akt, mTOR, and NF-κB pathways, and the JNK and p38-MAPK in human keratinocytes.
Materials and methods
Materials
Human tumor necrosis factor (TNF)-α (recombinant E. coli), Bay 11-7085 ((2E)-3-[[4-(1,1-Dimethylethyl)phenyl]sulfonyl]-2-propenenitrile), Akt inhibitor (type II, SH-5), rapamycin, SB203580 (a selective inhibitor of p38-MAPK), and horseradish peroxidase-conjugated anti-mouse IgG were purchased from EMD-Calbiochem. Co. (La Jolla, CA, USA). Human epidermal growth factor protein was purchased from Sino Biological Inc. (Beijing, People’s Republic of China and), Enzyme-linked immunosorbent assay (ELISA) kits for human CXCL1/IL1β, human IL6, human thymus and activation-regulated chemokine CCL17/TARC, human CTACK/CCL27, and human/mouse/rat phospho-Akt (Pan) were purchased from R&D systems, Inc. (Minneapolis, MN, USA). All antibodies used for Western blot assay were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). TransAM™ NF-κB assay kit was purchased from Active Motif® (Carlsbad, CA, USA). SP600125 (a selective inhibitor of c-JNK), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and other chemicals were purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA).
Extraction, isolation, and structural identification of rotundarpene
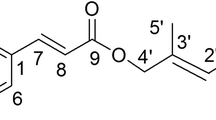
The rotundarpene (M.W. = 264) used in this study was isolated from the stems of I. rotunda (IR) Thunb, and the structural identity of the compound was characterized by spectral analyses, previously described [18]. Rotundarpene was provided from Prof. Min Won Lee (College of Pharmacy, Chung-Ang University, Seoul, South Korea).
Keratinocyte culture
Human keratinocytes (HEK001, tissue: skin; morphology: epithelial; cell type: human papillomavirus 16 E6/E7 transformed) were purchased from American Type Culture Collection (Manassas, VA) and cultured in keratinocyte SFM supplemented with bovine pituitary extract, recombinant epidermal growth factor, 100 U/ml penicillin, and 100 µg/ml streptomycin (GIBCO®, Invitrogen Co., Grand Island, NY, USA).
Normal human keratinocytes were provided by the Department of Urology, Chung-Ang University Hospital (Seoul, Korea). Keratinocytes were obtained and prepared from neonatal foreskin discarded after circumcision [22] in accordance with the Declaration of Helsinki Principles and the ethical guidelines of Chung-Ang University Hospital. Neonatal foreskin was chopped and split overnight in sucrose-trypsin solution (0.1% sucrose, 0.25% trypsin, and 1 mM EDTA) at 4 °C. Keratinocyte suspension was cultured in EpiLife® medium supplemented with growth factor (Cascade Biologics™, Portland, OR, USA).
Immunoassays for cytokines and chemokines
Keratinocytes (1 × 105 cells/300 μl for the cytokine assay and 5 × 105 cells/400 μl for the chemokine assay in a 24-well plate) were treated with 10 ng/ml TNF-α for 24 h. After centrifugation at 412×g for 10 min, the amounts of IL-1β, IL-6, and CCL27 in culture supernatants were analyzed using an enzyme-linked immunosorbent assay kit, according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a microplate reader (Spectra MAX 340, Molecular Devices Co., Sunnyvale, CA, USA).
Preparation of cytosolic and nuclear extracts
Keratinocytes (5 × 106 cells/ml) were treated with TNF-α for 15 min in the Western blot assay for NF-κB (4-h treatment with TNF-α for the Akt and mTOR assays) at 37 °C. Keratinocyte cytosolic and nuclear extracts were prepared according to the previously reported method [23]. Keratinocytes were harvested by centrifugation at 412×g for 10 min and washed twice with PBS. The cells were suspended in 400 μl lysis buffer (10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 0.5 mM PMSF, 1 mM sodium orthovanadate, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 10 mM HEPES–KOH, pH 7.8) and were allowed to swell on ice for 15 min. Afterward, 25 μl of a 10% Nonidet NP-40 solution (final approximately 0.6%) was added, and the tubes were vigorously vortexed for 10 s. The homogenates were centrifuged at 12,000×g for 10 min at 4°C. The supernatants were stored as cytoplasmic extracts and kept at -70 °C. The nuclear pellets were resuspended in 50-μl ice-cold hypertonic solution containing 5% glycerol and 0.4 M NaCl in lysis buffer. The tubes were incubated on ice for 30 min and then centrifuged at 12,000×g for 15 min at 4°C. The supernatants were collected as the nuclear extracts and stored at −70°C. Protein concentration was determined by the method of Bradford according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot for the levels of phophorylated-Akt1, mTOR, NF-κB, phosphorylated-JNK, and phosphorylated-p38
Treatment time with TNF-α for the assays of NF-κB, phosphorylated-Iκ,B and binding of NF-κB p65 to DNA was 15 min, and the assays of phophorylated-Akt1, phosphorylation of Akt, mTOR, phosphorylated-JNK, and phosphorylated-p38 were 4 h. Treatment time was determined according to the previous reports [24–26] and manufacturer’s instructions. TNF-α concentration used in the present study also was determined according to the previous reports.
The cytosolic fraction for the mTOR assay and the cytosolic and nuclear extracts for the NF-κB assay was mixed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and boiled for 5 min. Samples (20–0 μg protein/well) were loaded onto each lane of a 10–12% SDS–polyacrylamide gel, electrophoresis was carried out, and the proteins were transferred onto polyvinylidene difluoride membranes (GE Healthcare Chalfont St. Giles, Buckinghamshire, UK). Membranes were blocked for 2 h in TBS (50 mM Tris–HCl, pH 7.5, and 150 mM NaCl) containing 0.1% Tween 20. Each membrane was labeled with the appropriate antibody (NF-κB p65, NF-κB p50, phosphorylated-IκB-α, Akt1, phosphorylated-Akt, mTOR, JNK, phosphorylated-JNK, p38, and phosphorylated-p38). Membranes were treated with horseradish peroxidase-conjugated anti-mouse IgG for 2 h at room temperature. Membranes were treated with SuperSignal® West Pico chemiluminescence substrate and protein bands were visualized by detecting the enhanced chemiluminescence in an appropriate image analyzer (ImageQuantTM LAS4000, GE Healthcare Bio-Sciences AB, Björkgatan 30, 751 84 Uppsala Sweden).
The densities of protein bands were determined using TINA 2.10 g software licensed for Seoul National University (SNU and SNUMD, Seoul, South Korea) and were expressed as a fold increase compared to the control density.
Assay for DNA-binding activity of NF-κB p65
Biding of NF-κB p65 to DNA was determined according to the user’s manual for the transAM™ NF-κB kit. Keratinocytes (2 × 106 cells/ml) were treated with 10 ng/ml TNF-α for 15 min. Nuclear extracts were prepared according to the procedure described in the Active Motif® protocol and added to a 96-well plate to which oligonucleotides containing an NF-κB consensus-binding site (5′-GGGACTTTCC-3′) are immobilized. The active NF-κB p65 bound to DNA was exposed to primary antibody for NF-κB p65 and then reacted with anti-rabbit horseradish peroxidase-conjugated IgG. At this point, the color developing and stop solution were added to the plate. Absorbance of the samples was measured at 450 nm with a reference wavelength of 655 nm in a microplate reader.
Enzyme-linked immunosorbent assays for phosphorylation of Akt
Keratinocytes (1 × 106 cells/ml) were treated with 10 ng/ml TNF-α for 4 h. Cells were harvested by centrifugation at 412×g for 10 min, washed twice with PBS, and suspended in lysis buffer provided from R&D systems for whole cell lysates. The homogenates were centrifuged at 2000×g for 5 min and the supernatant was used for ELISA. The amount of phosphorylated-Akt was determined according to the manufacturer’s instructions for the immunoassays. The supernatants were sequentially reacted with antibodies for the phosphorylated forms of the kinases, biotinylated detection antibodies, and streptavidin-horseradish-peroxidase. Absorbance was measured at 405 nm.
Measurement of intracellular reactive oxygen species production
The dye 2′,7′-dichlorofluorescin diacetate (DCFH2-DA), which is oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF) by hydroperoxides, was used to measure relative levels of cellular peroxides [27]. Keratinocytes (1 × 105 cells/400 μl in 24-well plate) were treated with TNF-α for 24 h at 37 °C. Cells were washed, suspended in fetal bovine serum-free RPMI-1640, incubated with 50 μM dye for 30 min at 37 °C, and washed with phosphate-buffered saline. The cell suspensions were centrifuged at 412×g for 10 min and medium was removed. Cells were dissolved with 1% Triton X-100 and fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence microplate reader (SPECTRAFLUOR, TECAN, Salzburg, Austria).
Measurement of cell viability
Cell viability was measured by using the MTT reduction assay, which is based on the conversion of MTT to formazan crystals by mitochondrial dehydrogenases [28]. Cells (3 × 104) were treated with rotundarpene for 24 h at 37°C. The medium (200 μl) was incubated with 10 μl of 10 mg/ml MTT solution for 2 h at 37°C. After centrifugation at 412×g for 10 min, culture medium was removed and 100 μl of dimethyl sulfoxide was added to each well to dissolve the formazan. Absorbance was measured at 570 nm using a microplate reader (Spectra MAX 340, Molecular Devices Co., Sunnyvale, CA, USA). Cell viability was expressed as a percentage of the value measured from the control cultures in the cells that did not receive any treatments.
Statistical analysis
The data are expressed as the mean ± SEM. Data from these experiments were analyzed using randomized block design analysis of variance (ANOVA) with Tukey’s post hoc test. A probability value of P < 0.05 was considered statistically significant.
Results
Rotundarpene inhibits production of inflammatory mediators
The inhibitory effect of rotundarpene on the production of cytokines in keratinocytes exposed to TNF-α was investigated. TNF-α significantly caused the production of cytokine IL-1β and IL-6 in HEK001 keratinocytes and primary keratinocytes. Rotundarpene reduced the TNF-α-induced productions of IL-1β and IL-6 with approximately 71–79% inhibition at 15 μM in keratinocytes (Fig. 1a). Whether TNF-α-induced production of cytokines was mediated by the Akt, mTOR, and NF-kB pathways, we examined the effect of signaling inhibitors. The addition of 2.5 µM Bay 11-7085 (an irreversible inhibitor of TNF-α-activated IkBα phosphorylation), 0.5 µM Akt inhibitor or 0.5 µM rapamycin (an mTOR inhibitor) reduced the TNF-α-induced production of IL-1β and IL-6 in HEK001 keratinocytes and primary keratinocytes (Fig. 1b). They alone did not induce the cytokine production. Whether TNF-α-induced production of cytokines was mediated by oxidative stress was examined. Treatment with 1 mM thiol antioxidant N-acetylcysteine reduced the TNF-α-induced production of cytokines (Fig. 1b). It did not induce the cytokine production.

Rotundarpene reduces TNF-α-induced cytokine production. In a, HEK001 keratinocytes were pre-treated with 1–25 μM rotundarpene (RTD) for 20 min and exposed to 10 ng/ml TNF-α in combination with rotundarpene for 24 h. Then the amounts of IL-1β and IL-6 were determined using ELISA kits. In b and c, HEK001 keratinocytes were pre-treated with compounds (2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, 0.5 μM rapamycin, and 1 mM N-acetylcysteine (NAC), 20 µM SP600125 and 10 µM SB203580) for 20 min and exposed to 10 ng/ml TNF-α in combination with compounds for 24 h. Data represent the mean ± SEM (n = 6). + P < 0.05 compared to control; *P < 0.05 compared to TNF-α alone
We examined whether the rotundarpene effect on TNF-α-induced production of inflammatory mediators was accomplished by suppressing activation of the JNK and p38-MAPK. The addition of 20 µM JNK inhibitor SP600125 and 10 µM p38-MAPK inhibitor SB203580 reduced the production of IL-1β in HEK001 keratinocytes treated with TNF-α (Fig. 1c).
Using primary keratinocytes, we confirmed that the TNF-α-induced production of cytokines was mediated by the Akt, mTOR, and NF-kB pathways. TNF-α treatment induced production of IL-1β (Fig. 2a). The addition of 2.5 µM Bay 11-7085, 0.5 µM Akt inhibitor, or 0.5 µM rapamycin reduced the TNF-α-induced production of IL-1β in primary keratinocytes (Fig. 2b). They alone did not induce the cytokine production.

Rotundarpene inhibits TNF-α-induced IL-1β production. In a, primary keratinocytes were pre-treated with 1–25 μM rotundarpene (RTD) for 20 min and exposed to 10 ng/ml TNF-α in combination with rotundarpene for 24 h. Then the amount of IL-1β was determined using ELISA kits. In b, primary keratinocytes were pre-treated with compounds (2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, and 0.5 μM rapamycin) for 20 min and exposed to 10 ng/ml TNF-α in combination with compounds for 24 h. Data represent the mean ± SEM (n = 6). + P < 0.05 compared to control; *P < 0.05 compared to TNF-α alone
We further examined the rotundarpene effect on the TNF-α-induced production of chemokines. TNF-α significantly caused the production of chemokine CCL27 and CCL17 in HEK001 keratinocytes. Rotundarpene reduced the TNF-α-induced production of CCL27 and CCL17, with approximately 75% inhibition at 15 μM (Fig. 3a). We examined whether TNF-α-induced production of chemokines was mediated by the Akt, mTOR, and NF-kB pathways. The addition of Bay 11-7085, Akt inhibitor, or rapamycin reduced the TNF-α-induced production of CCL27 and CCL17 (Fig. 3b). They alone did not induce the chemokine production. We examined whether TNF-α-induced production of chemokine was mediated by oxidative stress. Treatment with N-acetylcysteine reduced the TNF-α-induced production of CCL27 (Fig. 3b). It did not induce the chemokine production.

Rotundarpene attenuates TNF-α-induced chemokine production. In a, HEK001 keratinocytes were pre-treated with 1–25 μM rotundarpene (RTD) for 20 min and exposed to 10 ng/ml TNF-α in combination with rotundarpene for 24 h. In b and c, HEK001 keratinocytes were pre-treated with compounds (2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, 0.5 μM rapamycin, 1 mM N-acetylcysteine (NAC), 20 µM SP600125, and 10 µM SB203580) for 20 min and exposed to 10 ng/ml TNF-α in combination with compounds for 24 h. Then the amounts of CCL27 and CCL17 were determined using ELISA kits. Data represent the mean ± SEM (n = 5–6). + P < 0.05 compared to control; *P < 0.05 compared to TNF-α alone
We examined whether the rotundarpene effect on TNF-α-induced production of inflammatory mediators was mediated by the effect on activation of the JNK and p38-MAPK . The addition of 20 µM JNK inhibitor SP600125 and 10 µM p38-MAPK inhibitor SB203580 reduced the production of CCL27 in HEK001 keratinocytes treated with TNF-α (Fig. 3c).
Rotundarpene inhibits activation of NF-κB
We examined whether the rotundarpene effect on the TNF-α-induced production of inflammatory mediators in keratinocytes was mediated by suppressing the NF-κB activation. Treatment with TNF-α increased the cytosolic levels of NF-κB p65, NF-κB p50, and phospho-IκB, and the nuclear levels of NF-κB p65 in keratinocytes (Fig. 4a). The addition of rotundarpene, Bay 11-7085, Akt inhibitor, and rapamycin inhibited the TNF-α-induced phosphorylation of IkBα and activation of NF-κB (Fig. 4a).

Rotundarpene reduces TNF-α-induced activation of NF-κB and of JNK and p38. HEK001 keratinocytes were pre-treated with compounds (15 μM rotundarpene (RTD), 1 mM N-acetylcysteine (NAC), 2.5 μM Bay 11-7085, 0.5 μM Akt inhibitor, and 0.5 μM rapamycin) for 20 min and then treated with 10 ng/ml TNF-α in the presence of compounds for 15 min. In a, the levels of NF-κB p65, NF-κB p50, and phosphorylated-IκB-α were analyzed by Western blotting with specific antibodies. The densities of protein bands were determined using TINA 2.10 g software, and data represent a fold increase from the control density. Data are representative of three to four different experiments. In b, the NF-κB to DNA-binding activity was measured by assay kit. Data represent the mean ± SEM (n = 5). + P < 0.05 compared to control; *P < 0.05 compared to TNF-α alone. In c, HEK001 keratinocytes were pre-treated with compounds (15 μM rotundarpene (RTD), 1 mM N-acetylcysteine (NAC), 20 µM SP600125 or 10 µM SB203580) for 20 min and then treated with 10 ng/ml TNF-α in the presence of compounds for 4 h. Data are representative of three to four different experiments. In d, HEK001 keratinocytes were pre-treated with compounds (20 µM SP600125 or 10 µM SB203580) for 20 min and then treated with 10 ng/ml TNF-α in the presence of compounds for 15 min. The NF-κB to DNA-binding activity was measured by assay kit. Data represent the mean ± SEM (n = 5). + P < 0.05 compared to control; *P < 0.05 compared TNF-α alone
We confirmed the inhibitory effect of rotundarpene on the TNF-α-induced NF-κB activation by monitoring the effect on the binding of NF-κB to DNA. Non-stimulated cells exhibited a small increase in the NF-κB-DNA-binding activity. TNF-α produced a marked increase in the NF-κB-DNA-binding activity, which was attenuated by the addition of rotundarpene, Bay 11-7085, Akt inhibitor, rapamycin, and N-acetylcysteine (Fig. 4b).
The rotundarpene effect on TNF-α-induced activation of the JNK and p38-MAPK was examined. TNF-α induced phosphorylation of the JNK and p38-MAPK in HEK001 keratinocytes. The addition of rotundarpene, N-acetylcysteine, 20 µM JNK inhibitor SP600125 or 10 µM p38-MAPK inhibitor SB203580 reduced the TNF-α-induced phosphorylation of the JNK and p38-MAPK in HEK001 keratinocytes (Fig. 4c). Then we investigated the regulatory effect of JNK and p38 on the TNF-α-induced increase in the NF-κB-DNA-binding activity. The addition of 20 µM JNK inhibitor SP600125 and 10 µM p38-MAPK inhibitor SB203580 reduced the TNF-α-induced increase in the NF-κB-DNA-binding activity in HEK001 keratinocytes (Fig. 4d).
Rotundarpene inhibits Akt activation and changes in the levels of mTOR
We examined whether TNF-α-induced NF-κB activation-mediated inflammatory mediator production was regulated by Akt pathway. To define the inhibitory effect of rotundarpene, we assessed the rotundarpene effect on the Akt level changes at a 4-h exposure time of TNF-α. The inhibitory effect of rotundarpene on changes in the levels of phosphorylated-Akt was observed by Western blot analysis. Rotundarpene and Akt inhibitor attenuated the TNF-α-induced increase in the levels of phospho-Akt1 (Fig. 5a). Treatment with TNF-α and stated compounds did not affect the levels of Akt1. We further examined the inhibitory effect of rotundarpene on phosphorylation of Akt. The addition of rotundarpene, Akt inhibitor, and N-acetylcysteine inhibited the TNF-α-induced phosphorylation of Akt (Fig. 5b). Rotundarpene, Akt inhibitor, and N-acetylcysteine alone did not increase the Akt phosphorylation.

Rotundarpene reduces TNF-α-induced activation of Akt and changes in the levels of mTOR. In a, HEK001 keratinocytes were treated with 10 ng/ml TNF-α in combination with 5–15 μM rotundarpene (RTD) and 0.5 μM Akt inhibitor for 4 h, and the level of phosphorylated (activated)-Akt was determined by Western blotting with specific antibodies or by ELISA. The densities of protein bands were determined using TINA 2.10 g software, and data represent a fold increase from the control density. Data are representative of three to four different experiments. In b and c, HEK001 keratinocytes were treated with 10 ng/ml TNF-α (or 100 ng/ml epidermal growth factor) in combination with 5–15 μM rotundarpene (RTD) and 0.5 μM Akt inhibitor for 4 h, and the phosphorylation of Akt was determined by ELISA. In upper part in d, HEK001 keratinocytes were treated with TNF-α for the 30 min to 6 h. In lower part in d, keratinocytes were treated with TNF-α in the presence of compounds (15 µM rotundarpene, 0.5 μM Akt inhibitor, and 0.5 μM rapamycin) for 4 h. The level of mTOR was determined by Western blotting with specific antibody. Data are representative of three to four different experiments
To assess whether rotundarpene could regulate signaling pathways other than pathway that affected by TNF-α, we examined whether the rotundarpene and Akt inhibitor may influence EGF-induced phosphorylation of Akt. HEK001 keratinocytes treated with EGF exhibited an increase in the Akt phosphorylation. The addition of rotundarpene and Akt inhibitor reduced EGF-induced Akt phosphorylation, respectively. Further combination almost completely prevented the Akt phosphorylation. The rotundarpene and Akt inhibitor alone did not affect the Akt phosphorylation (Fig. 5c).
To examine whether TNF-α-induced production of inflammatory mediators was regulated by mTOR signaling, we measured changes in the levels of mTOR. In HEK001 keratinocytes treated with TNF-α, the levels of mTOR increased up to 4-h exposure time and then the levels declined. The addition of rotundarpene, Akt inhibitor, and rapamycin inhibited the TNF-α-induced increase in the levels of mTOR (Fig. 5d). Compounds alone did not induce changes in the levels of mTOR (data not shown).
Rotundarpene inhibits production of reactive oxygen species
The production of reactive oxygen species within cells was determined by monitoring a conversion of DCFH2-DA to DCF. Keratinocytes treated with TNF-α showed a significant increase in DCF fluorescence. The production of reactive oxygen species in keratinocytes treated with TNF-α was confirmed with the effect of oxidant scavengers. The addition of rotundarpene, N-acetylcysteine, 50 μM trolox (a cell-permeable, water-soluble derivative of vitamin E; a scavenger of hydroxyl radicals and peroxynitrite) and 100 μM ascorbic acid inhibited TNF-α-induced increase in DCF fluorescence (Fig. 6).

Rotundarpene reduces TNF-α-induced production of reactive oxygen species. HEK001 keratinocytes were treated with 10 ng/ml TNF-α in the presence of compounds (15 μM rotundarpene (RTD), 1 mM N-acetylcysteine (NAC), 30 μM trolox, and 100 μM ascorbic acid) for 24 h. Changes in DCF fluorescence were measured and data are expressed as arbitrary units (a.u.) of fluorescence. Data represent the mean ± SEM (n = 6). + P < 0.05 compared to control; *P < 0.05 compared TNF-α alone
Rotundarpene up to 25 µM does not alter cell viability
To assess whether the inhibitory effect of rotundarpene on the stimulated keratinocyte responses was attributed to its effect on cell viability, we examined the cytotoxic effect of rotundarpene using the MTT assay that provides rapid and precise results for cellular growth and survival. As the data showed, rotundarpene up to 25 µM did not significantly affect the cell viability in keratinocytes. When HEK001 keratinocytes were treated with 25 µM rotundarpene for 24 h, approximately 5% cell death was observed, which is not statistically significant (Fig. 7a).

Rotundarpene up to 25 μM does not alter cell viability. In a, HEK001 keratinocytes were treated with 1–25 μM rotundarpene (RTD) for 24 h and then cell viability was determined using a MTT reduction assay. In b, HEK001 keratinocytes were treated with 1–25 ng/ml TNF-α for 24 h. Data represent the mean ± SEM (n = 6). + P < 0.05 compared to control (percentage of control)
Then we examined the cytotoxic effect of TNF-α on HEK001 keratinocytes. When keratinocytes were treated with 1–25 µM TNF-α for 24 h, cell viability declined in a dose-dependent manner. Approximately 10 and 36% cell death were detected at 10 and 25 ng/ml TNF-α, respectively, for 24 h (Fig. 7b).
Discussion
HEK001 keratinocytes treated with TNF-α exhibited significant production of IL-1β, IL-6, and CCL27. The extract from the barks of and Hemiterpene glycosides from the leaves of I. rotunda Thunb have been shown to have anti-inflammatory and antioxidant properties [15, 18]. We assessed the rotundarpene effect on TNF-α-induced inflammatory mediator production in keratinocytes in relation to activation of the Akt, NF-κB, and mTOR pathways. TNF-α binds to the TNF receptors and activates the NF-κB, which regulates the transcription genes involved in inflammation and immune responses [10, 29]. Aberrant activation of NF-κB in both keratinocytes and lymphocytes is considered to be involved in the development of inflammatory skin disease [30]. TNF-α induces production of cytokines and chemokines in keratinocytes by activating NF-κB [25, 31]. TNF-α induces the phosphorylation and proteolytic degradation of IκB and the subsequent release of NF-κB dimers [32]. The translocation of the active NF-κB dimers to the nucleus elicits activation of specific target genes, such as transcription of proinflammatory genes, leading to the production of mRNA responsible for synthesis of cytokines and chemokines [10, 33]. TNF-α treatment increased the phosphorylated-IκB and NF-κB p65/50 levels, and the binding of NF-κB to DNA in keratinocytes. The results suggest that the TNF-α effect on the cytokine and chemokine production is mediated by translocation of NF-κB dimers to the nucleus and binding to specific DNA sites. We measured whether the suppressive effect of rotundarpene on the TNF-α-induced production of inflammatory mediators in keratinocytes was due to inhibition of NF-κB activation. Rotundarpene and an NF-kB activation inhibitor Bay 11-7085 reduced the TNF-α-induced production of inflammatory mediators, increase in the levels of phosphorylated-IκB and NF-κB p65/50, and binding of NF-κB to DNA. Thus, rotundarpene may reduce the TNF-α-induced production of inflammatory mediators by suppressing the activation of NF-κB.
The PI3 K/Akt and mTOR pathways regulate the activation of NF-κB [8, 9]. TNF-α induces phosphorylation of PI3K, Akt, and mTOR in colonic epithelial cell lines HT-29 and U937 cells [34]. In keratinocytes, we assessed whether TNF-α-induced production of inflammatory mediators and activation of NF-κB were mediated by the activation of Akt and mTOR. Treatment with TNF-α induced the phosphorylation of Akt, increase in the levels of mTOR, and activation of BAY11-7085 in keratinocytes. Rotundarpene, Akt inhibitor, and rapamycin inhibited the production of inflammatory mediators and reduced the changes in the levels and activities in signal transduction proteins in TNF-α-stimulated keratinocytes. Thus, rotundarpene appears to reduce TNF-α-induced production of inflammatory mediators by suppressing activation of Akt, mTOR, and NF-κB pathways.
EGF treatment stimulates the phosphorylation (activation) of EGF receptor-mediated activation of PI3K, Akt, and mTOR signalings [36–38]. EGF induces the MAPK/ERK and MAPK/JNK-mediated signal transduction [37]. However, EGF stimulates adipophilin synthesis in HC11 mouse mammary epithelial cells via the PI3-kinase/Akt/mTOR pathway and p38 but not JNK [39]. To assess whether rotundarpene could modulate signaling pathways other than pathway that affected by TNF-α, we examined that rotundarpene may regulate the EGF-induced phosphorylation of Akt. HEK001 keratinocytes treated with EGF exhibited an increase in the Akt phosphorylation. The addition of rotundarpene and Akt inhibitor reduced the EGF-induced Akt phosphorylation, respectively. Further combination almost completely prevented the phosphorylation of Akt. The rotundarpene and Akt inhibitor alone did not affect the Akt phosphorylation. These results suggest that rotundarpene could regulate activation of signaling pathways triggered by stimulants other than TNF-α.
Reactive oxygen species play a critical role in inflammation process and in transduction of signals from receptors to IL-1β or TNF-α [35]. Reactive oxygen species cause the activation of NF-κB [35, 40, 41]. Reactive oxygen species activate NF-κB through PI3-k/Akt pathway [35]. The oxidant scavenger N-acetylcysteine inhibited the changes in the levels and activities in the Akt, mTOR, and NF-κB, and the production of reactive oxygen species in TNF-α-stimulated keratinocytes. These results suggest that the TNF-α-induced activation of the Akt, NF-κB, and mTOR pathways is mediated by production of reactive oxygen species. Connectively, rotundarpene appears to reduce the activation of the Akt, mTOR, and NF-κB pathways by suppressing production of reactive oxygen species.
We examined whether the inhibitory effect of rotundarpene on the stimulated keratinocyte responses was due to the effect on cell viability. When keratinocytes were treated with rotundarpene for 24 h, approximately 3 and 5% cell death at 15 and 25 µM, respectively, were detected. Therefore, the inhibitory effect of rotundarpene less than 25 µM on the inflammatory mediator production and signaling pathways may not be affected by changes in cell viability. Further, we investigated TNF-α may affect cell viability in HEK001 keratinocytes. TNF-α treatment for 24 h induced approximately 10% cell death at 10 ng/ml, which was statistically significant. Western blot analysis and ELISA indicate that TNF-α induces apoptotic cell death. The concentration of TNF-α used in the present study was based on reports in discussion section and may be necessarily to assess action mechanism of test materials in relation to the TNF-α-mediated process. Although TNF-α had a toxic effect at 10 ng/ml, TNF-α significantly stimulated production of cytokines and chemokines in keratinocytes. Thus, the TNF-α concentration has been proper to define the effect of rotundarpene on the inflammatory mediator production and signaling pathways.
The mitogen-activated protein (MAP) kinases, including c-Jun N-terminal kinases (JNKs) and p38-MAPKs, are involved in the maintenance of intracellular functions such as inflammation and innate immunity [12, 42–44]. The JNK and p38 MAPK signaling pathways are activated by various types of cellular stress such as oxidative stress and proinflammatory cytokines TNF-α and IL-1β [12]. The binding of TNF-α to cell surface receptors triggers activation of JNK and p38-MAPK. The MAP kinase signaling pathways induce a secondary response by increasing the expression of several inflammatory cytokines [11]. TNF-α induces activation of the NF-κB, JNK, and p38-MAPK in HaCaT keratinocytes resulted in skin inflammation [13, 14, 45]. In this respect, we examined whether the rotundarpene effect on the TNF-α-induced production of inflammatory mediators was mediated by suppressing the activation of the JNK and p38-MAPK. HEK001 keratinocytes treated with TNF-α exhibited an increase in the cytosolic levels of phosphorylated-JNK and phosphorylated-p38. Rotundarpene, JNK inhibitor (SP600125), and p38-MAPK inhibitor (SB203580) reduced the TNF-α-induced production of cytokines and chemokines and increased the cytosolic levels of phosphorylated-JNK and phosphorylated-p38. Thus, rotundarpene may reduce the TNF-α-induced production of inflammatory mediators in keratinocytes by suppressing the activation of JNK and p38-MAPK. In addition, the inhibitory effect of N-acetylcysteine on the activation of the JNK and p38-MAPK indicates that the TNF-α-induced activation of the JNK and p38-MAPK may be mediated by reactive oxygen species in keratinocytes. The suppressive effect of rotundarpene on activation of the JNK and p38-MAPK appears to be attributed to the inhibition of reactive oxygen species production.
A previous report suggests there is a mutual cross-talk reaction between formation of reactive oxygen species and activation of NF-κB [46]. It has been also shown that inhibition of NF-κB may attenuate oxidative stress and improve cardiac mitochondrial structural integrity [47]. Thus, previous reports suggest that the TNF-α-induced production of reactive oxygen species appears to depend on the activation of Akt, mTOR, and NF-κB pathways, and the JNK and p38.
In additionally, rotundarpene suppressed all the TNF-α-induced activation of the Akt, mTOR, and NF-κB pathways, activation of JNK and p38-MAPK, and production of reactive oxygen species, which may be involved in the production of inflammatory mediators. These findings suggest that rotundarpene may directly inhibit the stability of TNF-α or TNF-α receptor or the interaction between TNF-α and TNF-α receptor. To investigate precise action of rotundarpene, thus, the study on the stability of TNF-α or TNF-α receptor will be required.
Overall, the results show that rotundarpene may reduce the TNF-α-stimulated production of inflammatory mediators in keratinocytes by suppressing the reactive oxygen species-mediated activation of the Akt, mTOR, and NF-κB pathways, and the JNK and p38-MAPK. Additionally, rotundarpene appears to reduce the Akt, mTOR, and NF-κB pathways and the JNK- and p38-MAPK-mediated inflammatory skin diseases.
Abbreviations
- TNF-α:
-
Tumor necrosis factor-α
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- mTOR:
-
Mammalian target of rapamycin
- NF-κB:
-
Nuclear factor-κB
- MAPKs:
-
Mitogen-activated protein kinases
- ERKs:
-
Extracellular signal-regulated kinases
- JNKs:
-
c-Jun NH2-terminal kinases
- p38-MAPK:
-
p38-Mitogen-activated protein kinase
- EGF:
-
Epidermal growth factor
References
Mazza J, Rossi A, Weinberg JM (2010) Innovative uses of tumor necrosis factor alpha inhibitors. Dermatol Clin 28:559–575. doi:10.1016/j.det.2010.03.009
Pastore S, Mascia F, Girolomoni G (2006) The contribution of keratinocytes to the pathogenesis of atopic dermatitis. Eur J Dermatol 16:125–131
Tsuruta D (2009) NF-κB links keratinocytes and lymphocytes in the pathogenesis of psoriasis. Recent Pat Inflamm Allergy Drug Discov 3:40–48
Faurschou A, Gniadecki R, Calay D, Wulf HC (2008) TNF-α impairs the S-G2/M cell cycle checkpoint and cyclobutane pyrimidine dimer repair in premalignant skin cells: role of the PI3K-Akt pathway. J Investig Dermatol 128:2069–2077. doi:10.1038/jid.2008.19
Li J, Li J, Yue Y, Hu Y, Cheng W, Liu R, Pan X, Zhang P (2014) Genistein suppresses tumor necrosis factor α-induced inflammation via modulating reactive oxygen species/Akt/nuclear factor κB and adenosine monophosphate-activated protein kinase signal pathways in human synoviocyte MH7A cells. Drug Des Devel Ther 8:315–323. doi:10.2147/DDDT.S52354
Sung HC, Liang CJ, Lee CW, Yen FL, Hsiao CY, Wang SH, Jiang-Shieh YF, Tsai JS, Chen YL (2015) The protective effect of eupafolin against TNF-α-induced lung inflammation via the reduction of intercellular cell adhesion molecule-1 expression. J Ethnopharmacol 170:136–147
Faurschou A (2010) Role of tumor necrosis factor-α in the regulation of keratinocyte cell cycle and DNA repair after ultraviolet-B radiation. Dan Med Bull 57:B4179
Thomson AW, Turnquist HR, Raimondi G (2009) Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 9:324–337
Zhong LM, Zong Y, Sun L, Guo JZ, Zhang W, He Y, Song R, Wang WM, Xiao CJ, Lu D (2012) Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS One 7:e32195
Ghosh S, Hayden MS (2008) New regulators of NF-κB in inflammation. Nat Rev Immunol 8:837–848. doi:10.1038/nri2423
Sabio G, Davis RJ (2014) TNF and MAP kinase signalling pathways. Semin Immunol 26:237–245
Kim EK, Choi EJ (2015) Compromised MAPK signaling in human diseases: an update. Arch Toxicol 89:867–882. doi:10.1007/s00204-015-1472-2
Cho JW, Lee KS, Kim CW (2007) Curcumin attenuates the expression of IL-1beta, IL-6, and TNF-α as well as cyclin E in TNF-alpha-treated HaCaT cells; NF-κB and MAPKs as potential upstream targets. Int J Mol Med 19:469–474. doi:10.3892/ijmm.19.3.469
Yano C, Saeki H, Komine M, Kagami S, Tsunemi Y, Ohtsuki M, Nakagawa H (2015) Mechanism of macrophage-derived chemokine/CCL22 production by HaCaT keratinocytes. Ann Dermatol 27:152–156
Jiang J, Xu S, Kong Y (2000) Pharmacological studies of trilex on treatment of pharyngitis. Zhong Yao Cai 23:630–632
Zhao LC, He Y, Deng X, Xia XH, Liang J, Yang GL, Zhao LC (2012) Ultrasound-assisted extraction of syringin from the bark of Ilex rotunda thumb using response surface methodology. Int J Mol Sci 13:7607–7616
Choi J, Shin KM, Park HJ, Jung HJ, Kim HJ, Lee YS, Park HJ, Shin KM (2004) Anti-inflammatory and antinociceptive effects of sinapyl alcohol and its glucoside syringin. Planta Med 70:1027–1032. doi:10.1055/s-2004-832642
Kim MH, Park KH, Oh MH, Kim HH, Choe KI, Park SH, Lee MW (2012) Two new hemiterpene glycosides from the leaves of Ilex rotunda. Thunb. Arch Pharm Res 35:1779–1784. doi:10.1007/s12272-012-1010-1
Wu Y, Zhou C, Song L, Li X, Shi S, Mo J, Chen H, Bai H, Wu X, Zhao J, Zhang R, Hao X, Sun H, Zhao Y (2006) Effect of total phenolics from Laggera alata on acute and chronic inflammation models. J Ethnopharmacol 108:243–250
Kim SS, Park RY, Jeon HJ, Kwon YS, Chun W (2005) Neuroprotective effects of 3,5-dicaffeoylquinic acid on hydrogen peroxide-induced cell death in SH-SY5Y cells. Phytother Res 19:243–245. doi:10.1002/ptr.1652
Park KH, Park M, Choi SE, Jeong MS, Kwon JH, Oh MH, Choi HK, Seo SJ, Lee MW (2009) The anti-oxidative and anti-inflammatory effects of caffeoyl derivatives from the roots of Aconitum koreanum R. RAYMOND. Biol Pharm Bull 32:2029–2033. doi:10.1248/bpb.32.2029
Kim JE, Kim BJ, Jeong MS, Seo SJ, Kim MN, Hong CK, Ro BI (2005) Expression and modulation of LL-37 in normal human keratinocytes, HaCaT cells and inflammatory skin diseases. J Korean Med Sci 20:649–654. doi:10.3346/jkms.2005.20.4.649
Schreiber E, Matthias P, Müller MM, Schaffner W (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 17:6419
Coward WR, Okayama Y, Sagara H, Wilson SJ, Holgate ST, Church MK (2002) NF-κB and TNF-alpha: a positive autocrine loop in human lung mast cells? J Immunol 169:5287–5293. doi:10.4049/jimmunol.169.9.5287
Lee CS, Jeong EB, Kim YJ, Lee MS, Seo SJ, Park KH, Lee MW (2013) Quercetin-3-O-(2″-galloyl)-α-l-rhamnopyranoside inhibits TNF-α-activated NF-κB-induced inflammatory mediator production by suppressing ERK activation. Int Immunopharmacol 16:481–487. doi:10.1016/j.intimp.2013.05.001
Zhou C, Huang M, Xie L, Shen J, Xiao T, Wang R (2015) IVIG inhibits TNF-α-induced MMP9 expression and activity in monocytes by suppressing NF-κB and P38 MAPK activation. Int J Clin Exp Pathol 8:15879–15886 eCollection 2015
Fu W, Luo H, Parthasarathy S, Mattson MP (1998) Catecholamines potentiate amyloid β-peptide neurotoxicity: involvement of oxidative stress, mitochondrial dysfunction, and perturbed calcium homeostasis. Neurobiol Dis 5:229–243. doi:10.1006/nbdi.1998.0192
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi:10.1016/0022-1759(83)90303-4
Diamant G, Dikstein R (2013) Transcriptional control by NF-κB: elongation in focus. Biochim Biophys Acta 1829:937–945. doi:10.1016/j.bbagrm.2013.04.007
Rebholz B, Haase I, Eckelt B, Paxian S, Flaig MJ, Ghoreschi K, Nedospasov SA, Mailhammer R, Debey-Pascher S, Schultze JL, Weindl G, Förster I, Huss R, Stratis A, Ruzicka T, Röcken M, Pfeffer K, Schmid RM, Rupec RA (2007) Crosstalk between keratinocytes and adaptive immune cells in an IκBα protein-mediated inflammatory disease of the skin. Immunity 27:296–307. doi:10.1016/j.immuni.2007.05.024
Young CN, Koepke JI, Terlecky LJ, Borkin MS, Boyd SL, Terlecky SR (2008) Reactive oxygen species in tumor necrosis factor-α-activated primary human keratinocytes: implications for psoriasis and inflammatory skin disease. J Investig Dermatol 128:2606–2614
Napetschnig J, Wu H (2013) Molecular basis of NF-κB signaling. Annu Rev Biophys 42:443–468. doi:10.1146/annurev-biophys-083012-130338
Vestergaard C, Johansen C, Otkjaer K, Deleuran M, Iversen L (2005) Tumor necrosis factor-alpha-induced CTACK/CCL27 (cutaneous T-cell-attracting chemokine) production in keratinocytes is controlled by nuclear factor κB. Cytokine 29:49–55
Park S, Regmi SC, Park SY, Lee EK, Chang JH, Ku SK, Kim DH, Kim JA (2014) Protective effect of 7-O-succinyl macrolactin A against intestinal inflammation is mediated through PI3-kinase/Akt/mTOR and NF-κB signaling pathways. Eur J Pharmacol 735C:184–192. doi:10.1016/j.ejphar.2014.04.024
Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D (2013) The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol 64:409–421
Xu Z, Zhang Y, Jiang J, Yang Y, Shi R, Hao B, Zhang Z, Huang Z, Kim JW, Zhang G (2010) Epidermal growth factor induces HCCR expression via PI3K/Akt/mTOR signaling in PANC-1 pancreatic cancer cells. BMC Cancer 10:161. doi:10.1186/1471-2407-10-161
Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu M, Tao Y, Wang Y, Qian H, Gu L (2013) PKG II inhibits EGF/EGFR-induced migration of gastric cancer cells. PLoS One 8(4):e61674. doi:10.1371/journal.pone.0061674
Wu M, Chen Y, Jiang L, Li Y, Lan T, Wang Y, Qian H (2013) Type II cGMP-dependent protein kinase inhibits epidermal growth factor-induced phosphatidylinositol-3-kinase/Akt signal transduction in gastric cancer cells. Oncol Lett 6:1723–1728. doi:10.3892/ol.2013.1630
Pauloin A, Chanat E (2012) Prolactin and epidermal growth factor stimulate adipophilin synthesis in HC11 mouse mammary epithelial cells via the PI3-kinase/Akt/mTOR pathway. Biochim Biophys Acta 823(5):987–996. doi:10.1016/j.bbamcr.2012.02.016
Gloire G, Legrand-Poels S, Piette J (2006) NF-κB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72:1493–1505. doi:10.1016/j.bcp.2006.04.011
Poli G, Biasi F, Leonarduzzi G (2013) Oxysterols in the pathogenesis of major chronic diseases. Redox Biol 1:125–130. doi:10.1016/j.redox.2012.12.001
Arthur JS, Ley SC (2013) Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 13:679–692. doi:10.1038/nri3495
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H (2014) p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 344:174–179
Kim AR, Lee B, Joung EJ, Gwon WG, Utsuki T, Kim NG, Kim HR (2016) 6,6′-Bieckol suppresses inflammatory responses by down-regulating nuclear factor-κB activation via Akt, JNK, and p38 MAPK in LPS-stimulated microglial cells. Immunopharmacol Immunotoxicol 38:244–252. doi:10.3109/08923973.2016.1173060
Bahar-Shany K, Ravid A, Koren R (2010) Upregulation of MMP-9 production by TNFα in keratinocytes and its attenuation by vitamin D. J Cell Physiol 222:729–737. doi:10.1002/jcp.22004
Bubuci C, Papa S, Dean K, Franzoso G (2006) Mutual cross-talk between reactive oxygen species and NF-κB: molecular basis and biological significance. Oncogene 25:6731–6748. doi:10.1038/sj.onc.1209936
Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J (2010) NF-κB-induced oxidative stress contributes to mitochondrial and cardiac function in type II diabetes. Cardiovasc Res 85:473–483. doi:10.1093/cvr/cvp305
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kim, A., Nam, Y.J., Shin, Y.K. et al. Rotundarpene inhibits TNF-α-induced activation of the Akt, mTOR, and NF-κB pathways, and the JNK and p38 associated with production of reactive oxygen species. Mol Cell Biochem 434, 113–125 (2017). https://doi.org/10.1007/s11010-017-3041-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3041-x