
Abstract
The aryl hydrocarbon receptor (AhR) was initially identified as the receptor that binds and mediates the toxic effects induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and structurally related halogenated aromatics. Other toxic compounds including some polynuclear aromatic hydrocarbons act through the AhR; however, during the last 25 years, it has become apparent that the AhR plays an essential role in maintaining cellular homeostasis. Moreover, the scope of ligands that bind the AhR includes endogenous compounds such as multiple tryptophan metabolites, other endogenous biochemicals, pharmaceuticals and health-promoting phytochemicals including flavonoids, indole-3-carbinol and its metabolites. It has also been shown that like other receptors, the AhR is a drug target for multiple diseases including cancer, where both AhR agonists and antagonists effectively block many of the critical hallmarks of cancer in multiple tumor types. This review describes the anti-cancer activities of AhR ligands and demonstrates that it is time to separate the AhR from TCDD and exploit the potential of the AhR as a novel target for cancer chemotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cancer statistics and background
Regulatory and cancer research agencies carefully monitor changes in cancer statistics each year and determine both incidence and death rates for all tumor types (Miller et al. 2016; Siegel et al. 2015; Torre et al. 2016). Despite scientific and medical advances in detection, treatment and understanding the unique features of each tumor type, the overall progress in terms of decreased incidence and mortality has been limited (Miller et al. 2016). For example, cancer is still the leading cause of death worldwide and the total number of cancer cases and deaths are increasing along with population growth (Torre et al. 2016). The statistics in the USA are more encouraging and from 2007 to 2011, cancer incidence rates had decreased by 1.8% in men but were unchanged in women; cancer death rates decreased by 1.8 and 1.4% in men and women, respectively (Siegel et al. 2015). Improvements in cancer incidence were not only tumor specific, but also dependent on age, sex, race, socioeconomic status and region. Some of the most dramatic changes in cancer incidence have been correlated with lifestyle changes such as decreased smoking in males from the 1990s, leading to a significant decline in this disease (Siegel et al. 2015). The success of cancer therapies in contributing to improved survival of cancer patients is due, in part, to the extensive use of combination drug therapy regimens and the limited, but impressive effects of targeted mechanism-based therapies for treatment of some tumors. For example, the use of BCR–ABL tyrosine kinase inhibitors such as imatinib has increased the 5-year survival of chronic myeloid leukemia patients from 31 to 60% (Ferdinand et al. 2012; Miller et al. 2016). Unfortunately, “wonder” drugs for most other cancers have not been developed.
The basic science of cancer initiation, promotion, progression and metastasis has been extensively studied, and the progress made at the organismal, cellular and genomic levels have been remarkable and will form the future basis for successful development of new targeted therapies. Hanahan and Weinberg (2000) organized thinking about cancer based on their initial proposal of six hallmarks of cancer including “sustained proliferative signaling, evading growth suppressors, resisting cell death, enabling reproductive mortality, inducing angiogenesis, activating invasion and metastasis”. Two additional hallmarks, reprogramming of energy metabolism and evading immune destruction, have been added (Hanahan and Weinberg 2011), and these hallmarks now serve not only to define critical features of cancer cells but also as a framework for development of new targeted therapies. The complexity of cancer cells and tumors is apparent from the continuing efforts by pathologists and oncologists to divide tumors from each site into various subclasses based on their unique pathologies and stages (early to late) and their biochemical/molecular characteristics, since these classifications are not only related to outcomes (e.g., survival times) but to specific treatment regimens. Not surprisingly, tumor classifications are continually changing based on the acquisition of new information on various cellular and molecular characteristics of each tumor type. Breast cancer classifications initially relied on expression of the estrogen receptor (ERα) in the presence or absence of the progesterone receptor (PR); this was subsequently expanded to include expression of the oncogenic epidermal growth factor receptor 2 (HER2, ErbB2) which could be targeted by antibodies such as Herceptin (trastuzumab), an antibody that binds HER2 and blocks its function. Breast cancer classifications continue to evolve and include molecular characteristics, staging, pathology and other factors (Perou et al. 2000; Sinn and Kreipe 2013; Viale 2012). Thus, tumors from the same site are highly heterogenous and provide enormous problems for designing stage-specific therapies and for overcoming subsequent drug-resistance problems associated with activation of the alternative pro-oncogenic pathway.
The AhR and its physiological role
The AhR was initially identified as the receptor that bound the environmental toxicant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and structurally related toxic halogenated aromatic industrial compounds and by-products (Poland et al. 1976; Poland and Knutson 1982) (Fig. 1). The development of AhR knockout mice confirmed that this receptor was necessary to mediate the toxic effects of TCDD and other dioxin-like compounds (DLCs) (Fernandez-Salguero et al. 1996; Mimura et al. 1997). Unfortunately, this has been and continues to be a major problem in exploiting the AhR as a drug target, whereas other receptors such as the ER that plays a role in breast cancer and other hormone-dependent diseases is a major target for selective ER modulators that are extensively used for clinical applications (Jordan 2007, 2009). Over the past 25 years, it has been well established that multiple different classes of compounds including biochemicals that are possible endogenous AhR ligands, health-promoting phytochemicals and AhR-active pharmaceuticals bind the AhR (Denison and Nagy 2003; Denison et al. 2011; Hu et al. 2007; Safe et al. 2012; Soshilov and Denison 2014) (Fig. 1). Moreover, there is increasing evidence that the AhR plays a prominent role in physiology and pathophysiology including important roles in the immune function, autoimmunity, gastrointestinal function, inflammation and cancer (Benson and Shepherd 2011; Boitano et al. 2010; Ehrlich et al. 2016; Esser 2012; Kerkvliet et al. 2009; Marshall and Kerkvliet 2010; Murray et al. 2010; Punj et al. 2014; Quintana et al. 2008; Veldhoen et al. 2008), and development of selective AhR modulators is a promising new area of pharmacological research, particularly for cancer chemotherapy (Murray et al. 2014; Safe et al. 2013).

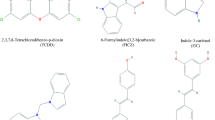
AhR ligands. 2,3,7,8-TCDD and benzo[a]pyrene are classified as “toxic” AhR ligands. FICZ and kynurenine are endogenous ligands. CH223191 is an AhR antagonist and omeprazole is an AhR-active pharmaceutical
Cancer chemotherapies and a role for the AhR
The standard first-line chemotherapies for most cancers include a range of cytotoxic drugs that target critical functions more highly expressed in tumor versus non-tumor tissues/cells (Masui et al. 2013). Some of the genes/pathways that are targeted in cancer cells include membrane receptors (tyrosine kinases) and their ligands, oncogenes such as Ras and other pro-oncogenic factors, transcription factors and nuclear receptors. Members of the nuclear receptor superfamily are ligand-activated nuclear transcription factors that include the estrogen receptor and androgen receptor which are targeted by selective receptor modulators (SRMs) for treatment of early stage receptor-positive breast and prostate cancer (Aesoy et al. 2015; Baek and Kim 2014; Burris et al. 2013; Tice and Zheng 2016). Over 80 drugs targeting 18 different nuclear receptors have been approved for various uses (Tice and Zheng 2016). In contrast, compounds targeting the aryl hydrocarbon receptor (AhR), which is also a ligand-activated nuclear transcription factor and a member of the basic helix-loop-helix (bHLH) family, have not been approved for any pharmacologic applications. There are only a few AhR ligands including aminoflavone and laquinimod that have been used in clinical trials for treatment of breast cancer and multiple sclerosis, respectively (Haggiag et al. 2013; Loaiza-Perez et al. 2004).
The AhR and its ligand in tumorigenesis and cancer chemotherapy
Most initial studies on the AhR and its ligands focused on the effects of TCDD on tumor formation after long-term rodent feeding studies, and there was general consensus that TCDD was a hepatocarcinogen in most studies [reviewed in (Bock and Kohle 2005; Knerr and Schrenk 2006)]. TCDD-induced tumors were also observed in multiple sites; however, in a lifetime feeding study in Sprague–Dawley rats, there was a decrease in spontaneous mammary and uterine tumors (Kociba et al. 1978). The AhR has been characterized in multiple cell lines and human tumors (Safe et al. 2013) and, with the development of selective AhR modulators (SAhRMs) (Safe et al. 1999) including AhR-active pharmaceuticals, the AhR has emerged as a drug target for cancer and other diseases. In this review, we will outline the role of the AhR in cancer cell and mouse models and also the opportunities for novel approaches of using SAhRMs as cancer therapeutics. It is also apparent that the AhR and its ligands can act as agonists or antagonists to block many of the hallmarks of cancer (Fig. 2) and these results will be apparent in the following summaries.

Targeting the hallmarks of cancer via the AhR
Genitourinary cancers
Table 1 summarizes the effects of several AhR ligands on various genitourinary-derived tumors and also the endogenous role of the AhR in prostate cancer using the TRAMP mouse model (Fritz et al. 2009). TCDD and related compounds and also omeprazole and tranilast inhibit pancreatic cancer cell invasion; however, there is evidence for different mechanisms of action dependent on the cell classification (Jin et al. 2015; Koliopanus et al. 2002). For example, in Panc1 cells which are highly invasive, the mechanism of omeprazole-mediated inhibition of invasion is due to a non-genomic AhR pathway (Jin et al. 2015). The role of the AhR and its ligands in prostate cancer cells are dependent on androgen receptor (AR) expression. There is evidence that AhR ligands are anti-androgenic in AR-expressing prostate cancer cells, and the AhR itself is growth inhibitory (Gluschnaider et al. 2010). In contrast, knockdown of the AhR in AR-negative prostate cancer cells decreases proliferation (Tran et al. 2013), multiple AhR ligands induce pro-invasion MMP9 (Haque et al. 2005) and the AhR antagonist CH223191 inhibits growth (Richmond et al. 2014). In TRAMP mice which are AR-positive, the evidence suggests that the AhR and its ligands are tumor growth inhibitory, although some mixed results were observed for TCDD (Fritz et al. 2007, 2009; Moore et al. 2016). The results of limited studies in urinary tract tumors suggest that the AhR and its ligands increase invasion (Ishida et al. 2010), whereas in kidney cancer cell lines the results are contradictory and may be cell context dependent (Callero et al. 2012; Ishida et al. 2015).
Neurological cancers
Glioblastoma is a highly lethal tumor in which survival times are low and treatment options are limited and not very effective. Initial studies showed that the AhR was expressed in human tumors and glioblastoma cell lines, and the pro-oncogenic activity of the AhR was linked to regulation of TGFβ signaling (Gramatzki et al. 2009). Moreover, this study showed that AhR knockdown or the AhR antagonist CH223191 inhibited clonal survival and migration of glioblastoma cells. A subsequent study by this group demonstrated that tryptophan-2,3-dioxygenase-mediated metabolism of tryptophan to give kynurenine was a key pro-carcinogenic event, since kynurenine promotes AhR-dependent tumor cell survival and motility (Opitz et al. 2011). A recent report indicates that AhR–integrin–TGFβ cross talk is also involved in glioblastoma (Silginer et al. 2016). It is clear that these studies demonstrate a potential clinical role for AhR antagonists in the treatment of glioblastoma. Other neurological cancers including medulloblastoma and pituitary adenomas also express an AhR that is pro-oncogenic (Dever and Opanashuk 2012; Jaffrain-Rea et al. 2009), whereas the AhR enhances differentiation in neuroblastoma cells (Huang et al. 2011) and TCDD induces apoptosis in PC12 cells (Sanchez-Martin et al. 2010). These studies suggest different roles for the AhR and its ligand in brain cancers (Table 2).
Lung, head and neck, esophageal, melanoma, leukemia and lymphoma
In lung cancer cells, there is evidence from most studies that PAHs and other ligands are growth promoters and induce growth-promoting genes, and the constitutive AhR is also involved in lung cancer cell growth (Chuang et al. 2012; Shimba et al. 2002; Wang et al. 2009) (Table 3). The major exception to these results was observed in CL1-5 cells which express low AhR levels; however, in an AhR-inducible cell line overexpression of the AhR protected against sidestream smoke-induced ROS (Cheng et al. 2012). This “protective” effect may be significant; however, AhR overexpression was also associated with increased anchorage-independent growth and cell proliferation and this is consistent with other studies in lung cancer cells. The AhR is also pro-oncogenic in head and neck and oral cancers and AhR agonists enhance cell growth and survival, whereas AhR antagonists exhibit anti-cancer activity, demonstrating a possible role for these compounds in clinical applications (DiNatale et al. 2011, 2012; Stanford et al. 2016a). The AhR is expressed in esophageal cancer and leukemia/lymphomas; however, the function of the AhR and its ligands are not well defined, although one study showed that β-naphthoflavone significantly inhibited invasion of esophageal cancer cells. Contradictory data have also been reported for melanoma. Loss of the AhR enhanced tumorigenicity in vivo and leflunomide inhibited melanoma cell proliferation (Contador-Troca et al. 2013; O’Donnell et al. 2012); however, it was also reported that AhR knockdown decreased growth (Barretina et al. 2012) and TCDD increased invasion and expression of MMPs (Villano et al. 2006). Differences in these data may be cell context dependent and mouse model specific and need further investigation.
Colon and gastric cancer
The functions of AhR ligands in colon cancer cells are cell context and ligand dependent (Table 4). Several different ligands, including 3-methylcholanthrene (MC) (Caco-2, LS174T) and TCDD (H508, SN7-C4), exhibit pro-oncogenic responses including induction of cell growth and genes associated with migration (MMP9) and drug transport (ABCG2) (Tompkins et al. 2010; Villard et al. 2007; Xie et al. 2012). However, in several other colon cancer cell lines, the AhR ligands FICZ (LoVo) and chrysin (HCT116, DLD-1 and SW837) inhibited cell growth (Ronnekleiv-Kelly et al. 2016; Yin et al. 2016). In contrast, several reports demonstrate that the loss of the AhR in wild-type and APCmin/+ mice enhances colon/cecum carcinogenesis and in APCmin/+ and wild-type mice I3C/DIM inhibit carcinogenesis (Diaz-Diaz et al. 2016; Ikuta et al. 2013; Kawajiri et al. 2009). Thus, the in vivo mouse model clearly demonstrates tumor suppressor-like activity for the AhR in colon/cecum cancer and specific AhR ligands can inhibit tumorigenesis. In MNK5 gastric cancer cells ± AhR, in vitro and in vivo (xenograft-AhR) studies indicate that the AhR promotes cell growth, migration and survival (Lai et al. 2014; Yin et al. 2013). TCDD induced proliferation and invasion of AGS cells (Peng et al. 2009), whereas DIM decreased SGC-7901 cell growth (Yin et al. 2012); however, it is not clear if the growth-inhibitory effects of DIM are AhR dependent. Expression of constitutively active AhR (CA-AhR) in mice results in gastric tumor formation, suggesting pro-oncogenic function of the receptor (Andersson et al. 2002; Kuznetsov et al. 2005). Future studies are needed to determine whether AhR agonists or antagonists will be effective for treatment for gastric cancer.
Liver cancer
Liver cancer is a leading cause of cancer-related mortality worldwide, accounting for more than 600,000 deaths each year. Although liver cancer is much more common in Southeast Asia, liver cancer cases worldwide including in the USA have been on the rise. The prognosis for liver cancer patients is quite poor, with a 5-year survival rate of approximately 15% (American Cancer Society 2016). This poor outcome is explained in large part by the ability of hepatocellular carcinoma (HCC), which accounts for 90% of liver cancers, to become resistant to chemotherapy and lack of existing targeted therapies. The only targeted therapy for liver cancer is sorafenib, a kinase inhibitor that extends patient survival, on average, by only 3 months (Bruera et al. 2014). Thus, there is a dire need to make bold moves and identify effective treatment options for liver cancer patients. Based on the recent evidence summarized below, we propose that the AhR is a viable molecular target for liver cancer. The function of the AhR in liver cancer is somewhat contradictory and the role of AhR and its ligands in both in vitro and in vivo model systems is summarized in Table 5.
AhR plays a significant role in development, presumably due to its ability to regulate cell growth and differentiation. AhR null mice have much smaller livers and display defects in development of vasculature (Fernandez-Salguero et al. 1996; Lahvis and Bradfield 1998; Mimura et al. 1997). Genes required for proper growth and development often play significant roles in cancer, functioning as oncogenes or tumor suppressors and sometimes both as tumor suppressor and oncogene depending on the context and stimuli. The genetic background or the expression of other co-regulatory proteins plays a role in the function of a gene. AhR null mice do not develop spontaneous tumors in liver suggesting that the AhR is not a classical tumor suppressor gene. Tumorigenesis is still a rare event and it is often kept under control by checks and balances in the system regulated by multiple genes that eliminate abnormal cells. The endogenous AhR functions as a tumor modifier gene in liver cancer in the absence of any exogenous ligand stimulation. The identification of a tumor modifier role for the AhR was investigated by crossing the AhR knockout mice with mice that express oncogenes or by exposure to chemical carcinogens that predispose mice to cancer. Puga and colleagues utilized genotoxic carcinogen diethylnitrosamine (DEN) to induce liver tumors in wild-type mice expressing the AhR and knockout mice lacking the AhR (Fan et al. 2010). In this study, the absence of the AhR expression was associated with increased BrdU incorporation, a marker used to identify proliferating cells. In addition, decreased expression of known tumor suppressor genes in this study strongly demonstrated a tumor-suppressive modifier role for the AhR.
The AhR is highly expressed in liver cancer cells (O’Donnell et al. 2012) and several AhR ligands inhibit cancer cell proliferation and/or induce liver cancer cell death. Some of these effects have been shown to be dependent on AhR expression. Recent evidence including results from our laboratories supports the possibility that the AhR can also be transformed to yield biological responses that can be exploited for the treatment of cancer (Jin et al. 2014, 2015; Koch et al. 2015; O’Donnell et al. 2012, 2014; Safe et al. 2013). Chemical libraries were screened to identify AhR ligands that have anti-cancer effects. The specificity and selectivity of the identified small molecules for the AhR were validated in well-characterized cell systems. Furthermore, these compounds were tested for AhR-dependent growth-inhibitory effects in cancer cells. This resulted in the identification of promising AhR ligands with potential anti-cancer effects, one of which was raloxifene. Raloxifene is a selective estrogen receptor modulator used in the clinic for prevention of osteoporosis. Raloxifene directly bound the AhR, promoted cytosol to nuclear translocation of the AhR, strongly activated AhR-driven reporter gene activity and endogenous AhR target genes (Bisson et al. 2009; O’Donnell et al. 2014). AhR-dependent programmed cell death in breast and liver cancer cells that do not express estrogen receptor contributed to raloxifene-induced growth inhibition. Despite the ability of TCDD to strongly activate AhR signaling, TCDD did not induce apoptosis suggesting the unique activity of certain AhR ligands such as raloxifene (O’Donnell et al. 2014). Unlike TCDD, raloxifene is not a high affinity ligand and it is important to understand ligand-selective AhR signaling that drive AhR-dependent anti-cancer activities. Raloxifene is well tolerated in humans and this compound or new raloxifene-based molecules with improved AhR binding affinity need to be identified for future clinical applications.
Humans exposed to high levels of TCDD did not exhibit higher incidences of cancer (Collins et al. 2009; McBride et al. 2009). Analysis of TOXcast chemicals and their activation of nuclear receptors including AhR revealed that there was no association between AhR activation and progression of hepatic lesions (Shah et al. 2011). Human HCCLM3 hepatoma cells were inhibited both in vitro and in vivo (xenograft) by the AhR ligand ITE (Zhao et al. 2015). The FDA-approved drug and anti-androgen, flutamide, is also an AhR ligand, and the growth-suppressive effects of flutamide are due to AhR-dependent induction of TGFβ1 in human HCC cells (Koch et al. 2015). AhR-mediated activation of TGFβ1 signaling resulted in activation of cell cycle inhibitory proteins p15 and p27, and knockdown of AhR or TGFβ1 abrogated the anti-proliferative effects of flutamide. This is an example of an AhR-active approved pharmaceutical that could be repurposed for treatment of hepatocellular carcinomas.
Breast cancer
Breast cancer is the most common cancer among women worldwide and metastasis is responsible for most of the deaths associated with breast cancer. Breast cancer is composed of multiple subtypes with distinct molecular markers. The three major classes of breast cancers are (i) hormone receptor-positive cancers that express estrogen receptor (ER) and progesterone receptor (PR), (ii) human epidermal growth factor receptor 2 (HER2)-positive cancers meaning cancers with overexpression of Her2 and (iii) triple-negative breast cancers (TNBC) that do not express ER or PR with normal or no expression of Her2 (American Cancer Society 2016; Santagata et al. 2014). Approximately, 20% of breast cancers are classified as TNBC, which is composed of at least six subclasses (Lehmann et al. 2011). TNBCs are the most difficult to treat with very limited options and poor prognosis.
The AhR is expressed in both hormone receptor-positive and -negative breast cancers including in TNBC (O’Donnell et al. 2010). Higher expression of AhR correlates with better prognosis including increased overall survival and distant metastasis-free survival in different forms of breast cancer (O’Donnell et al. 2014). Targeting AhR expressing breast cancer patient subsets with AhR-based therapeutics is an exciting possibility for patients with limited treatment options and a recent paper elucidating the role of AhR in breast cancer is summarized in Table 6. Many studies presented in this table strongly support the role of AhR as an anti-cancer target in breast cancer.
TCDD pretreatment inhibited chemical carcinogen 7,12-dimethylbenz[a]anthracene-induced mammary tumors in CB6F1 mice (Wang et al. 2011a). Diindolylmethane (DIM), a dietary AhR ligand, also inhibited DMBA-induced mammary tumors in Sprague–Dawley rats (Chen et al. 1998). TCDD exposure reduced breast tumor metastasis to the lung and to other mammary glands in a syngeneic mouse model of breast cancer metastasis (Wang et al. 2011b). Interestingly, TCDD treatment did not influence primary tumor growth in these mice or affect proliferation in in vitro assays. The data from these studies support testing of AhR-targeting anti-cancer compounds independently both in vitro and in vivo studies. Most of the breast cancer deaths are due to complications in distant organ metastasis, and systematic testing of different classes of AhR modulators will likely identify those that effectively inhibit metastasis.
The proton pump inhibitor omeprazole activates AhR transcription and also decreases metastasis of triple-negative breast cancer cells (Jin et al. 2014). Activation of the AhR by certain agonists including omeprazole downregulated G-protein coupled receptor CXCR4, which is implicated in the promotion of metastasis of breast tumors (Hall et al. 2010; Hsu et al. 2007, 2008; Jin et al. 2014; Wang et al. 2011b). AhR–regulated microRNAs also have roles in breast cancer metastasis. TCDD and MCDF induced the expression of miR-335 in BT474 and MDA-MD-231 cells (Zhang et al. 2012a) resulting in the inhibition of the prometastatic SOX4 gene and inhibition of lung metastasis in vivo. The antiestrogen raloxifene induced apoptosis in TNBC cells, indicating that this compound or its analogs also have potential as AhR-targeted therapeutics for breast cancer therapy (O’Donnell et al. 2014). Focused virtual ligand screening utilizing AhR ligand binding pocket models may help to identify such compounds (Bisson et al. 2009; Perkins et al. 2014).
Cancer stem cells
There is also evidence that the AhR plays a role in stem cell functions and this includes an early study showing that AhR antagonists promoted the expansion of hematopoietic stem cells (Bock 2017; Boitano et al. 2010; Casado et al. 2011; Hou et al. 2013; Rentas et al. 2016; Singh et al. 2009). Cancer stem cells are often drug resistant and are important for maintaining and expanding individual tumor types. There is also evidence that the AhR can be targeted in cancer stem cells; for example, the AhR-active pharmaceutical tranilast significantly inhibits breast cancer stem cell growth and metastasis in vivo using MDA-MB-231 drug-surviving cancer stem cells (Prud’homme et al. 2010). Another study characterized the Ah-responsiveness of triple-negative Hs578T breast cancer-derived stem cells and showed that AhR ligands induce AhR interactions with Sox2, a regulator of self-renewal and this study clearly demonstrated a role for the AhR and its agonists as enhancers of cancer stem cells (Stanford et al. 2016b). These results differ from those observed using tranilast suggesting some cell context-dependent differences in AhR function in breast cancer stem cells, and this may be related to differential expression of the AhR, Arnt, HIF-1α and other cofactors. Cheng et al. (2015) investigated the effects of several tryptophan-derived AhR ligands including 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) and demonstrated that these compounds suppressed transcription of Oct4 in stem-like cancer cells. ITE induced an AhR-dependent decrease in Oct4, a stem cell marker, and also decreased the tumorigenicity of stem-like leukemia (U87) cancer cells. In contrast, AhR antagonists enhanced leukemia stem cell activity (Pabst et al. 2014) and this corresponded to their effects reported in hematopoietic stem cells (Boitano et al. 2010). These results and other studies (Kim et al. 2016; Tsai et al. 2015) demonstrate that the AhR and AhR-regulated genes such as Oct4 are important in cancer stem cells, indicating that AhR ligands (agonists or antagonists) are a unique set of agents for targeting cancer stem cells.
Concluding remarks
The endogenous function of the AhR as a tumor modifier and the anti-cancer effects stimulated by distinct classes of AhR ligands with diverse pharmacologies offer an opportunity to pursue AhR signaling holistically beyond TCDD-induced responses. The effects of TCDD and AhR functions have been interlinked for a long time resulting in decreased support by major funding agencies and biotech companies for developing AhR-based cancer therapeutics. The reason for the cautionary approach to target AhR in cancers is understandable, when there are other treatment options or clearly targetable molecular pathways. However, for difficult to treat cancers and for cancers where treatment options are very limited or non-existent, such as pancreatic, liver and hormone-independent breast and prostate cancers, the time is ripe to exploit the potential of AhR signaling to develop a new class of anti-cancer therapeutics. It is important to define the modes of AhR function that contributes to its anti-cancer actions and some common themes have emerged including regulation of cell cycle genes (Hall et al. 2010; Huang and Elferink 2005; Jin et al. 2014; Kolluri et al. 1999; Levine-Fridman et al. 2004; Zhang et al. 2009), interaction with distinct co-regulatory molecules (Barhoover et al. 2010; Huang and Elferink 2005; Kang et al. 2006; Safe et al. 2013) and non-genomic pathways that contribute to the anti-cancer activities of the AhR (Jin et al. 2015) (see summary; Fig. 3). Design and selection of AhR ligands based on a given anti-cancer mechanism of action will allow discovery of molecules with therapeutic value. There are numerous successful examples from the nuclear receptor field where therapeutics targeting the retinoid X receptor (bexarotene), ER (tamoxifen and raloxifene), AR (flutamide, enzalutamide) and glucocorticoid receptor (fluticasone) (Bambury and Scher 2015; Helsen et al. 2014; le Maire et al. 2012; McDonnell and Wardell 2010; Su et al. 2016) have been identified and used in clinical applications. It will be fascinating to see FDA-approved AhR-targeted compounds added to this list and this is strongly supported by the increasing number of studies showing that ligands for this receptor target many of the hallmarks of cancer (Fig. 2) through activating/inactivating various genes and pathways (Fig. 3).

A summary of the role of the AhR and its ligands (agonists or antagonists) as inhibitors of carcinogenesis
References
Aesoy R, Clyne CD, Chand AL (2015) Insights into orphan nuclear receptors as prognostic markers and novel therapeutic targets for breast cancer. Front Endocrinol 6:115. doi:10.3389/fendo.2015.00115
American Cancer Society (2016) Cancer facts and figures 2016. American Cancer Society, Atlanta
Andersson P, McGuire J, Rubio C et al (2002) A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci USA 99(15):9990–9995. doi:10.1073/pnas.152706299
Andersson P, Rubio C, Poellinger L, Hanberg A (2005) Gastric hamartomatous tumours in a transgenic mouse model expressing an activated dioxin/Ah receptor. Anticancer Res 25(2A):903–911
Baek SH, Kim KI (2014) Emerging roles of orphan nuclear receptors in cancer. Annu Rev Physiol 76:177–195. doi:10.1146/annurev-physiol-030212-183758
Bambury RM, Scher HI (2015) Enzalutamide: development from bench to bedside. Urol Oncol 33(6):280–288. doi:10.1016/j.urolonc.2014.12.017
Barhoover MA, Hall JM, Greenlee WF, Thomas RS (2010) Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol 77(2):195–201. doi:10.1124/mol.109.059675
Barnes-Ellerbe S, Knudsen KE, Puga A (2004) 2,3,7,8-Tetrachlorodibenzo-p-dioxin blocks androgen-dependent cell proliferation of LNCaP cells through modulation of pRB phosphorylation. Mol Pharmacol 66(3):502–511. doi:10.1124/mol.104.000356
Barretina J, Caponigro G, Stransky N et al (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483(7391):603–607. doi:10.1038/nature11003
Becker RA, Patlewicz G, Simon TW, Rowlands JC, Budinsky RA (2015) The adverse outcome pathway for rodent liver tumor promotion by sustained activation of the aryl hydrocarbon receptor. Regul Toxicol Pharmacol 73(1):172–190. doi:10.1016/j.yrtph.2015.06.015
Benson JM, Shepherd DM (2011) Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol Sci 120(1):68–78. doi:10.1093/toxsci/kfq360
Bisson WH, Koch DC, O’Donnell EF et al (2009) Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem 52(18):5635–5641. doi:10.1021/jm900199u
Bock KW (2017) From dioxin toxicity to putative physiologic functions of the human Ah receptor in homeostasis of stem/progenitor cells. Biochem Pharmacol 123:1–7. doi:10.1016/j.bcp.2016.06.015
Bock KW, Kohle C (2005) Ah receptor- and TCDD-mediated liver tumor promotion: clonal selection and expansion of cells evading growth arrest and apoptosis. Biochem Pharmacol 69(10):1403–1408. doi:10.1016/j.bcp.2005.02.004
Boitano AE, Wang J, Romeo R et al (2010) Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329(5997):1345–1348. doi:10.1126/science.1191536
Brinkman AM, Wu J, Ersland K, Xu W (2014) Estrogen receptor alpha and aryl hydrocarbon receptor independent growth inhibitory effects of aminoflavone in breast cancer cells. BMC Cancer 14:344. doi:10.1186/1471-2407-14-344
Bruera G, Cannita K, Giordano AV et al (2014) Multidisciplinary management of hepatocellular carcinoma in clinical practice. Biomed Res Int 2014:806391. doi:10.1155/2014/806391
Bunaciu RP, Yen A (2011) Activation of the aryl hydrocarbon receptor AhR promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res 71(6):2371–2380. doi:10.1158/0008-5472.CAN-10-2299
Burris TP, Solt LA, Wang Y et al (2013) Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev 65(2):710–778. doi:10.1124/pr.112.006833
Callero MA, Suarez GV, Luzzani G, Itkin B, Nguyen B, Loaiza-Perez AI (2012) Aryl hydrocarbon receptor activation by aminoflavone: new molecular target for renal cancer treatment. Int J Oncol 41(1):125–134. doi:10.3892/ijo.2012.1427
Casado FL, Singh KP, Gasiewicz TA (2011) Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol Pharmacol 80(4):673–682. doi:10.1124/mol.111.071381
Chang JT, Chang H, Chen PH, Lin SL, Lin P (2007) Requirement of aryl hydrocarbon receptor overexpression for CYP1B1 up-regulation and cell growth in human lung adenocarcinomas. Clin Cancer Res 13(1):38–45. doi:10.1158/1078-0432.CCR-06-1166
Chen I, McDougal A, Wang F, Safe S (1998) Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis 19:1631–1639
Cheng YH, Huang SC, Lin CJ, Cheng LC, Li LA (2012) Aryl hydrocarbon receptor protects lung adenocarcinoma cells against cigarette sidestream smoke particulates-induced oxidative stress. Toxicol Appl Pharmacol 259(3):293–301. doi:10.1016/j.taap.2012.01.005
Cheng J, Li W, Kang B et al (2015) Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat Commun 6:7209. doi:10.1038/ncomms8209
Chuang CY, Chang H, Lin P et al (2012) Up-regulation of osteopontin expression by aryl hydrocarbon receptor via both ligand-dependent and ligand-independent pathways in lung cancer. Gene 492(1):262–269. doi:10.1016/j.gene.2011.10.019
Collins JJ, Bodner K, Aylward LL, Wilken M, Bodnar CM (2009) Mortality rates among trichlorophenol workers with exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Am J Epidemiol 170(4):501–506. doi:10.1093/aje/kwp153
Contador-Troca M, Alvarez-Barrientos A, Barrasa E et al (2013) The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis 34(12):2683–2693. doi:10.1093/carcin/bgt248
D’Amato NC, Rogers TJ, Gordon MA et al (2015) A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res 75(21):4651–4664. doi:10.1158/0008-5472.CAN-15-2011
de Tomaso Portaz AC, Caimi GR, Sanchez M et al (2015) Hexachlorobenzene induces cell proliferation, and aryl hydrocarbon receptor expression (AhR) in rat liver preneoplastic foci, and in the human hepatoma cell line HepG2. AhR is a mediator of ERK1/2 signaling, and cell cycle regulation in HCB-treated HepG2 cells. Toxicology 336:36–47. doi:10.1016/j.tox.2015.07.013
Denison MS, Nagy SR (2003) Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 43:309–334. doi:10.1146/annurev.pharmtox.43.100901.135828
Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B (2011) Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci 124(1):1–22. doi:10.1093/toxsci/kfr218
Dever DP, Opanashuk LA (2012) The aryl hydrocarbon receptor contributes to the proliferation of human medulloblastoma cells. Mol Pharmacol 81(5):669–678. doi:10.1124/mol.111.077305
Dewa Y, Nishimura J, Jin M et al (2009) Molecular expression analysis of beta-naphthoflavone-induced hepatocellular tumors in rats. Toxicol Pathol 37(4):446–455. doi:10.1177/0192623309335062
Diaz-Diaz CJ, Ronnekleiv-Kelly SM, Nukaya M et al (2016) The aryl hydrocarbon receptor is a repressor of inflammation-associated colorectal tumorigenesis in mouse. Ann Surg 264(3):429–436. doi:10.1097/SLA.0000000000001874
DiNatale BC, Schroeder JC, Perdew GH (2011) Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol Carcinog 50(3):173–183. doi:10.1002/mc.20702
DiNatale BC, Smith K, John K, Krishnegowda G, Amin SG, Perdew GH (2012) Ah receptor antagonism represses head and neck tumor cell aggressive phenotype. Mol Cancer Res 10(10):1369–1379. doi:10.1158/1541-7786.MCR-12-0216
Ehrlich AK, Pennington JM, Wang X et al (2016) Activation of the aryl hydrocarbon receptor by 10-Cl-BBQ prevents insulitis and effector T cell development independently of Foxp3+ regulatory T cells in nonobese diabetic mice. J Immunol 196(1):264–273. doi:10.4049/jimmunol.1501789
Esser C (2012) Biology and function of the aryl hydrocarbon receptor: report of an international and interdisciplinary conference. Arch Toxicol 86(8):1323–1329. doi:10.1007/s00204-012-0818-2
Fan Y, Boivin GP, Knudsen ES, Nebert DW, Xia Y, Puga A (2010) The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res 70(1):212–220. doi:10.1158/0008-5472.CAN-09-3090
Ferdinand R, Mitchell SA, Batson S, Tumur I (2012) Treatments for chronic myeloid leukemia: a qualitative systematic review. J Blood Med 3:51–76. doi:10.2147/JBM.S33380
Fernandez-Salguero P, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ (1996) Aryl hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol 140:173–179
Fritz WA, Lin TM, Cardiff RD, Peterson RE (2007) The aryl hydrocarbon receptor inhibits prostate carcinogenesis in TRAMP mice. Carcinogenesis 28(2):497–505. doi:10.1093/carcin/bgl179
Fritz WA, Lin TM, Safe S, Moore RW, Peterson RE (2009) The selective aryl hydrocarbon receptor modulator 6-methyl-1,3,8-trichlorodibenzofuran inhibits prostate tumor metastasis in TRAMP mice. Biochem Pharmacol 77(7):1151–1160. doi:10.1016/j.bcp.2008.12.015
Fukasawa K, Kagaya S, Maruyama S et al (2015) A novel compound, NK150460, exhibits selective antitumor activity against breast cancer cell lines through activation of aryl hydrocarbon receptor. Mol Cancer Ther 14(2):343–354. doi:10.1158/1535-7163.MCT-14-0158
Gluschnaider U, Hidas G, Cojocaru G, Yutkin V, Ben-Neriah Y, Pikarsky E (2010) beta-TrCP inhibition reduces prostate cancer cell growth via upregulation of the aryl hydrocarbon receptor. PLoS ONE 5(2):e9060. doi:10.1371/journal.pone.0009060
Goode GD, Ballard BR, Manning HC, Freeman ML, Kang Y, Eltom SE (2013) Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int J Cancer 133(12):2769–2780. doi:10.1002/ijc.28297
Gramatzki D, Pantazis G, Schittenhelm J et al (2009) Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 28(28):2593–2605. doi:10.1038/onc.2009.104
Gu A, Ji G, Jiang T et al (2012) Contributions of aryl hydrocarbon receptor genetic variants to the risk of glioma and PAH-DNA adducts. Toxicol Sci 128(2):357–364. doi:10.1093/toxsci/kfs158
Haggiag S, Ruggieri S, Gasperini C (2013) Efficacy and safety of laquinimod in multiple sclerosis: current status. Ther Adv Neurol Disord 6(6):343–352. doi:10.1177/1756285613499424
Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS (2010) Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol Endocrinol 24(2):359–369. doi:10.1210/me.2009-0346
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Hanieh H (2015) Aryl hydrocarbon receptor-microRNA-212/132 axis in human breast cancer suppresses metastasis by targeting SOX4. Mol Cancer 14:172. doi:10.1186/s12943-015-0443-9
Haque M, Francis J, Sehgal I (2005) Aryl hydrocarbon exposure induces expression of MMP-9 in human prostate cancer cell lines. Cancer Lett 225(1):159–166. doi:10.1016/j.canlet.2004.11.043
Harrill JA, Parks BB, Wauthier E, Rowlands JC, Reid LM, Thomas RS (2015) Lineage-dependent effects of aryl hydrocarbon receptor agonists contribute to liver tumorigenesis. Hepatology 61(2):548–560. doi:10.1002/hep.27547
Hayashibara T, Yamada Y, Mori N et al (2003) Possible involvement of aryl hydrocarbon receptor (AhR) in adult T-cell leukemia (ATL) leukemogenesis: constitutive activation of AhR in ATL. Biochem Biophys Res Commun 300(1):128–134
Helsen C, Van den Broeck T, Voet A et al (2014) Androgen receptor antagonists for prostate cancer therapy. Endocr Relat Cancer 21(4):T105–T118. doi:10.1530/ERC-13-0545
Hou P, Li Y, Zhang X et al (2013) Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341(6146):651–654. doi:10.1126/science.1239278
Hruba E, Vondracek J, Libalova H et al (2011) Gene expression changes in human prostate carcinoma cells exposed to genotoxic and nongenotoxic aryl hydrocarbon receptor ligands. Toxicol Lett 206(2):178–188. doi:10.1016/j.toxlet.2011.07.011
Hsu EL, Yoon D, Choi HH et al (2007) A proposed mechanism for the protective effect of dioxin against breast cancer. Toxicol Sci 98(2):436–444. doi:10.1093/toxsci/kfm125
Hsu EL, Chen N, Westbrook A et al (2008) CXCR4 and CXCL12 down-regulation: a novel mechanism for the chemoprotection of 3,3′-diindolylmethane for breast and ovarian cancers. Cancer Lett 265(1):113–123. doi:10.1016/j.canlet.2008.02.033
Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR (2007) Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol Pharmacol 71(6):1475–1486
Huang G, Elferink CJ (2005) Multiple mechanisms are involved in Ah receptor-mediated cell cycle arrest. Mol Pharmacol 67(1):88–96. doi:10.1124/mol.104.002410
Huang TC, Chang HY, Chen CY et al (2011) Silencing of miR-124 induces neuroblastoma SK-N-SH cell differentiation, cell cycle arrest and apoptosis through promoting AHR. FEBS Lett 585(22):3582–3586. doi:10.1016/j.febslet.2011.10.025
Iida K, Mimura J, Itoh K et al (2010) Suppression of AhR signaling pathway is associated with the down-regulation of UDP-glucuronosyltransferases during BBN-induced urinary bladder carcinogenesis in mice. J Biochem 147(3):353–360. doi:10.1093/jb/mvp169
Ikuta T, Kobayashi Y, Kitazawa M et al (2013) ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis 34(7):1620–1627. doi:10.1093/carcin/bgt083
Ishida M, Mikami S, Kikuchi E et al (2010) Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by upregulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. Carcinogenesis 31(2):287–295. doi:10.1093/carcin/bgp222
Ishida M, Mikami S, Shinojima T et al (2015) Activation of aryl hydrocarbon receptor promotes invasion of clear cell renal cell carcinoma and is associated with poor prognosis and cigarette smoke. Int J Cancer 137(2):299–310. doi:10.1002/ijc.29398
Jaffrain-Rea ML, Angelini M, Gargano D et al (2009) Expression of aryl hydrocarbon receptor (AHR) and AHR-interacting protein in pituitary adenomas: pathological and clinical implications. Endocr Relat Cancer 16(3):1029–1043. doi:10.1677/ERC-09-0094
Jana NR, Sarkar S, Ishizuka M, Yonemoto J, Tohyama C, Sone H (1999) Cross-talk between 2,3,7,8-tetrachlorodibenzo-p-dioxin and testosterone signal transduction pathways in LNCaP prostate cancer cells. Biochem Biophys Res Commun 256(3):462–468
Jin UH, Lee SO, Pfent C, Safe S (2014) The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer 14:498. doi:10.1186/1471-2407-14-498
Jin UH, Kim SB, Safe S (2015) Omeprazole inhibits pancreatic cancer cell invasion through a nongenomic aryl hydrocarbon receptor pathway. Chem Res Toxicol 28(5):907–918. doi:10.1021/tx5005198
Jordan VC (2007) SERMs: meeting the promise of multifunctional medicines. J Natl Cancer Inst 99(5):350–356. doi:10.1093/jnci/djk062
Jordan VC (2009) A century of deciphering the control mechanisms of sex steroid action in breast and prostate cancer: the origins of targeted therapy and chemoprevention. Cancer Res 69(4):1243–1254. doi:10.1158/0008-5472.CAN-09-0029
Kang HJ, Kim HJ, Kim SK et al (2006) BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J Biol Chem 281(21):14654–14662. doi:10.1074/jbc.M601613200
Kawajiri K, Kobayashi Y, Ohtake F et al (2009) Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci USA 106(32):13481–13486. doi:10.1073/pnas.0902132106
Kennedy GD, Nukaya M, Moran SM et al (2014) Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and TNF/IL-1 receptors. Toxicol Sci 140(1):135–143. doi:10.1093/toxsci/kfu065
Kerkvliet NI, Steppan LB, Vorachek W et al (2009) Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 1(4):539–547. doi:10.2217/imt.09.24
Kim HM, Kim JW, Choi Y et al (2016) Xeno-sensing activity of the aryl hydrocarbon receptor in human pluripotent stem cell-derived hepatocyte-like cells. Sci Rep 6:21684. doi:10.1038/srep21684
Knerr S, Schrenk D (2006) Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol Nutr Food Res 50(10):897–907. doi:10.1002/mnfr.200600006
Koch DC, Jang HS, O’Donnell EF et al (2015) Anti-androgen flutamide suppresses hepatocellular carcinoma cell proliferation via the aryl hydrocarbon receptor mediated induction of transforming growth factor-beta1. Oncogene 34(50):6092–6104. doi:10.1038/onc.2015.55
Kociba RJ, Keyes DG, Beyer JE et al (1978) Results of a 2-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicol Appl Pharmacol 46:279–303
Koliopanus A, Kleeff J, Xiao Y et al (2002) Increased aryl hydrocarbon receptor expression offers a potential therapeutic target in pancreatic cancer. Oncogene 21:6059–6070
Kolluri SK, Weiss C, Koff A, Gottlicher M (1999) p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev 13:1742–1753
Kolluri SK, Balduf C, Hofmann M, Gottlicher M (2001) Novel target genes of the Ah (dioxin) receptor: transcriptional induction of N-myristoyltransferase 2. Cancer Res 61(23):8534–8539
Kuznetsov NV, Andersson P, Gradin K et al (2005) The dioxin/aryl hydrocarbon receptor mediates downregulation of osteopontin gene expression in a mouse model of gastric tumourigenesis. Oncogene 24(19):3216–3222. doi:10.1038/sj.onc.1208529
Lahvis GP, Bradfield CA (1998) Ahr null alleles: distinctive or different? Biochem Pharmacol 56(7):781–787
Lai DW, Liu SH, Karlsson AI et al (2014) The novel aryl hydrocarbon receptor inhibitor biseugenol inhibits gastric tumor growth and peritoneal dissemination. Oncotarget 5(17):7788–7804. doi:10.18632/oncotarget.2307
le Maire A, Alvarez S, Shankaranarayanan P, Lera AR, Bourguet W, Gronemeyer H (2012) Retinoid receptors and therapeutic applications of RAR/RXR modulators. Curr Top Med Chem 12(6):505–527
Lehmann BD, Bauer JA, Chen X et al (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767. doi:10.1172/JCI45014
Levine-Fridman A, Chen L, Elferink CJ (2004) Cytochrome P4501A1 promotes G1 phase cell cycle progression by controlling aryl hydrocarbon receptor activity. Mol Pharmacol 65(2):461–469. doi:10.1124/mol.65.2.461
Li ZD, Wang K, Yang XW, Zhuang ZG, Wang JJ, Tong XW (2014) Expression of aryl hydrocarbon receptor in relation to p53 status and clinicopathological parameters in breast cancer. Int J Clin Exp Pathol 7(11):7931–7937
Loaiza-Perez AI, Kenney S, Boswell J et al (2004) Aryl hydrocarbon receptor activation of an antitumor aminoflavone: basis of selective toxicity for MCF-7 breast tumor cells. Mol Cancer Ther 3(6):715–725
Marshall NB, Kerkvliet NI (2010) Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci 1183:25–37. doi:10.1111/j.1749-6632.2009.05125.x
Masui K, Gini B, Wykosky J et al (2013) A tale of two approaches: complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 34(4):725–738. doi:10.1093/carcin/bgt086
McBride DI, Collins JJ, Humphry NF et al (2009) Mortality in workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin at a trichlorophenol plant in New Zealand. J Occup Environ Med 51(9):1049–1056. doi:10.1097/JOM.0b013e3181b571ae
McDonnell DP, Wardell SE (2010) The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol 10(6):620–628. doi:10.1016/j.coph.2010.09.007
McLean LS, Watkins CN, Campbell P et al (2015) Aryl hydrocarbon receptor ligand 5F 203 induces oxidative stress that triggers DNA damage in human breast cancer cells. Chem Res Toxicol 28(5):855–871. doi:10.1021/tx500485v
Miller KD, Siegel RL, Lin CC et al (2016) Cancer treatment and survivorship statistics. CA Cancer J Clin 66(4):271–289. doi:10.3322/caac.21349
Mimura J, Yamashita K, Nakamura K et al (1997) Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 2(10):645–654
Moennikes O, Loeppen S, Buchmann A et al (2004) A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res 64(14):4707–4710. doi:10.1158/0008-5472.CAN-03-0875
Moore RW, Fritz WA, Schneider AJ et al (2016) 2,3,7,8-Tetrachlorodibenzo-p-dioxin has both pro-carcinogenic and anti-carcinogenic effects on neuroendocrine prostate carcinoma formation in TRAMP mice. Toxicol Appl Pharmacol 305:242–249. doi:10.1016/j.taap.2016.04.018
Morrow D, Qin C, Smith Iii R, Safe S (2004) Aryl hydrocarbon receptor-mediated inhibition of LNCaP prostate cancer cell growth and hormone-induced transactivation. J Ster Biochem Mol Biol 88:27–36
Mulero-Navarro S, Carvajal-Gonzalez JM, Herranz M et al (2006) The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis 27(5):1099–1104. doi:10.1093/carcin/bgi344
Murray IA, Morales JL, Flaveny CA et al (2010) Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol Pharmacol 77(2):247–254. doi:10.1124/mol.109.061788
Murray IA, Patterson AD, Perdew GH (2014) Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer 14(12):801–814. doi:10.1038/Nrc3846
Nukaya M, Walisser JA, Moran SM, Kennedy GD, Bradfield CA (2010) Aryl hydrocarbon receptor nuclear translocator in hepatocytes is required for aryl hydrocarbon receptor-mediated adaptive and toxic responses in liver. Toxicol Sci 118(2):554–563. doi:10.1093/toxsci/kfq305
O’Donnell EF, Saili KS, Koch DC et al (2010) The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS ONE 5(10):e13128. doi:10.1371/journal.pone.0013128
O’Donnell EF, Kopparapu PR, Koch DC et al (2012) The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PLoS ONE 7(7):e40926. doi:10.1371/journal.pone.0040926
O’Donnell EF, Koch DC, Bisson WH, Jang HS, Kolluri SK (2014) The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis 5:e1038. doi:10.1038/cddis.2013.549
Opitz CA, Litzenburger UM, Sahm F et al (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478(7368):197–203. doi:10.1038/nature10491
Pabst C, Krosl J, Fares I et al (2014) Identification of small molecules that support human leukemia stem cell activity ex vivo. Nat Methods 11(4):436–442. doi:10.1038/nmeth.2847
Parks AJ, Pollastri MP, Hahn ME et al (2014) In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol Pharmacol 86(5):593–608. doi:10.1124/mol.114.093369
Peng TL, Chen J, Mao W, Song X, Chen MH (2009) Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biology 10:27. doi:10.1186/1471-2121-10-27
Perkins A, Phillips JL, Kerkvliet NI et al (2014) A structural switch between agonist and antagonist bound conformations for a ligand-optimized model of the human aryl hydrocarbon receptor ligand binding domain. Biology 3(4):645–669. doi:10.3390/biology3040645
Perou CM, Sorlie T, Eisen MB et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752. doi:10.1038/35021093
Pesatori AC, Consonni D, Rubagotti M, Grillo P, Bertazzi PA (2009) Cancer incidence in the population exposed to dioxin after the “Seveso accident”: twenty years of follow-up. Environ Health 8:39. doi:10.1186/1476-069X-8-39
Poland A, Knutson JC (1982) 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons. Examinations of the mechanism of toxicity. Ann Rev Pharmacol Toxicol 22:517–554
Poland A, Glover E, Kende AS (1976) Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol: evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem 251:4936–4946
Portal-Nunez S, Shankavaram UT, Rao M et al (2012) Aryl hydrocarbon receptor-induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. Cancer Res 72(22):5790–5800. doi:10.1158/0008-5472.CAN-12-0818
Prud’homme GJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S (2010) Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS ONE 5(11):e13831. doi:10.1371/journal.pone.0013831
Punj S, Kopparapu P, Jang HS et al (2014) Benzimidazoisoquinolines: a new class of rapidly metabolized aryl hydrocarbon receptor (AhR) ligands that induce AhR-dependent Tregs and prevent murine graft-versus-host disease. PLoS ONE 9(2):e88726. doi:10.1371/journal.pone.0088726
Quintana FJ, Basso AS, Iglesias AH et al (2008) Control of Treg and T(9)H17 cell differentiation by the aryl hydrocarbon receptor. Nature 453(7191):65–71
Rentas S, Holzapfel NT, Belew MS et al (2016) Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. Nature 532(7600):508–511. doi:10.1038/nature17665
Richmond O, Ghotbaddini M, Allen C, Walker A, Zahir S, Powell JB (2014) The aryl hydrocarbon receptor is constitutively active in advanced prostate cancer cells. PLoS ONE 9(4):e95058. doi:10.1371/journal.pone.0095058
Rignall B, Grote K, Gavrilov A et al (2013) Biological and tumor-promoting effects of dioxin-like and non-dioxin-like polychlorinated biphenyls in mouse liver after single or combined treatment. Toxicol Sci 133(1):29–41. doi:10.1093/toxsci/kft034
Ronnekleiv-Kelly SM, Nukaya M, Diaz-Diaz CJ et al (2016) Aryl hydrocarbon receptor-dependent apoptotic cell death induced by the flavonoid chrysin in human colorectal cancer cells. Cancer Lett 370(1):91–99. doi:10.1016/j.canlet.2015.10.014
Safe S, Qin C, McDougal A (1999) Development of selective aryl hydrocarbon receptor modulators (SAhRMs) for treatment of breast cancer. Expert Opin Investig Drugs 8:1385–1396
Safe S, Chadalapaka G, Jutooru I (2012) AHR-reactive compounds in the human diet. In: Pohjanvirta R (ed) The Ah receptor in biology and toxicology. Wiley, Hoboken, pp 331–342
Safe S, Lee SO, Jin UH (2013) Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol Sci 135(1):1–16. doi:10.1093/toxsci/kft128
Sanchez-Martin FJ, Fernandez-Salguero PM, Merino JM (2010) 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptosis in neural growth factor (NGF)-differentiated pheochromocytoma PC12 cells. Neurotoxicology 31(3):267–276. doi:10.1016/j.neuro.2010.03.005
Santagata S, Thakkar A, Ergonul A et al (2014) Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J Clin Invest 124(2):859–870. doi:10.1172/JCI70941
Schreck I, Deigendesch U, Burkhardt B, Marko D, Weiss C (2012) The Alternaria mycotoxins alternariol and alternariol methyl ether induce cytochrome P450 1A1 and apoptosis in murine hepatoma cells dependent on the aryl hydrocarbon receptor. Arch Toxicol 86(4):625–632. doi:10.1007/s00204-011-0781-3
Shah I, Houck K, Judson RS et al (2011) Using nuclear receptor activity to stratify hepatocarcinogens. PLoS ONE 6(2):e14584. doi:10.1371/journal.pone.0014584
Shimba S, Komiyama K, Moro I, Tezuka M (2002) Overexpression of the aryl hydrocarbon receptor (AhR) accelerates the cell proliferation of A549 cells. J Biochem 132(5):795–802
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics. CA Cancer J Clin 65(1):5–29. doi:10.3322/caac.21254
Silginer M, Burghardt I, Gramatzki D et al (2016) The aryl hydrocarbon receptor links integrin signaling to the TGF-beta pathway. Oncogene 35(25):3260–3271. doi:10.1038/onc.2015.387
Singh KP, Wyman A, Casado FL, Garrett RW, Gasiewicz TA (2009) Treatment of mice with the Ah receptor agonist and human carcinogen dioxin results in altered numbers and function of hematopoietic stem cells. Carcinogenesis 30(1):11–19. doi:10.1093/carcin/bgn224
Sinn HP, Kreipe H (2013) A brief overview of the WHO classification of breast tumors, 4th Edition, focusing on issues and updates from the 3rd Edition. Breast Care 8(2):149–154. doi:10.1159/000350774
Soshilov AA, Denison MS (2014) Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol Cell Biol 34(9):1707–1719. doi:10.1128/MCB.01183-13
Stanford EA, Ramirez-Cardenas A, Wang Z et al (2016a) Role for the aryl hydrocarbon receptor and diverse ligands in oral squamous cell carcinoma migration and tumorigenesis. Mol Cancer Res 14(8):696–706. doi:10.1158/1541-7786.MCR-16-0069
Stanford EA, Wang Z, Novikov O et al (2016b) The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol 14:20. doi:10.1186/s12915-016-0240-y
Su Y, Zeng Z, Zhang W, Chen Z, Xu D, Zhang XK (2016) Recent progress in the design and discovery of RXR modulators targeting alternate binding sites of the receptor. Curr Top Med Chem 16:1–13
Sun F, Indran IR, Zhang ZW et al (2015) A novel prostate cancer therapeutic strategy using Icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis 36(7):757–768. doi:10.1093/carcin/bgv040
Tice CM, Zheng YJ (2016) Non-canonical modulators of nuclear receptors. Bioorg Med Chem Lett 26(17):4157–4164. doi:10.1016/j.bmcl.2016.07.067
To KK, Yu L, Liu S, Fu J, Cho CH (2012) Constitutive AhR activation leads to concomitant ABCG2-mediated multidrug resistance in cisplatin-resistant esophageal carcinoma cells. Mol Carcinog 51(6):449–464. doi:10.1002/mc.20810
Tomblin JK, Salisbury TB (2014) Insulin like growth factor 2 regulation of aryl hydrocarbon receptor in MCF-7 breast cancer cells. Biochem Biophys Res Commun 443(3):1092–1096. doi:10.1016/j.bbrc.2013.12.112
Tompkins LM, Li H, Li L et al (2010) A novel xenobiotic responsive element regulated by aryl hydrocarbon receptor is involved in the induction of BCRP/ABCG2 in LS174T cells. Biochem Pharmacol 80(11):1754–1761. doi:10.1016/j.bcp.2010.08.016
Torre LA, Siegel RL, Ward EM, Jemal A (2016) Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev 25(1):16–27. doi:10.1158/1055-9965.EPI-15-0578
Tran C, Richmond O, Aaron L, Powell JB (2013) Inhibition of constitutive aryl hydrocarbon receptor (AhR) signaling attenuates androgen independent signaling and growth in (C4-2) prostate cancer cells. Biochem Pharmacol 85(6):753–762. doi:10.1016/j.bcp.2012.12.010
Tsai CF, Hsieh TH, Lee JN et al (2015) Curcumin suppresses phthalate-induced metastasis and the proportion of cancer stem cell (CSC)-like cells via the inhibition of AhR/ERK/SK1 signaling in hepatocellular carcinoma. J Agric Food Chem 63(48):10388–10398. doi:10.1021/acs.jafc.5b04415
Veldhoen M, Hirota K, Westendorf AM et al (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453(7191):106–109. doi:10.1038/nature06881
Viale G (2012) The current state of breast cancer classification. Ann Oncol 23(Suppl 10):x207–x210. doi:10.1093/annonc/mds326
Villano CM, Murphy KA, Akintobi A, White LA (2006) 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in A2058 melanoma cells. Toxicol Appl Pharmacol 210(3):212–224. doi:10.1016/j.taap.2005.05.001
Villard PH, Caverni S, Baanannou A et al (2007) PPARa transcriptionally induces AhR expression in Caco-2, but represses AhR pro-inflammatory effects. Biochem Biophys Res Commun 364(4):896–901. doi:10.1016/j.bbrc.2007.10.084
Vogel CF, Li W, Sciullo E et al (2007) Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol 171(5):1538–1548. doi:10.2353/ajpath.2007.070406
Volkov MS, Bolotina NA, Evteev VA, Koblyakov VA (2012) Ah-receptor-independent stimulation of hepatoma 27 culture cell proliferation by polycyclic aromatic hydrocarbons. Biochemistry 77(2):201–207. doi:10.1134/S0006297912020125
Wang CK, Chang H, Chen PH et al (2009) Aryl hydrocarbon receptor activation and overexpression upregulated fibroblast growth factor-9 in human lung adenocarcinomas. Int J Cancer 125(4):807–815. doi:10.1002/ijc.24348
Wang T, Gavin HM, Arlt VM et al (2011a) Aryl hydrocarbon receptor activation during pregnancy, and in adult nulliparous mice, delays the subsequent development of DMBA-induced mammary tumors. Int J Cancer 128(7):1509–1523. doi:10.1002/ijc.25493
Wang T, Wyrick KL, Meadows GG, Wills TB, Vorderstrasse BA (2011b) Activation of the aryl hydrocarbon receptor by TCDD inhibits mammary tumor metastasis in a syngeneic mouse model of breast cancer. Toxicol Sci 124(2):291–298. doi:10.1093/toxsci/kfr247
Weiss C, Kolluri SK, Kiefer F, Gottlicher M (1996) Complementation of Ah receptor deficiency in hepatoma cells: negative feedback regulation and cell cycle control by the Ah receptor. Exp Cell Res 226(1):154–163. doi:10.1006/excr.1996.0214
Weiss C, Faust D, Durk H et al (2005) TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38-MAPK-dependent pathway. Oncogene 24(31):4975–4983. doi:10.1038/sj.onc.1208679
Xie G, Peng Z, Raufman JP (2012) Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am J Physiol Gastrointest Liver Physiol 302(9):G1006–G1015. doi:10.1152/ajpgi.00427.2011
Yin XF, Chen J, Mao W, Wang YH, Chen MH (2012) A selective aryl hydrocarbon receptor modulator 3,3′-diindolylmethane inhibits gastric cancer cell growth. J Exp Clin Cancer Res 31:46. doi:10.1186/1756-9966-31-46
Yin XF, Chen J, Mao W, Wang YH, Chen MH (2013) Downregulation of aryl hydrocarbon receptor expression decreases gastric cancer cell growth and invasion. Oncol Rep 30(1):364–370. doi:10.3892/or.2013.2410
Yin J, Sheng B, Han B et al (2016) The AhR is involved in the regulation of LoVo cell proliferation through cell cycle-associated proteins. Cell Biol Int 40(5):560–568. doi:10.1002/cbin.10592
Zhang S, Lei P, Liu X et al (2009) The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocr Relat Cancer 16(3):835–844. doi:10.1677/ERC-09-0054
Zhang J, Zong H, Li S, Zhang D, Zhang L, Xia Q (2012a) Activation of aryl hydrocarbon receptor suppresses invasion of esophageal squamous cell carcinoma cell lines. Tumori 98(1):152–157. doi:10.1700/1053.11514
Zhang S, Kim K, Jin UH et al (2012b) Aryl hydrocarbon receptor agonists induce microRNA-335 expression and inhibit lung metastasis of estrogen receptor negative breast cancer cells. Mol Cancer Ther 11(1):108–118. doi:10.1158/1535-7163.MCT-11-0548
Zhao QW, Zhou YW, Li WX et al (2015) Akt-mediated phosphorylation of Oct4 is associated with the proliferation of stemlike cancer cells. Oncol Rep 33(4):1621–1629. doi:10.3892/or.2015.3752
Acknowledgements
The Grant support of the National Institutes of Health (P30-ES023512, R01-ES025839, R01-CA202697), Texas AgriLife Research, and the Sid Kyle endowment are gratefully appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00204-017-2026-6.
Rights and permissions
About this article

Cite this article
Kolluri, S.K., Jin, UH. & Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch Toxicol 91, 2497–2513 (2017). https://doi.org/10.1007/s00204-017-1981-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-1981-2