
Abstract
Mammalian aldehyde oxidases (AOXs; EC1.2.3.1) are a group of conserved proteins belonging to the family of molybdo-flavoenzymes along with the structurally related xanthine dehydrogenase enzyme. AOXs are characterized by broad substrate specificity, oxidizing not only aromatic and aliphatic aldehydes into the corresponding carboxylic acids, but also hydroxylating a series of heteroaromatic rings. The number of AOX isoenzymes expressed in different vertebrate species is variable. The two extremes are represented by humans, which express a single enzyme (AOX1) in many organs and mice or rats which are characterized by tissue-specific expression of four isoforms (AOX1, AOX2, AOX3, and AOX4). In vertebrates each AOX isoenzyme is the product of a distinct gene consisting of 35 highly conserved exons. The extant species-specific complement of AOX isoenzymes is the result of a complex evolutionary process consisting of a first phase characterized by a series of asynchronous gene duplications and a second phase where the pseudogenization and gene deletion events prevail. In the last few years remarkable advances in the elucidation of the structural characteristics and the catalytic mechanisms of mammalian AOXs have been made thanks to the successful crystallization of human AOX1 and mouse AOX3. Much less is known about the physiological function and physiological substrates of human AOX1 and other mammalian AOX isoenzymes, although the importance of these proteins in xenobiotic metabolism is fairly well established and their relevance in drug development is increasing. This review article provides an overview and a discussion of the current knowledge on mammalian AOX.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mammalian aldehyde oxidases (AOXs; EC 1.2.3.1) are proteins belonging to the family of molybdoenzymes along with xanthine dehydrogenase (XDH) also known as xanthine oxidoreductase (XOR) (Carpani et al. 1990; Cazzaniga et al. 1994; Falciani et al. 1992, 1994; Hille 1996; Ichida et al. 1993; Nishino et al. 2008; Terao et al. 1992), sulfite oxidases (Cohen and Fridovich 1971a, b; Lee et al. 2002; Rajapakshe et al. 2012), mitochondrial amidoxime reducing component (mARC) (Gruenewald et al. 2008; Havemeyer et al. 2011; Ott et al. 2015), and a number of other prokaryotic enzymes (Leimkuhler and Iobbi-Nivol 2015). Molybdoenzymes require the molybdenum cofactor (Moco) for their catalytic activity (Mendel 2013; Mendel and Kruse 2012; Mendel and Leimkuhler 2015; Rajagopalan and Johnson 1992; Schwarz and Mendel 2006; Schwarz et al. 2009). AOXs and XDHs (xanthine oxidase subfamily) are classified as molybdo-flavoenzymes, since they require FAD besides Moco to oxidize their substrates (Garattini et al. 2008). The primary structure of AOXs and XDHs is very similar and the catalytically active form of both types of enzymes is a homodimer consisting of two 150 kDa subunits (Fig. 1a). The monomeric subunit of both AOXs and XDHs is divided into three distinct regions: an amino-terminal 25-kDa domain containing two non-identical 2Fe/2S redox centers, a central 40-kDa domain where the FAD-binding site is located and a carboxy-terminal 85-kDa domain consisting of the Moco-binding site mapping in close proximity to the substrate pocket. As detailed in one of the next sections, the structural similarities between vertebrate XDHs and AOXs proteins (approximately 50 % average amino acid identity; Fig. 2) indicate that the two types of molybdo-flavoproteins have a common evolutionary origin. The idea is supported by the fact that vertebrate AOX and XDH genes are characterized by very similar exon structures and by strict conservation of the exon/intron junctions. Indeed, the data available indicate that the AOX genes evolved from an ancestral XDH via an initial gene duplication event. As detailed in the first section of the review article, the number of AOX enzymes is highly dependent on the mammalian species considered. Rodents, such as mice and rats, are characterized by the largest number of AOXs, synthesizing the so called AOX1, AOX2, AOX3, and AOX4 isoenzymes (Garattini et al. 2009). In contrast, humans and the majority of primates produce a single AOX isoform, i.e., the orthologue of mouse AOX1.

General structure and catalytic activity of vertebrate AOXs. a The panel shows that all vertebrate AOXs consist of two identical subunits consisting of an N-terminal domain containing the 2Fe/2S redox centers (orange). The N-terminal domain is connected to the FAD-containing intermediate domain (green) via an unstructured and poorly conserved stretch of amino acids. A second unstructured hinge region links the intermediate domain with the C-terminal Moco domain which contains the substrate pocket. b The catalytic cycle of the AOX enzymes is schematically represented. A generic type of substrate, RH, is oxidized to the corresponding product, ROH with concomitant reduction of MoVI to MoIV. The electrons generated are subsequently transferred to FAD, with the production of FADH2, and molecular O2 which is the final electron acceptor giving rise to H2O2

Sequence alignment of human AOX1, mouse AOXs, and bovine XDH. The indicated amino acid sequences were aligned with the CLUSTAL-W algorithm. The bovine XDH (bXDH) sequence is shown as a reference to highlight the sequence similarities between mammalian AOXs and XDH. The dark blue line above the sequences indicates the 25-kDa domain I containing the two 2Fe/2S redox centers whose eight cystein residues chelating the iron atoms are indicated by a black solid circle. The red line indicates the 40-kDa domain II which contains the FAD-binding site. The light blue line indicates the 85-kDa domain III which is endowed with the Moco-binding site (Moco fingerprint) and the substrate pocket as shown. The Glu residue playing a fundamental role in the mechanisms of catalysis is marked by a black solid circle. The color code of the amino acids is defined by the CLC Main Workbench (www.clcbio.com)
A major difference between XDHs and AOXs is represented by substrate specificity. XDHs act preferentially on the two purines, hypoxanthine and xanthine, oxidizing them to xanthine and uric acid, respectively (Asai et al. 2007; Coughlan and Rajagopalan 1980; Hille and Sprecher 1987; Nishino 1994; Yamaguchi et al. 2007). Given its ability to oxidize hypoxanthine and xanthine, XDH is a key enzyme in the catabolism of purines. In contrast, the substrate specificity of AOXs is broad, and the physiological function of these enzymes is still largely undefined. The enzymes oxidize different types of organic aldehydes into the corresponding carboxylic acid, hydroxylate various hetero-aromatic rings, and catalyze the reduction of amino- and sulfo-groups. Clearly, AOXs are not enzymes acting specifically on aldehydes. Thus, the general “aldehyde oxidase” term adopted to define EC 1.2.3.1 enzymes is misleading. The broad substrate specificity of AOXs is at the basis of the recognized role played by these enzymes in phase I xenobiotic metabolism (Garattini et al. 2008, 2009; Garattini and Terao 2011, 2012, 2013).
As for the mechanisms of catalysis, XDH can use both NAD+ and molecular oxygen as the final acceptors of the reducing equivalents generated during substrate oxidation depending on the enzyme being under the dehydrogenase or oxidase form. Indeed, the enzyme can be reversibly or irreversibly converted from the dehydrogenase to the oxidase form, which uses solely molecular oxygen as the electron acceptor. In contrast, the classic reactions catalyzed by AOXs produce electrons, which reduce only molecular oxygen and generate superoxide anions or hydrogen peroxide. A typical AOX catalytic cycle is represented in Fig. 1b. The generic substrate, RH, is oxidized into its product, ROH, at the molybdenum center. The reducing equivalents generated are conveyed to FAD, generating FADH2. FADH2 is subsequently re-oxidized by molecular oxygen. The two 2Fe/2S redox centers mediate the transfer of electrons between Moco and FAD/FADH2 serving as the electron sinks responsible for the storage of the reducing equivalents during the catalytic cycle.
Moco is central to the catalytic activity of AOXs and XDH. Moco synthesis and assembly into vertebrate AOX and XDH apo-proteins is a complex process controlled by multiple enzymes. Unlike various other cofactors, Moco is not a dietary component and it needs to be synthesized de novo by the living organism (Fig. 3). The first step in Moco biosynthesis is radical-mediated cyclization of GTP to (8S)-3′,8-cyclo-7,8-dihydroguanosine 5′-triphosphate (3′,8-cH2GTP) which is catalyzed by Moco Synthesis protein 1A (MOCS1A) in humans (Reiss et al. 1998, 1999). This is followed by the biotransformation of 3′,8-cH2GTP into cyclic pyranopterin monophosphate (cPMP) which is carried out by MOCS1B (Hover et al. 2013, 2015a, b). The second step is represented by the conversion of cPMP into molybdopterin (MPT) by MOCS2, which is a heterodimeric protein consisting of the two subunits, MOCS2A and MOCS2B generated by alternative splicing of the same MOCS2 gene (Hahnewald et al. 2006; Reiss et al. 1999) and by the adenylyltransferase MOCS3 protein (Matthies et al. 2005). Gephyrin catalyzes the last step of the biosynthetic pathway which consists of molybdate insertion into MPT to generate Moco (Stallmeyer et al. 1999). The complexity of Moco biosynthetic pathway is at the basis of the fact that the presence of detectable levels of AOX mRNAs and proteins in specific tissues and cell types does not necessarily indicate the presence of a catalytically active holoenzyme. Deficits in single components of the biosynthetic pathway may also be responsible for the difficulties often encountered in over-expressing active mammalian AOXs and XDH holo-enzymes in many eukaryotic cell lines.

Molybdenum cofactor biosynthesis. The figure illustrates the last steps of the Moco biosynthetic pathway in humans. GTP guanosine triphosphate, cPMP cyclic pyranopterin monophosphate, MPT molybdopterin, Moco molybdenum cofactor. The enzymes involved in the sequential biosynthetic steps are indicated: MOCS1A molybdenum cofactor synthetase synthesis 1A, MOCS1B molybdenum cofactor synthetase synthesis 1B, MOCS2A molybdenum cofactor synthetase synthesis 2A, MOCS2B molybdenum cofactor synthesis 2B, MOCS3 molybdenum cofactor synthetase 3, HMCS human Moco sulfurase
This review article is aimed at providing an overview of the current knowledge on mammalian AOXs. The first section presents a summary of the information available on the structure, evolution and mechanisms of catalysis of mammalian AOXs. The second chapter focuses on a discussion of the few data available relating to the possible physiological significance of AOXs. The emerging relevance of human AOX1 and mammalian AOXs in xenobiotic metabolism is covered in the third section, which is followed by a chapter containing a series of concluding remarks and future perspectives.
Mammalian AOXs: evolution, structure, and mechanisms of catalysis
Although the physiological function of mammalian AOXs is still largely obscure, the studies conducted in the last few years have provided a rather detailed picture of the evolution and structure of this group of enzymes. Indeed, sequencing of an increasing number of prokaryotic and eukaryotic genomes has resulted in the prediction of the amino acid sequences of an ever increasing number of AOX proteins, providing the necessary basic information on the primary structure of these enzymes and corresponding genes across many vertebrate species. Gathering and analysis of the sequencing data have generated a first draft of AOXs evolutionary history (Kurosaki et al. 2013). As for the definition of the structural characteristics of AOXs, the crystallization of the first mammalian AOX protein, i.e., mouse AOX3 (Coelho et al. 2012; Mahro et al. 2011), has represented a seminal advancement in the field. Further important insights into the structure of catalytically active AOXs have been recently provided by the crystallization and structure determination of human AOX1 (Coelho et al. 2015). The development of a reliable method for the expression of recombinant and catalytically active molybdo-flavoproteins of mammalian origin in E. coli has been instrumental in the conduction of these last structural studies. In addition the expression system is an invaluable tool for direct functional studies aimed at establishing the role played by single amino acids in the catalytic activity of mammalian AOXs.
The number of active mammalian AOX isoforms is species specific as a result of gene duplication and gene deletion/inactivation events during vertebrate evolution
AOXs are present throughout evolution from bacteria to man, and they are likely to have originated from a primordial gene duplication event involving an ancestral XDH gene (Kurosaki et al. 2013). Subsequently, two distinct lines of AOXs evolved, as indicated by the phylogenetic tree shown in Fig. 4. Bacteria, algae, plant, and insects are characterized by a similar set of AOXs which map to one side of the phylogenetic tree whose central portion is occupied by XDHs. Conversely, vertebrate AOXs cluster together on the opposite side of the dendrogram relative to XDHs. Vertebrate AOXs and XDHs are characterized not only by high levels of similarity in terms of the amino acid sequence, but also as far as the structure of the corresponding genes is concerned. In fact, all vertebrate AOX and XDH genes consist of 35 and 36 exons, respectively, and the position of the 35 exon–intron junctions within the protein-coding regions are strictly conserved throughout vertebrate evolution (Garattini et al. 2008; Kurosaki et al. 2013).

Unrooted phylogenetic tree of AOXs and XDHs. Phylogenesis of AOX isoenzymes and XDH proteins in eukaryotes and prokaryotes. The phylogenetic tree is constructed aligning a large number of publicly available sequences using a CLUSTAL omega algorithm
A single and transcriptionally active XDH gene is observed in practically all vertebrate species. In contrast, sequencing of the available vertebrate genomes predicts the presence of one or more AOX genes, each coding for a distinct AOX isoenzyme. This is the consequence of a process of asynchronous AOX gene duplication events which occurred during the evolution of vertebrates, starting from fishes. Indeed, while the oldest vertebrate for which genomic data are available, i.e., the lamprey, a jawless fish, is characterized by a typical XDH gene, the animal is devoid of sequences with similarity to AOXs. Ray-finned fishes are the first vertebrates endowed with AOX genes and contain two such genetic loci, which we named AOXα and AOXβ (Garattini et al. 2008). On the basis of a series of considerations, we propose that fish AOXα is the product of the first gene duplication event from vertebrate XDH, while AOXβ arose from a subsequent duplication involving AOXα.
Mammals are characterized by a maximum of four AOX genes coding for a corresponding number of AOX isoenzymes. The two extremes are represented by humans, which express a single AOX1 protein and rodents, such as mice and rats, which synthesize the four AOX1, AOX2 (also known as AOX3l1), AOX3 and AOX4 isoenzymes. In mice, rats and all the other mammals characterized by more than one AOX gene, the AOX loci form a small cluster, as they map to a short region of the same chromosome, namely chromosome 1 in the case of mice (Kurosaki et al. 2004). The four mouse genes are transcribed from the same DNA strand and are located in close proximity to each other in a head-to-tail configuration, according to the following order, AOX1, AOX3, AOX4, and AOX2 (Fig. 5). Although it is currently impossible to establish the exact order of appearance of the four genes during vertebrate evolution, AOX1 is likely to be the most ancient gene.

AOX genes in humans and selected experimental animals. A schematic representation of AOX genes and pseudogenes in humans and selected primates or mammals used for studies of drug and xenobiotic metabolism. Orthologous AOX genes and pseudogenes are marked in the same color. Pseudogenes are crossed and marked with an asterisk. When determined, the chromosomal location is indicated on the right. CHR chromosome
The presence of a single AOX isoform in humans and <4 AOX enzymes in different mammalian species is due to species-specific gene deletion or pseudogenization events. These deletions and pseudogenizations characterize the evolution of AOX genes in mammals. For instance, human chromosome 2 presents with the vestiges of the mouse AOX3 and AOX2 orthologous genes which underwent a process of pseudogenization involving the disappearance of several exons. No trace of nucleotide sequences corresponding to the mouse Aox4 ortholog is evident on chromosome 2 in the region separating the AOX3 and AOX2 pseudogenes or in any other part of the human genome. As active AOX4 genes can be predicted in Rhesus monkeys, the results are consistent with an AOX4 deletion event along the process of evolution from primates to humans.
In conclusion, the evolutionary history of vertebrate AOXs involved a first phase which is characterized by a species-specific increase in the number of iso-enzymatic forms which may have been associated with the necessity to acquire new physiological functions. This was followed by a second evolutionary phase which resulted in a progressive and species-specific inactivation of one or more of the AOXs which is particularly evident in primates and led to the maintenance of a single isoform in humans. Interestingly, humans maintain AOX1, which is the most ancient AOX isoform, suggesting a non-redundant and common functional role of this enzyme in developing and/or adult vertebrates.
Crystallization of mouse AOX3 and human AOX1 provides new insights into the structure and catalytic activity of mammalian AOXs: identification of a common inhibitor site in AOX and XDH enzymes
The first data on the structure of mammalian AOXs derive from the crystallization of native mouse liver AOX3 (Coelho et al. 2012; Mahro et al. 2011) (PDB ID: 3ZYV; 2.9 Å resolution). Very recently the crystal structures of human AOX1, in its substrate-free form (PDB ID: 4UHW; 2.6 Å resolution), as well as in complex with the substrate, phthalazine, and the inhibitor, thioridazine (PDB ID: 4UHX; 2.7 Å resolution), were solved (Coelho et al. 2015). In both cases, crystals were prepared using the recombinant protein expressed in E. coli, although better AOX3 crystals were produced with the protein isolated from mouse liver. Comparison of the human AOX1 and mouse AOX3 crystal structures provides fundamental information as to the common and specific structural features of the two main AOXs present in rodent and mouse liver. These data have far-reaching implications in drug metabolism and drug development, given the importance of the two enzymes in these areas of research.
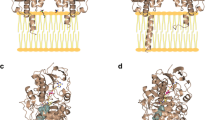
The structural data indicate that both mouse AOX3 and human AOX1 are homodimers of the following approximate dimensions: 150 Å × 90 Å × 65 Å. The results confirm that each monomer can be divided into three different domains involved in cofactor binding: a small N-terminal domain I (25 kDa) which harbors the two distinct 2Fe/2S clusters: the FAD domain II (40 kDa); the large C-terminal domain III (85 kDa) containing the Moco- and substrate-binding sites (Fig. 6). These different domains are connected by two linker regions bridging domains I/II and domains II/III. The crystal structures of human AOX1 and mouse AOX3 are characterized by high overall similarity, although they show marked differences in the FAD-binding region: the Moco active site and the substrate funnel.

Crystal structure the human AOX1 protein. Ribbon representation of the hAOX1 crystal structure. Monomer A is in gray and monomer B shows the three different protein domains colored as: domain I in red, domain II in green and domain III in blue. Domain III is separated from the FAD domain by linker 2. The protein cofactors (Moco, FeSI, FeSII, and FAD) are represented on the right in space-filling mode, with the shortest distances between them. The phthalazine and thioridazine (Pht and Thi) are also represented. The figure was created using PyMOL v1.7.2
In all members of the molybdo-flavoprotein xanthine oxidase subfamily, the access of substrates to the active site is determined by the structural features of a wide and deep funnel. The crystal structure of substrate-free human AOX1 indicates that several residues lining up the entrance of the funnel are mobile. These residues are located in two flexible loops which influence the entrance of substrates and the release of products (Fig. 7a, b).

Structural features of the AOX active site. Close up of the human AOX1 (blue) and the AOX3 (pink) active site. a Superposition of hAOX1 and mAOX3 residues surrounding the Moco cofactor. The residues which account for the major differences between the human and mouse proteins are marked with (asterisk). The Na+ ion is represented in a gray sphere and is present only in the mAOX3 crystal structure. b Superposition of the residues from gate 1 and gate 2 in hAOX1 (blue) and mAOX3 (pink) with the missing residues represented in dashed lines. The figure was created using PyMOL v1.7.2
Thioridazine is an antipsychotic drug belonging to the phenothiazine group which possesses a ring system similar to that of other antidepressants, and it is known to inhibit AOX activity in liver extracts (Obach and Walsky 2005; Rani Basu et al. 2005). The crystal structure of the human AOX1-phthalazine-thioridazine ternary complex provides important information regarding the modifications in the active site induced by substrate/inhibitor binding. The side chain of Phe-885 is mobile, and one of the two gates becomes better ordered upon ligand interaction. In addition, the possibility of generating human AOX1 crystals containing both phthalazine and thioridazine reveals distinct binding sites for the substrate and the inhibitor. The thioridazine inhibitor-binding region is located far away from the active site. This binding site has not been reported previously, and it is structurally conserved in other members of the xanthine oxidase family of molybdo-flavoproteins, i.e., mouse AOXs and bovine XDH. However, local structural differences in XDH and each mammalian AOX isoenzymes may account for differences in the affinity of thioridazine for each of these proteins. Complementation of the crystallographic data with steady state kinetic measurements shows a non-competitive inhibition pattern for human AOX1, mouse AOX3, and a mixed inhibition pattern for bovine XDH. The inhibition studies further suggest a more tight binding of the inhibitor thioridazine to human AOX1 than to mouse AOX3 or bovine XDH. In human AOX1 and mouse AOX3, thioridazine binding to the inhibitor pocket occurs in both the substrate-bound and free enzyme to the same extent. By contrast, the thioridazine inhibition constants calculated for the substrate-bound and free enzyme are different in bovine XDH. The mixed inhibition mode observed in XDH suggests that substrate binding to the active site modulates the affinity of thioridazine for the inhibitor-binding site of this molybdo-flavoenzyme.
The crystal structure of mouse AOX3 is a useful model in computational studies aimed at evaluating the factors that modulate substrate specificity and activity of the other mouse AOX isoforms, i.e. AOX1, AOX2, and AOX4 (Cerqueira et al. 2015). Combinations of homology modeling and molecular dynamics (MD) simulations demonstrate major differences among mouse AOX1, AOX2, AOX3, and AOX4 at the protein surface and in the substrate-binding region (Fig. 8). The differences found in some loops at the protein surface, far from the substrate funnel, should not affect the enzymatic activity of the four AOXs. In contrast, changes in the substrate-binding site are likely to be responsible for differences in the catalytic activity, substrate and inhibitor specificities previously described for the different mouse AOX isoforms (Vila et al. 2004). On the basis of all these data, we propose that the substrate-binding site consists of two different regions. The first region contains the inner active site consisting of conserved amino acid residues (in mouse AOX3: Gln-772, Ala-807, Phe-909, and Phe-1014) as well as Lys-889 and Glu-1266 which are relevant for the catalytic activity of all AOX isoforms (Table 1). The second one is an isoform-specific region located in the distal half of the catalytic tunnel which shows remarkable differences in the four mouse AOX isoforms (Fig. 8). The conserved sequence is likely to be responsible for the direction and correct alignment of the substrates into the active site of all AOXs. The variable and isoform-specific region is likely to determine the nature and chemical shape of as yet undetermined ligands recognized selectively by AOX1, AOX2, AOX3, and AOX4.

Substrate-binding site and tunnel in mouse AOX isoforms. Representation of the substrate-binding sites in mouse AOX1, AOX2, AOX3, and AOX4 as well as residues in the conserved region of the active site and in the non-conserved region which dictate substrate specificity. Residues in black indicate the amino acids whose nature is conserved in all mouse AOX isoforms
The above-mentioned computational studies suggest that mouse AOX1 is endowed with the largest isoform-specific region, which is able to accept a wide range of substrates with variable shape, size, and nature. By converse, mouse AOX4 is characterized by the narrowest isoform-specific region, which is predicted to bind only small and very hydrophobic substrates (Fig. 8). In addition, mouse AOX3 and AOX2 seem to be very similar to each other. In conclusion, the crystal and computer simulation data predict that all isoforms are likely to be endowed with equivalent catalytic activities, although the observed differences in the binding site are foreseen to influence the substrate specificity of each enzyme.
The expression system developed in E. coli provides information on the catalytic activity of mammalian AOXs
The development of an efficient system for the production and isolation of recombinant mammalian AOX proteins in their catalytically active forms (Alfaro et al. 2009; Hartmann et al. 2012; Mahro et al. 2011) has been a fundamental step in the structural characterization of this class of enzymes, as demonstrated by the results described in the previous section. In addition, the system has provided initial insights into the amino acid residues involved in the mechanisms of enzyme catalysis and inhibition.
One of the main problems in obtaining significant amounts of catalytically active mammalian AOXs in E. coli is related to the low saturation of sulfurated and active Moco (Hartmann et al. 2012; Mahro et al. 2011). In fact, the enzymatic system necessary for Moco sulfuration of mammalian AOXs is not present in E. coli. Sulfuration is the last important step in the biosynthesis of the mono-oxo Moco present in the various members of the xanthine oxidase family of molybdoenzymes. The active enzyme Moco is fully sulfurated, with a sulfur ligand bound to the molybdenum atom at the equatorial position. In mammalian AOX and XDH, addition of the terminal inorganic sulfur ligand is catalyzed by a Moco sulfurase, which is known as HMCS (Human Moco Sulfurase) (Mendel and Leimkuhler 2015). The plant homologue, ABA3, has been intensively studied, and it is a homodimeric two-domain protein (Bittner et al. 2001) with its N-terminal domain sharing structural and functional homologies to bacterial l-cysteine desulfurases, such as SufS or IscS. In a pyridoxal phosphate-dependent manner, the N-terminal domain of ABA3 decomposes l-cysteine to yield alanine and elemental sulfur (Heidenreich et al. 2005), the latter being bound as a persulfide to a highly conserved cysteine residue of ABA3. The C-terminal domain of ABA3 binds Moco, which further receives the terminal sulfur via an intramolecular persulfide relay from the N-terminal l-cysteine desulfurase domain (Lehrke et al. 2012; Wollers et al. 2008). HMCS is likely to catalyze the sulfuration of Moco in the same manner. To overcome the sulfuration problem in E. coli, the original approach considered was based on the overexpression of human Moco sulfurase to increase the sulfuration levels of recombinant AOXs. However, all the attempts in this direction failed, due to expression in inclusion bodies. Thus, we introduced a chemical sulfuration step successfully used to increase the catalytic activity of native XDH (Massey and Edmondson 1970; Wahl and Rajagopalan 1982). Chemical sulfuration increases the enzymatic activity of human recombinant AOX1 isolated from E. coli approximately tenfold and the activity of mouse AOX3 by a factor of three (Hartmann et al. 2012; Mahro et al. 2011).
The studies performed on the catalytically active recombinant forms of human and mouse AOX1 as well as mouse AOX3 expressed in E. coli have provided information on some of the amino acid residues critical for the enzymatic activity of these proteins, using site-directed mutagenesis approaches (Hartmann et al. 2012; Mahro et al. 2011; Schumann et al. 2009). As observed in XDH, the Glu residue contained in the active site of AOXs (Glu-1265 in mouse AOX1, Glu-1270 in human AOX1 and Glu-1266 in mouse AOX3) is essential for catalysis, since its substitution with a Gln residue in mouse and human AOX1 results in inactive enzymes (Mahro et al. 2013; Schumann et al. 2009). In mouse AOX3 the situation is more complex, as the Glu-1266-Gln variant shows residual activity with benzaldehyde (60 % reduction of activity relative to the wild-type enzyme), while it is inactive when challenged with N-heterocyclic compounds (Mahro et al. 2013). Residual activity is explained by the higher electrophilicity of the carbonyl carbon atom in aldehydes, as compared to N-heterocyclic compounds.
The presence of some highly conserved residues at the catalytic center (Glu-1270, Phe-923, Gln-776; human AOX1 numbering) suggests that the reaction mechanism for both AOXs and XDHs is similar, although the two types of enzymes act on different substrates (Cerqueira et al. 2015; Coelho et al. 2012; Mahro et al. 2013). One important difference between AOXs and XDH is represented by the presence of Lys-893 (human AOX1 numbering). Lys-893 is strictly conserved in all AOX enzymes, whereas it is replaced by an equally conserved His residue in XDH (His-884; bovine XDH numbering). Lys-893 lies ~10 Å away from the Mo center and ~6 Å away from Glu-1270. Molecular dynamics simulations performed with mouse AOX3 (Coelho et al. 2012) demonstrate that substrate docking into the active site causes this Lys residue (Lys-889; mouse AOX3 numbering) to move from its original position, establishing new interactions with the residue equivalent to human Glu-1270 (Glu-1266; mouse AOX3 numbering) and/or the substrate itself. This illustrates the importance of human AOX1 Lys-893 and equivalent Lys residues in other mammalian AOXs for the interaction between the above-mentioned Glu residue and the hydroxyl group of the molybdenum atom.
The proposed reaction mechanism for AOXs (Coelho et al. 2012) (Fig. 9) starts with a nucleophilic attack of the activated Mo-OH ligand (activated by Glu-1270) on the carbon atom of the substrate (carbon atom adjacent to an aromatic nitrogen atom for N-heterocycles). Concomitantly, a hydride is transferred to the sulfido ligand with the formation of an intermediate species. This intermediate state is stabilized by hydrogen bonding interactions with residues of the active site, i.e., Glu-1270 and Lys-893, and it replaces the labile OH ligand. This concerted mechanism is supported by experimental evidence obtained with substituted N-heterocycles as well as by density functional theory (DFT) calculations (Cerqueira et al. 2015; Coelho et al. 2012; Mahro et al. 2013). In a subsequent step, the reaction product is released from the reduced Mo site, and a water molecule reoccupies the vacant coordination position. The reaction cycle is completed once Mo is re-oxidized, and the two reducing equivalents are transferred to molecular oxygen via the two [2Fe–2S] centers and the FAD cofactor. The proton transfer from the hydroxyl ligand of Mo to Glu-1270 disrupts the interaction of Glu-1270 with Lys-893. This forces Lys-893 to move away from Glu-1270 and from the Mo center to interact with the product before the latter is released from the active site.

Reaction mechanism for AOXs. Representation of the currently proposed reaction mechanism for aldehyde oxidases, exemplified for the substrate phthalazine and including, on the left, the interaction with the non-competitive inhibitor thioridazine forming the ESI complex
The residues suggested to be involved in substrate binding and orientation in bovine XDH, Glu-802, and Arg-880 are replaced by Val and Met, respectively, in human AOX1, as well as Ala and Tyr in mouse AOX3 (Schumann et al. 2009) (Table 1). In human and R. capsulatus XDHs, substitution of these Glu and Arg residues to Val and Met, by site-directed mutagenesis, results in loss of activity toward purines as substrates and gain of activity toward aldehydes (Schumann et al. 2009; Yamaguchi et al. 2007). This indicates that the two residues contribute to substrate binding and determine substrate specificity. Specular exchanges of Val-806 to Glu and Met-884 to Arg in mouse AOX1 abolish catalytic activity when both purines and aldehydes are used as substrates (Schumann et al. 2009). However, it should be noticed that these last experiments are biased, since the activity of recombinant mouse AOX1 is generally very low and the complete loss of activity with both types of substrates may be the result of the restricted detection limit of the steady state kinetic determinations.
In a more recent report (Mahro et al. 2011), we studied the influence of the residues involved in substrate binding at the active site, using mouse AOX3 and site-directed mutagenesis of two residues conserved in all AOX1 enzymes (Ala-807 into Val and Tyr-885 into Met). The data obtained indicate that the Ala-807-Val mutation does not affect the kinetic constants when the enzyme is challenged with small substrates, like benzaldehyde or phthalazine. In contrast, the affinity for bulky substrates, such as phenanthridine, is decreased and the catalytic efficiency is slightly increased. As for the Tyr-885-Met mutation, the kinetic constants are also left largely unchanged in the presence of small hydrophobic substrates like benzaldehyde and phthalazine. Relative to the native enzyme, the variant oxidizes these substrates with the same rate constants and with only a minor increase in KM (possibly due to the smaller hydrophobic pocket in the Tyr-885-Met variant). On the other hand, bulkier substrates, such as phenanthridine, and charged substrates, like N1-methylnicotinamide, are converted by the Tyr-885-Met variant more efficiently. This is due to the greater flexibility of the Met side chain in comparison with Tyr, which may facilitate binding of these substrates. All these effects are much more pronounced in the double site-directed mutants of mouse AOX3.
In a further study, the basis of mouse AOX3 substrate selectivity and catalytic activity were investigated in a site-directed mutant containing numerous amino acid substitutions of residues found in XDH and other AOX isoforms in and near the active site (Mahro et al. 2013). The Phe-776-Lys/Ala-807-Glu/Asp-878-Leu/Leu-881-Ser/Tyr-885-Arg/Pro-1015-Thr/Tyr-1019-Leu variant of mouse AOX3 shows drastically decreased activity with all the substrates tested. Introduction of a further Lys-889-His substitution rescues AOX enzymatic activity and results in XDH-like activity when challenged with hypoxanthine. This last finding confirms the importance of active site Lys-889, and it has potential implications for the reaction mechanism.
Toward the definition of mammalian AOXs physiological function
Except for the involvement of human AOX1 in xenobiotic metabolism and inactivation of environmental pollutants characterized by potential toxicity, the physiological functions, substrates, and products of mammalian AOXs are still largely unknown or just hypothesized. It is equally unclear why certain mammalian species are endowed with multiple and catalytically active AOX isoforms, while many primates and humans are characterized by a single AOX isoenzyme. As the decrease in the number of active AOX isoenzymes from rodents to humans is the result of progressive deletion/inactivation of the corresponding genes, it is speculated that the function played by the lost enzymes is not necessary for the homeostasis of the human organism. Alternatively, it is possible that the function carried out by rodent AOX1, AOX2, AOX3, and AOX4 is concentrated in human AOX1. This may have resulted from evolutionary changes in the structure of human AOX1 which resulted in an increase in the ability of the enzyme to recognize physiological substrates originally specific to AOX2, AOX3 and AOX4. It is equally possible that mouse AOX2, AOX3, and AOX4 exert specialized functions in organs and tissues which are no more active or have become dispensable in humans.
The tissue- and cell-specific expression profiles of human AOX1 and mouse AOXs
A careful analysis of the data available on the tissue- and cell distribution of human AOX1 and mouse AOX1, AOX2, AOX3, and AOX4 is a prerequisite to formulate hypotheses on the role played by AOXs in homeostatic processes other than xenobiotic metabolism. The following discussion is limited to data obtained in humans and mice. The mouse was selected to provide a comparative picture of the mRNA and protein distribution in a popular experimental animal characterized by the presence of the maximum number of AOX isoenzymes observed in vertebrates.
The profile of human AOX1 mRNA tissue-specific expression (Fig. 10) is available in the UNIGENE section of the NCBI site (UniGene Hs.406238), and it is based on EST (Expression Sequence Tags) data indicating that detectable levels of the transcript are observed in many tissues. The adrenal gland, adipose tissue, and liver are the richest sources of human AOX1, followed by trachea, bone, kidney and connective tissue. The tissue-specific distribution of human AOX1 mRNA is in line with many of the results obtained at the protein level using immunocytochemistry (Moriwaki et al. 2001). According to these last data, the richest source of human AOX1 protein is represented by the liver, although significant amounts of the protein are found in respiratory, digestive, urogenital, and endocrine organs. In the respiratory tract, the epithelial component of the trachea and bronchi as well as the lung alveolar cells contains relatively high levels of the protein. By the same token, the epithelial cells lining the cavity of the small and large intestine are endowed with detectable amounts of human AOX1. In the kidney, AOX1 expression is limited to the proximal, distal, and collecting tubules, while immunostaining of the glomeruli is not observed. The glandular epithelium of the prostate is also particularly rich of AOX1 protein. Consistent with the mRNA data, large amounts of the AOX1 protein are present in the adrenal gland cortex. This last observation is consistent with a potential role of AOX1 in the steroid hormone biosynthetic pathway. To the best of our knowledge, there is a single published study on the expression of human AOX1 in the central nervous system (CNS) which was spurred by the unconfirmed hypothesis that the corresponding gene is involved in the etiopathogenesis of amyotrophic lateral sclerosis, a lethal disease characterized by motor neuron degeneration (Berger et al. 1995). The study demonstrates the presence of the AOX1 mRNA in the glial cell population of the spinal cord, although this does not rule out the possibility the transcript is expressed in other cellular populations of the CNS.

Expression profiles of human AOX1 and mouse AOX1 to AOX4. The bar graphs indicate the normalized mRNA content of the indicated AOXs in the tissues considered. The values of the tissue mRNA contents were calculated using the data available in the UNIGENE section of the NCBI site: hAOX1 = human AOX1 (UniGene Hs.406238); mAOX1 = mouse AOX1 (UNIGENE Mm.26787); mAOX2 = mouse AOX2 (UNIGENE Mm.414292); mAOX3 = mouse AOX3 (UNIGENE Mm.20108); mAOX4 = mouse AOX4 (UNIGENE Mm.244525)
If the expression profiles of human and mouse AOX1 (UNIGENE Mm.26787) transcripts are compared (Fig. 10), it is clear that significant levels of the murine orthologue are observed in a much more restricted number of tissues. The largest amounts of mouse AOX1 mRNA are present in the inner ear and the seminal vesicles, although measurable amounts of the AOX1 transcript are expressed in liver and lung. Mouse AOX1 mRNA is also detectable in the central nervous system (Bendotti et al. 1997). Consistent with what was reported for the human orthologue (Berger et al. 1995), the mRNA is expressed in the motor neurons of the spinal cord and specific populations of neuronal cells in the brain. Nevertheless, the largest amounts of the transcript are located in the epithelial component of the choroid plexus, where AOX1 may play a role in the absorption/secretion of the cerebrospinal fluid. Interestingly, the major sources of human and mouse AOX1 are not overlapping with the notable exception of liver. Similar to mouse AOX1, mouse AOX3 expression (UNIGENE Mm.20108) is also relatively restricted (Fig. 10), as significant amounts of the corresponding transcript are found only in the oviduct, liver, lung and testis. The data indicate that mouse AOX3 and AOX1 are co-expressed only in liver and lung. The tissue distribution results obtained for mouse AOX1 and AOX3 mRNA levels are fully confirmed for the corresponding proteins in a number of tissues, with particular reference to liver and lung (Kurosaki et al. 1999; Terao et al. 2000). In mouse liver, AOX1 and AOX3 are synthesized by the hepatocyte, as demonstrated by in situ hybridization experiments. However, it is currently unknown whether the same or different hepatocytic population is responsible for the synthesis of AOX1 and AOX3. This specific point is of interest in terms of the physiological function exerted by hepatic AOX1 and AOX3, particularly in consideration of the fact that histological data obtained in rat liver indicate that the distribution of AOX enzymatic activity is uneven. In fact, rat AOX activity is much higher in hepatocytes located in the pericentral than the periportal area (Moriwaki et al. 1998a, b). From a functional perspective, it is currently difficult to understand the reasons as to why mouse and rat liver is endowed with AOX3 and AOX1, while the same organ in humans and various other mammalian species expresses only AOX1 (Vila et al. 2004). The point is particularly puzzling in consideration of the fact that mouse hepatic tissue contains much higher levels of AOX3 than AOX1.
Mouse AOX4 mRNA (UNIGENE Mm.244525) is very abundant in the fertilized ovum, the inner ear and tongue (Fig. 10). In the tongue, our in situ hybridization data indicate that the AOX4 transcript is predominantly expressed by the keratin-rich epithelial cells lining the taste papillae (Terao et al. 2001). AOX4 mRNA expression extends beyond the oral cavity as the keratinized epithelium lining the esophagus and the most proximal tract of the stomach contains significant amounts of the transcript. The presence of AOX4 mRNA in keratinized epithelia is a consistent finding, as demonstrated by the observed expression in the epidermal layer of the skin (Terao et al. 2009). The data obtained at the protein level with the use of specific antibodies confirm the presence of AOX4 in the oral cavity, digestive tube and skin (Terao et al. 2009). However, recent results indicate that, by far, the richest source of AOX4 is represented by a largely overlooked organ present in most vertebrates, but not in humans and many other primates, i.e., the Harderian gland. In this organ, approximately 2 % of all the cytosolic proteins consist of AOX4. The Harderian gland is a large exocrine gland located behind the eye bulb and characterized by the secretion of a lipid-rich fluid, which is believed to serve for the lubrication of the eye surface and the fur (Buzzell 1996; Hardeland et al. 2006; Payne 1994). In furred animals the fluid is purported to act as a thermic isolator and to contribute to regulation of the body temperature. It is interesting to notice that skin sebaceous glands, which are also involved in fur lubrication and thermoregulation, share the same developmental origin with the Harderian gland and contain significant amounts of AOX4 mRNA (Terao et al. 2009).
AOX2 is the mouse AOX characterized by the most restricted pattern of expression (Fig. 10). Indeed the corresponding transcript (UNIGENE Mm.414292) is detectable only in the nasal cavity (Terao et al. 2000). In this location, high levels of the transcript and protein are expressed in the Bowman’s gland, which is the principal exocrine gland present in the submucosal layer, and it is responsible for the production of the mucous fluid excreted in the nasal cavity. AOX2 is also detectable in sustentacular cells which are located in the apical layer of the nasal neuroepithelium. Interestingly, this small cell population has been reported to have the same embryonal origin as the Bowman’s gland (Huard et al. 1998). Given this localization, it is speculated that AOX2 may play a role in olfaction, which is one of the phenotypic traits we are currently investigating in the AOX2 knockout mouse which we have generated and stabilized on a homogeneous genetic background (Terao M., unpublished data). The olfactory function is much more developed in rodents and other mammalian species than in humans, possibly explaining pseudogenization and inactivation of the human AOX2 gene.
The expression of mammalian AOXs is regulated by various intrinsic and extrinsic factors
Indirect clues as to the physiological function of AOXs can be obtained from an analysis of the intrinsic and extrinsic factors controlling the expression of human AOX1 and the other mammalian AOX isoenzymes. There are few published studies focusing on the factors and mechanisms regulating the expression of human AOX1 and the other mammalian AOX isoforms.
Development and age are two factors controlling AOX expression, and their influence on AOX levels have been studied in humans and rodents. Given its importance in drug metabolism and the differences in the pharmacokinetics and pharmacodynamics of many drugs in the pediatric and adult population, studies on the profiles of human liver AOX1 enzymatic activity in children and adults are available. Young children (13 days to 4 months after birth) are characterized by very low levels of liver AOX1 activity. AOX1 activity increases substantially after 4 months and reaches adult levels by approximately 2 years of age. The levels of AOX1 activity correlate with the amounts of the corresponding protein (Tayama et al. 2012). The results obtained in the study are largely in line with the data obtained on a larger cohort of children using a noninvasive method for the detection of total AOX1 activity based on the determination of the N(1)-methylnicotinamide product, pyridine, in the urine (Tayama et al. 2007a). Limited data on the age-dependent expression of rat and mouse liver AOXs are available. In a study focusing on rat liver, AOX enzymatic activity is undetectable or barely detectable until the second week from birth. From day 14 on, a rapid increase in AOX activity is observed, and this increase reaches a plateau at 4 weeks. The increase in AOX activity correlates with AOX1 protein levels, suggesting increased gene expression (Tayama et al. 2007b). However, one serious limitation of this study is represented by the fact that the main AOX isoenzyme expressed in rats is AOX3, for which information is not available. Indeed, the age-related expression profiles of liver AOX1 and AOX3 are different, as indicated by the data obtained in mice which demonstrate that the appearance of liver AOX3 is more precocious (Terao et al. 2000). Mouse embryos are substantially devoid of AOX3 and AOX1 mRNAs as well as proteins. AOX3 mRNA and protein start to be visible in newborn animals and their levels plateau at 5 days, while the AOX1 counterparts are detectable only in adult animals. At present, no data on the effects that aging has on liver AOX1 and AOX3 expression are available. Similarly, the developmental profile of AOX2 and AOX4 expression in the tissues that express them has not been the object of any study.
A second and important determinant of AOX expression is gender which was shown to control the levels of liver AOX activity via sex hormones in a series of relatively old studies performed in mice (Holmes 1979; Ventura and Dachtler 1980, 1981; Yoshihara and Tatsumi 1997). In summary, these studies demonstrate that liver AOX activity is higher in male than female animals. Castration of male mice causes a reduction in liver AOX activity, which is reconstituted upon administration of testosterone. Testosterone administration to female animals increases the levels of liver AOX activity, while challenge of male animals with estrogen causes the opposite effect. Taken together, the results indicate that the androgen/estrogen ratio controls the levels of liver AOX enzymatic activity. We validated and extended these data, demonstrating that mouse AOX1 and AOX3 in liver as well as AOX4 in the Harderian Gland is under the control of sex via regulation by androgens and estrogens. In particular, testosterone administration to female animals increases the levels of AOX1 and AOX3 mRNAs, which results in an induction of the corresponding proteins and enzymatic activities (Kurosaki et al. 1999; Terao et al. 2000). The action of testosterone accounts for the much larger amounts of AOX3 and AOX1 proteins observed in male than female mouse liver. In the Harderian gland, AOX4 expression is generally similar in adult male and female animals, although there is a restricted time window during postnatal development in which the enzyme shows sexual dimorphism. In fact, at 9 weeks of age, the levels of AOX4 in this organ are higher in females than male animals (Terao et al. 2009). This is due to the levels of circulating testosterone, which has a suppressive action on AOX4 expression. Thus the effect of testosterone is the opposite on Harderian gland AOX4 and liver AOX1/AOX3. It is currently unknown whether gender and sex hormones are major factors influencing the levels of human AOX1 as observed in mice and other animal species. In a single study, the levels of the human AOX1 protein were determined in the liver of a small cohort of human donors using a specific mass spectrometry assay. No significant differences in the amounts of the protein were observed in female relative to male individuals, although males showed a significantly broader distribution of AOX1 levels than females (Fu et al. 2013).
Another intrinsic factor controlling the expression of mammalian AOXs is the circadian rhythm, as originally observed in guinea pig liver, where the diurnal variations in AOX and XDH activity are caused by the circadian variations in circulating melatonin (Beedham et al. 1989). Indeed both liver AOX and XDH enzymatic activities are stimulated by melatonin through as yet undefined mechanisms. Diurnal variations in liver AOX enzymatic activity are due to circadian oscillations in the levels of the AOX1 and AOX3 proteins, which tend to be induced during the dark phase of the diurnal cycle. Variations in the expression during the light/dark cycle are not a characteristic of mouse AOX1 and AOX3. In the Harderian gland, also AOX4 enzymatic activity varies with the diurnal cycle, being low during the light phase and high at the end of the dark phase (Terao M., unpublished observations). Interestingly, the Aox4 knockout mice are characterized by alterations in the expression of clock genes, supporting an unexpected role for AOX4 in the control of the circadian rhythms (Terao M., unpublished observations).
As for the extrinsic factors controlling AOXs, there are three studies indicating that AOX levels in humans and other animal species are modulated by components of the diet. In one study, the effect of vitamin E and selenium supplementation on AOX activity is investigated in liver, kidney, and heart of rats in which a diabetic state is induced by streptozotocin administration. Relative to control animals, diabetic rats are characterized by increased levels of AOX enzymatic activity. Oral administration of vitamin E and sodium selenite reduces AOX activity in the liver, although it does not affect it in the kidney and heart (Ghaffari et al. 2012). In line with these results, selenium deficiency in rats is associated with an increase in AOX enzymatic activity which is accompanied by induction of the AOX1 protein, but not of the corresponding mRNA, suggesting translational control. At present, however, it is unknown whether selenium deficiency exert similar effects on the other rat hepatic AOX, i.e., AOX3 (Itoh et al. 2009). Consumption of tea beverages may also cause a decrease in AOX activity, as challenge of human and rat liver cytosolic preparations with epicatechin and epicatechin gallate, two important components of green tea causes substantial inhibition of AOX enzymatic activity (Tayama et al. 2011).
Some environmental pollutants, toxic agents and drugs modulate AOX levels or activity. Administration of phthalazine or 1-hydroxyphthalazine to rabbits increases the specific activity of liver AOX (Johnson et al. 1984). Similarly, induction of liver AOX activity is observed in rats administered the alkylating agents, N-methyl-N′-nitro-N-nitrosoguanidine, N-methyl-N-nitrosourea, and methylmethansulfonate (Ohkubo et al. 1983a). In the hepatoma cell line Hepa-1, expression of the mouse Aox1 gene is induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (dioxin). Induction is the result of a transcriptional effect mediated by the ligand-dependent transcription factor, aryl-hydrocarbon-receptor (AHR), which is activated by dioxin. Dioxin-dependent induction of the AOX1 mRNA is also observed in mouse liver (Rivera et al. 2005). In the same tissue, induction of the AOX3 mRNA and protein has been reported. Increased expression of the AOX1 and AOX3 mRNAs translate into an increase in AOX activity. A transcriptional mechanism is at the basis of the AOX1 mRNA induction afforded by the chemopreventive agent, phenethyl isothiocyanate in mouse liver (Hu et al. 2006). In this case the transcription factor involved is NRF2, which seems to be a direct regulator of the mouse Aox1 gene. The rat and human AOX1 orthologous genes are also direct target genes of NRF2 (Maeda et al. 2012; Shintani et al. 2015). Interestingly, two Nrf2-binding consensus elements (antioxidant responsive element, ARE) are located in the 5′ upstream region of the rat AOX1 gene. Nrf2 binds to the promoter region of the rat AOX1 gene and strongly activates its expression.
AOXs participate in numerous metabolic pathways recognizing various endogenous substrates
More direct insights into the physiological function of AOXs can be obtained from the identification of their endogenous substrates. In fact, AOXs are enzymes, and it is entirely possible that they play a role in specific physiological processes via control of the local or circulating levels of substrates or products. There is very little knowledge on the potential endogenous substrates of human AOX1. The knowledge on common and specific substrates of the other mammalian AOX isoforms is even more limited.
The information available on the KEGG database (http://www.kegg.jp) implicates mammalian AOXs in a number of metabolic pathways. In most cases, the involvement of AOXs in these pathways is only presumed, and it is based on in vitro studies which demonstrate that selected endogenous molecule can be metabolized by AOX preparations of different origins. It should also be pointed out that KEGG annotations are based predominantly on the assumption that AOXs act only on aliphatic or aromatic aldehydes oxidizing them to carboxylic acids. As already mentioned in the introduction, this is not correct, since AOXs are characterized by broad substrate specificity and hydroxylate, a number of heterocyclic structures which do not contain aldehyde functionalities. Thus, the assumption is likely to underestimate the number and types of potential AOX physiological substrates and to overlook the involvement of AOXs in pathways other than the ones specified in the KEGG database. In addition, it must be stressed that there is no direct in vivo evidence supporting the role of AOXs in any of the metabolic steps and pathways hypothesized by KEGG annotations, with the exception of the few data obtained in the Aox4 knockout mouse which we generated (Terao et al. 2009). A final and major problem is represented by the virtual absence of data regarding the specificity of the proposed endogenous substrates for AOX1, AOX2, AOX3, or AOX4, which is a further source of biases for the various mammalian species presenting with multiple AOX isoenzymatic forms. In spite of the above shortcomings, AOXs, including human AOX1, have been implicated in the following metabolic pathways: (1) ec00280: valine, leucine, and isoleucine degradation; (2) ec00350: tyrosine metabolism; (3) ec00380: tryptophan metabolism; (4) ec00750: vitamin B6 metabolism; (5) ec00760: nicotinate and nicotinamide metabolism; (6) ec00830: retinol metabolism.
In ec00280, human AOX1 is purported to oxidize the l-valine catabolic product, (S)-methylmalonate semialdehyde, into methylmalonate. The same metabolic step is also predicted to be catalyzed by one of the NAD-dependent aldehyde dehydrogenases (EC 1.2.1.3). In ec00350, the predicted AOX substrate is gentisate aldehyde which is metabolized into gentisate. In this case, AOX may compete with aryl-aldehyde dehydrogenase (EC 1.2.1.29) for the substrate.
The involvement of AOXs in tryptophan metabolism (ec00380) is due to the possibility that the enzymes catalyze the oxidation of 5-hydroxy indolacetaldehyde, a catabolite of serotonin, into 5-hydroxy-indolacetic acid. With respect to this, it is interesting to notice that a metabolomic analysis, which we conducted, demonstrates that 5-hydroxy-indolacetic acid levels are much higher in the Harderian gland of Aox4 knockout than control mice (M. Terao, unpublished data). This suggests that AOX4 is indeed involved in tryptophan catabolism. Interestingly, data obtained with purified AOX4 and AOX3 proteins demonstrate that 5-hydroxy-indolacetic acid is not only a potential product, but it is also a substrate of the two enzymes, being metabolized into a mono-hydroxylation product (M. Terao, unpublished data). Further evidence for an involvement of AOXs in tryptophan metabolism comes from data obtained with the same approaches described for 5-hydroxy-indolacetic acid. These results demonstrate a substantial increase of tryptophan itself in the Harderian gland of Aox4 knockout mice relative to the control counterparts. In addition, they demonstrate that purified AOX4 and AOX3 hydroxylate the heteroaromatic structure of tryptophan, which is indeed likely to represent another physiological substrate of the two enzymes. Tryptophan and 5-hydroxy-indolacetic acid are paradigms of AOX physiological substrates devoid of an aldehyde function.
Pyridoxal is a well-known AOX substrate (Schwartz and Kjeldgaard 1951) and this justifies the inclusion of AOX in the ec00750 pathway. Pyridoxal (Vitamin B6) is biotransformed into 4-pyridoxate, by AOXs, and the metabolic step seems to be of physiological significance in insects (Cypher et al. 1982; Stanulovic and Chaykin 1971). However, it remains to be established whether pyridoxal is a physiologically relevant substrate also in the case of mammalian AOXs. In this context, it is worthwhile mentioning that mouse AOX1 and AOX3 recognize pyridoxal as a substrate, while the compound is not a good substrate for mouse AOX4. In line with this last observation, the phenotypes described in Aox4 knockout mice are not amenable to perturbations of vitamin B6 catabolism (Terao et al. 2009).
N1-methyl-nicotinamide, the most proximal nicotinamide catabolic product, can be oxidized into N1-methyl-2-pyridone-5-carboxamide and N1-methyl-4-pyridone-5-carboxamide by AOXs, which led to the inclusion of these enzymes into the ec00760 metabolic pathway. Along with tryptophan and 5-hydroxy-indolacetic acid, N1-methyl-nicotinamide is another example of AOX substrate devoid of an aldehyde function, and it is the only example of non-aldehydic AOX substrate present in the KEGG database. N1-methyl-nicotinamide is metabolized by AOX-enriched extracts obtained from human (Sugihara et al. 1997), monkey (Sugihara et al. 1997), rat (Ohkubo et al. 1983b), rabbit (Stoddart and Levine 1992) and guinea pig (Yoshihara and Tatsumi 1985) liver, which indicates that the AOX1 isoform is certainly recognizing it as a substrate. The finding is of significance for the human situation, as the AOX1 orthologous protein is the only active AOX in man.
A final endogenous substrate of AOXs that may be of interest in man is nicotinamide riboside. Indeed, human AOX1 is purported to be the major enzyme involved in the oxidation of nicotinamide riboside to 4-pyridone-3-carboxamide-1-β-d-ribonucleoside (4PYR) (Pelikant-Malecka et al. 2015) (Fig. 11). This metabolic step is of primary importance in patients suffering from chronic renal disease, as PYR is an endothelial toxin which is purported to contribute to kidney insufficiency in uremic patients (Pelikant-Malecka et al. 2015).

Endogenous compounds, drugs, and xenobiotics metabolized by AOXs. The panel illustrates endogenous compounds (green), drugs (red) and xenobiotics of toxicological interest (blue) or derived metabolites which can be oxidized by mammalian AOXs. When AOXs act on metabolic intermediates of the parental drug, the enzymes involved in the primary metabolic step are indicated: MAO monoamine oxidase, CYP450 cytochrome P-450-dependent mono-oxygenase, XDH xanthine dehydrogenase
The last KEGG pathway AOXs are involved in is ec00830 which controls vitamin A metabolism. In this pathway, AOXs are implicated in the oxidation of 9-cis and all-trans retinal (RAL) into the corresponding retinoic acid. All-trans retinoic acid (ATRA) is the active form of vitamin A, and it controls many aspects of vertebrate homeostasis in both the developing and adult organism. In vertebrate embryos, ATRA is a well-known morphogen, and it regulates the development of numerous tissues and organs, including the central nervous system. In adult organisms, the molecule influences numerous biological process, including vision as well as growth and differentiation of skin epithelial cells and hematopoietic cells. At present, RAL is the candidate endogenous substrate of AOXs for which the largest number of supporting evidence is available (Fig. 11). For this reason and given the relevance of ATRA biosynthesis for the homeostasis of vertebrates and mammals in particular, this metabolic pathway is discussed in more detail.
In humans and rodents, β-carotene is the major dietary precursor of vitamin A, while retinyl esters are the predominant storage products. Retinol and retinyl esters are the short- and long-term storage forms of vitamin A. Mobilization of vitamin A for ATRA synthesis is a two-step process. First, retinol is oxidized to RAL in a reversible reaction by a number of alcohol dehydrogenases, such as the ubiquitous alcohol dehydrogenase 3 (Molotkov et al. 2002). This is followed by an irreversible oxidation of RAL into ATRA. Three NAD-dependent aldehyde dehydrogenases, ALDH1A1, ALDH1A2, and ALDH1A3 are known to oxidize RAL into ATRA and are deemed to play a major role in this metabolic step (Niederreither et al. 1999). In mice and other mammals AOXs are likely to carry out the same metabolic step as ALDH1As, since RAL is an excellent substrate for most of the AOXs studied. In fact, early studies indicated that rabbit cytosolic fractions are capable of oxidizing RAL into ATRA in a NAD-independent fashion, implicating AOX activity in the process (Huang and Ichikawa 1994). Subsequently, it was demonstrated that semi-purified rabbit liver AOX is indeed the enzyme endowed with RAL oxidizing activity (Tomita et al. 1993; Tsujita et al. 1994). These original findings were extended to the mouse, with the demonstration that purified liver AOX1 (Huang et al. 1999; Vila et al. 2004) and AOX3 (Terao et al. 2001) as well as purified Harderian gland AOX4 (Terao et al. 2009) and nasal AOX2 (Kurosaki et al. 2004) recognize RAL as a substrate. The Vmax calculated for the RAL oxidizing activity of AOX1 (180 nmol/min/mg protein), AOX3 (57 nmol/min/mg protein), ALDH1A1 (57 nmol/min/mg protein), and ALDH1A2 (105 nmol/min/mg protein) are very similar. Instead the km values for AOX1 (31 µM) and AOX3 (31 µM) are significantly higher than those determined for the two dehydrogenases (ALDH1A1 = 1.4 µM; ALDH1A2 = 0.7 µM). It is unlikely that AOXs oxidize RAL in the developing mouse embryo, as AOX enzymatic activity is detectable only after birth. The low affinity of RAL for the two AOXs suggests that AOX1 and AOX3 are equally unlikely to play a major role in local ATRA production in adult mouse liver, since the hepatic tissue contains high levels of ALDH1A1 and cytosolic NAD-dependent RAL oxidizing activity (Terao et al. 2009). The same may be true for the majority of other mouse tissues showing no AOX expression or co-expression of AOXs with ALDH1As. Instead, in mouse organs expressing AOX4, like the Harderian gland, tongue and skin, the enzyme may exert a major role in local ATRA synthesis. In fact, the Harderian glands of normal C57Bl/6J mice are devoid of ALDH1A1-3- and NAD-dependent RAL oxidizing activity in their cytosol (Terao et al. 2009). Consistent with this, Aox4 knockout mice are characterized by a marked decrease in the levels of ATRA measurable in the Harderian gland and skin relative to what is observed in wild-type animals (Terao et al. 2009).
In conclusion, the data so far available support the idea that AOXs recognize multiple endogenous substrates and RAL is the physiological substrate for which supportive evidence is particularly strong. Further and more direct functional in vivo approaches are needed to establish whether AOX4 is the only isoform of relevance for the bio-transformation of RAL to ATRA in mammalian species endowed with multiple AOX isoforms. In addition, it would be important to establish whether RAL is a substrate of human AOX1, as this may shed new light into the physiological function of this enzyme which is unlikely to be limited to xenobiotic metabolism.
The available indicate that AOXs are likely to be involved in a diverse array of physiological functions
The number of studies focusing on the potential involvement of mammalian AOXs in specific cell or tissue homeostatic processes using direct approaches is limited. Nevertheless, these studies indicate a role for AOXs in a rather varied array of physiological processes.
A recent paper demonstrates that human AOX1 is involved in maintaining the epithelial barrier integrity of lung cells. In fact, treatment with dexamethasone of the 16HBE bronchial epithelial cell line causes a decrease in inter-cellular permeability which is reminiscent of what is happening in asthmatic patients, where inhaled corticosteroids enhance the airway epithelial barrier integrity. In these cells, dexamethasone causes a marked increase in AOX1 expression. AOX1 knockdown blocks the decrease in inter-cellular permeability afforded by dexamethasone indicating that AOX1 is involved in the control of cell-to-cell junctions (Shintani et al. 2015). The mouse AOX1 ortholog has been implicated in myocyte differentiation, which leads to the formation of myotubes. Indeed, myocyte differentiation is associated with an increase in the levels of AOX1 and AOX1 silencing inhibits differentiation blocking myotube fusion (Kamli et al. 2014). Interestingly, the process of myotube fusion involves an early increase in inter-cellular contacts, suggesting that mouse and human AOX1 serve a similar function in bronchial epithelial cells and developing myocytes, i.e., control of cell–cell interactions. The mechanisms underlying the phenomena observed in human bronchial and mouse muscular cells are unknown although it is possible that they involve H2O2, a by-product of AOX1 enzymatic activity, as suggested by the data obtained in the model of muscular differentiation (Kamli et al. 2014).
Other physiological functions mammalian AOXs seem to contribute to are adipogenesis and liver fat storage, two highly related processes. In the mouse preadipocyte 3T3L1 cell line, mouse AOX1 is up-regulated during adipocytic differentiation, and it is down-regulated by the hypocholesterolemic agent and PPARγ agonist, fenofibrate, in differentiated cells. AOX1 knockdown in preadipocytes suppresses lipid storage and secretion of adiponectin, a protein hormone-stimulating fatty acid oxidation, in the differentiated adipocytes (Weigert et al. 2008). The role of AOXs in adipogenesis is supported by the observations that human AOX1 mRNA and protein are detectable in human adipocytes, and the transcript is higher in visceral than subcutaneous human adipose tissue (Weigert et al. 2008). The effect of AOXs on lipid homeostasis is not limited to the adipose tissue and extends to liver. In fact, AOX activity is increased in steatotic livers of rats exposed to high-fat diets (Neumeier et al. 2006). In addition, administration of adiponectin or fenofibrate reduces the levels of hepatic AOX activity. These observations support the idea that human and mouse AOX1 are major determinants of adipocyte differentiation and fat deposition in liver. The action of human and mouse AOX1 in lipid homeostasis may be independent of their enzymatic activity as suggested by the results contained in two published studies (Graessler and Fischer 2007; Sigruener et al. 2007). The two studies suggest that the action of AOX1 is mediated by its capacity to interact with and modulate the activity of the ABCA-1 lipid transporter which is involved in the control of lipid efflux from the cell. Human and mouse AOX1 are not the only AOX isoenzymes involved in lipid homeostasis, as AOX4 plays a role in this process as well. Indeed, our Aox4 knockout mice are characterized by a deficit in fat deposition at the level of the visceral adipose tissue and show resistance to obesity upon exposure to diets rich in fat (Terao M., unpublished results). These effects are accompanied by reduced liver steatosis. AOX4 actions on adipose tissue and liver fat accumulation are due to systemic mechanisms, as mouse adipocytes and hepatocytes are completely devoid of this AOX isoenzyme. Interestingly, resistance to obesity is observed also in Aldh1a1 knockout mice exposed to high-fat diets (Ziouzenkova et al. 2007). As AOX4 and ALDH1a1 are both involved in the oxidation of RAL into ATRA, this suggests a possible involvement of the two retinoids in fat deposition and resistance to obesity.
A last biological process AOXs may be involved in is the production of nitric oxide, a major vasodilator and intracellular second messenger under hypoxic conditions. Although nitric oxide is biosynthesized from l-arginine, oxygen, and NADPH by various nitric oxide synthases, nitrite is deemed to be a nitric oxide “storage form” that can be made available, through its one-electron reduction, to maintain nitric oxide formation under hypoxia/anoxia. Mammalian AOXs and XDH can reduce nitrite into nitric oxide under hypoxic conditions (Li et al. 2008). A recent study performed in HepG2 hepatocytes and human mammary epithelial cells supports a major involvement of human AOX1 in the production of nitric oxide under hypoxic conditions (Maia et al. 2015).
The role played by mammalian AOXs in xenobiotic metabolism
Although human AOX1 and the other mammalian AOX isoenzymes are likely to be characterized by incompletely defined and tissue-specific functions, the role this class of enzymes in phase I metabolism of xenobiotics of both medical and toxicological relevance is established (Garattini and Terao 2011, 2012, 2013). The interest in AOXs as drug-metabolizing enzymes, particularly in the liver, is increasing given the emerging significance of these enzymes in drug development programs, as discussed below.
Mammalian AOXs are an emerging class of enzymes in the metabolism of drugs
Mammalian AOXs are characterized by broad substrate specificity and are capable of recognizing a large range of organic molecules regardless of the presence of an aldehyde function, as already mentioned. Mammalian AOXs hydroxylate the rings of various aza-, oxo-, and sulfo-heterocycles and oxidize iminium functions to cyclic lactames. There is also evidence that AOXs act not only as oxidases but also as reductases, reducing N-oxides, sufoxides, nitro-compounds, and heterocycles, particularly under hypoxic conditions (Dick et al. 2006; Kitamura and Tatsumi 1984a, b). All this along with the high levels of human AOX1 and mammalian AOX1 and/or AOX3 in liver is at the basis of the relevance that these enzymes have in the metabolism of xenobiotics (Coelho et al. 2012; Sanoh et al. 2015; Zientek and Youdim 2015). Indeed, bio-transformation/inactivation of drugs as well as toxicants is deemed to be the major physiological functions of AOXs in liver. Hepatic AOXs and cytochrome P-450-dependent mono-oxygenases (CYP450) represent major phase I drug-metabolizing enzymes. While CYP450 enzymes are predominantly located in the endoplasmic reticulum of liver cells, AOX1 and AOX3 are cytosolic proteins. Sometime AOXs and CYP450 s act in concert, as observed with citalopram, a non-tricyclic anti-depressant belonging to the family of selective serotonin re-uptake inhibitors (SSRI) (Fig. 11). In this case, the tertiary amino group of citalopram is oxidized to an aldehyde by CYP450. Subsequently, the aldehyde is bio-transformed into the corresponding carboxylic acid by AOX. The number of drugs metabolized by AOXs is large and includes antitumor, immunosuppressive, antimalarial, and antiviral agents as well as molecules acting in the central nervous system. Among the antitumor and immunosuppressive agents, methotrexate and 6-mercaptopurine are the oldest examples of drugs metabolized by AOXs. Methotrexate is a folic acid antagonist, and it is used in the treatment of lymphoma and rheumatoid arthritis. The drug is oxidized into 7-hydroxy-methotrexate (Fig. 11) by both human AOX1 and rabbit, rat, mouse and hamster AOXs (Chladek et al. 1997; Kitamura et al. 1999). The AOX-dependent hydroxylation step is of fundamental importance for the pharmacokinetics and side effects of methotrexate, as 7-hydroxy-metotrexate is endowed with cytotoxic activity. In humans a large inter-individual variability in the bio-transformation of methotrexate into the 7-hydroxy derivative is observed and the phenomenon is correlated with the levels of AOX1 enzymatic activity (Kitamura et al. 1999). Another example of immunosuppressant and cytotoxic agent which is a recognized substrate of AOXs is 6-mercaptopurine, a major metabolite of the other immunosuppressant azathioprine (Beedham et al. 1987; Ding and Benet 1979; Rooseboom et al. 2004). The imidazole ring of 6-mercaptopurine undergoes two hydroxylation steps which are catalyzed by XOR and AOXs in sequence (Fig. 11). AOX-dependent hydroxylation in the liver and other tissues leads to metabolic inactivation and contributes to the pharmacokinetic profile of the compound in man and experimental animals. A final example of immunosuppressant for which there is evidence that some the identified toxic metabolites are due to AOX1 activity is thiopurine, a drug used in the treatment of inflammatory bowel disease (Chouchana et al. 2012).
Other drugs that are metabolized by AOXs belong to the classes of antimalarial and antiviral agents. Cryptolepine (Fig. 11) is an antimalarial agent, and it is oxidized to cryptolepine-11-one by rabbit liver AOX (Stell et al. 2012). Quinine is another example of antimalarial drug whose bio-disposition is partially dependent on AOXs, although the molecule is a CYP450 substrate as well. The 2-quinone derivative is the major metabolite produced by AOX enzymatic activity and its levels are different in different experimental animals (Beedham et al. 1992), consistent with the different spectrum of AOXs present in liver (Beedham et al. 1987). Dog and rat livers contain undetectable and low levels of cytosolic quinine oxidase activity. The first observation is in line with the predicted absence of liver AOX1 and AOX3 proteins (Terao et al. 2006) due to pseudogenization/inactivation of the corresponding genes in dogs (Kurosaki et al. 2013). By converse, the finding in rats suggests that AOX3 is much less efficient than AOX1 in oxidizing quinine to 2-oxo-quinone. The idea is supported by the data obtained in the hepatic tissue of baboons, which are characterized by high levels of quinine oxidizing activity and pseudogenization/inactivation of the AOX3 gene (Kurosaki et al. 2013). In support of this hypothesis is also the observation that the liver of guinea pigs and marmosets, which show deletion and pseudogenization of the AOX3 gene (Kurosaki et al. 2013), oxidize quinine to the same extent as the human counterpart.
In discussing the role of human AOX1 and other mammalian AOXs in the metabolism of drugs, there are at least three issues that should be considered. First, metabolic inactivation of drugs by AOXs does not simply involve oxidation, as these enzymes are capable of reducing appropriate substrates. A significant example of the reducing ability of AOXs is represented by ziprasidone, a drug which is approved by the US Food and Drug Administration (FDA) for the treatment of schizophrenia, acute mania and mixed states associated with bipolar disorder. Indeed, the main metabolic pathway for ziprasidone in humans occurs via AOX1 catalyzed reduction, followed by thiol-methyl-transferase catalyzed methylation (Obach et al. 2012). Second, oxidation of drugs by AOXs does not necessarily lead to metabolic inactivation. Metabolic activation of pro-drugs by human AOX1 has been exploited to circumvent pharmacokinetic problems as exemplified by the clinical development of the 5-iodo-2-deoxyuridine precursor, 5-iodo-2-pyrimidinone-2-deoxyribose (Kinsella et al. 1994, 1998, 2000). In prospective, human AOX1-dependent activation of pro-drugs may be a strategy to be pursued in the oncologic field to increase the therapeutic index of antitumor agents by increasing the tumor selectivity of their pharmacological action. In fact there are a number of tumor types which are characterized by the expression of detectable levels of the human AOX1 mRNA, as observed in the case of glioblastoma, esophageal, and gastric cancer (Garattini and Terao 2011). A last point that must be mentioned regards the fact that AOX-dependent metabolism of drugs may be relevant in tissues and organs other than the liver. An appropriate example is represented by the skin, as human skin explants has been shown to metabolize carbarazepan and zoniporide, two well-known AOX substrates (Manevski et al. 2014). The presence of the AOX1 mRNA (Fig. 10) and the AOX4 protein (Terao et al. 2009) in human and mouse skin, respectively, indicates that AOXs may represent a problem for drugs and cosmetics which are administered topically, as they may be metabolized and inactivated very rapidly in this location.
The importance of AOXs in drug development has increased over the course of the last few years with the development of organic synthesis strategies designed to avoid CYP450-dependent metabolism. In fact, this has resulted in the enrichment of chemical structures with a high probability to be metabolized by human AOX1 and related enzymes (Barr et al. 2014; Fratelli et al. 2013; Hutzler et al. 2013) and it has led to the development of new medicinal chemistry approaches to avoid AOX metabolism and inactivation of potential drugs (Pryde et al. 2012). In addition, the enrichment of potential substrates for human AOX1 underscores the necessity of adequate cell-free and whole-cell in vitro screening assays to confirm AOX-dependent metabolism. These assays are likely to become invaluable tools in any drug development program (Garattini and Terao 2011, 2013). A further problem in drug development is now represented by the selection of appropriate in vivo experimental models to study the pharmacokinetics and pharmacodynamics of drug candidates with the potential to be metabolized by human AOX1 to avoid failures in the clinics because of clearance and toxicity problems. This is becoming a major issue given the already discussed differences between humans and many mammalian in the complement of active AOX genes expressed in the liver and other tissues (Figs. 5, 10).
AOXs metabolize compounds of toxicological interest
The role of AOXs in xenobiotic metabolism is not limited to drugs, as it extends to various molecules of toxicological interest. Phthalazines are major environmental pollutants and are classic AOX substrates. Indeed, several types of mammalian AOXs oxidize phthalazine into 1-hydroxy-phthalazine which undergoes rapid and largely irreversible isomerization into 1-phthalazinone (Beedham et al. 1990; Coelho et al. 2015; Stubley et al. 1979). Oral administration of phthalazine causes an increase in rabbit liver AOX activity, indicating that the compound is not only a substrate for but it is also a bona fide AOX inducer (Johnson et al. 1984). Another example of AOX substrate of potential significance in the toxicological field is represented by the xanthine, caffeine, which is rapidly metabolized to an oxo-derivative by both AOXs and XDH (Castro et al. 2001) (Fig. 11). Vanillin, a widely used sweetener, is an aromatic aldehyde of nutritional interest which is rapidly metabolized by mammalian AOXs into the corresponding carboxylic acid (Fig. 11). A last example of toxicant which is deemed to be an AOX substrate is acetaldehyde, the primary product of ethanol metabolism. In this case, AOX has been proposed to be implicated in acute and chronic liver toxicity associated with alcohol consumption. The hypothesized mechanism underlying this AOX-dependent action is related to the production of reactive oxygen species, like superoxide anions or hydrogen peroxide, during the oxidation of acetaldehyde into the corresponding carboxylic acid (Shaw and Jayatilleke 1990). However, experiments conducted in the liver of AOX deficient DBA/2 and AOX proficient CD1 mice do not support this idea. In fact, CD1 and DBA/2 mice, which are characterized by undetectable levels of AOX3 and markedly reduced amounts of AOX1 relative to the CD1 counterpart, show the same amounts of liver acetaldehyde after chronic administration of ethanol (Vila et al. 2004). This indicates that neither liver AOX1 nor AOX3 contributes to acetaldehyde metabolism in vivo. The observation is consistent with the fact that acetaldehyde is a poor substrate for both enzymes, although it does not rule out the possibility that AOX1 and AOX3 play a role in acetaldehyde induced hepatotoxicity via other mechanisms. With respect to this, an alternative mechanism is suggested by an old study (Kundu et al. 2012; Mira et al. 1995), which indicates that NADH is an AOX substrate and oxidation of the dinucleotide by the enzyme produces toxic superoxide anions. According to the hypothesis proposed in the article, NAD+-dependent alcohol dehydrogenase produces NADH and acetaldehyde from ethanol. NADH is oxidized to NAD+ by AOXs with the consequent generation of toxic oxygen radicals. All this ignites a vicious circle between AOXs and alcohol dehydrogenase which is at the basis of oxygen radical dependent liver toxicity.
Another interesting class of toxicants which can be metabolized by different mammalian AOXs is represented by compounds containing a nitro function, as they exemplify the ability of these enzymes to act not only as oxidases, but also as reductases in the presence of electron donors, such as 2-hydroxypyrimidine (Tatsumi et al. 1986; Ueda et al. 2003, 2005). In fact, like XDH, AOXs have been shown to carry out the nitro-reduction of the environmental pollutants, 2-nitrofluorene (Fig. 11), 1-nitro-pyrene, and 4-nitrobiphenyl into the corresponding amines. Noticeably, AOX-dependent nitro-reduction is observed not only in cytosolic fractions obtained from the liver (Tatsumi et al. 1986) but also from the skin of different experimental animals, such as mice (Ueda et al. 2003, 2005). As mouse liver expresses AOX1 and AOX3, while mouse skin synthesizes predominantly AOX4, the data indicate that nitro-reduction is not a characteristic of specific AOX isoenzymes. The presence of AOX-dependent nitroreductase activity in the skin and the ability of mammalian AOX1 to carry out this reaction have implications for the human situation. In fact, human skin explants contain AOX1 enzymatic activity (Manevski et al. 2014), and AOX-dependent reduction of environmental nitro-compounds may represent a general defensive mechanism active in man as well as various mammalian species. A last example of AOX-dependent nitro-reductive metabolism is represented by the neonicotinoid, imidacloprid, which provides support to the notion this type of enzymatic reaction has in vivo relevance. Neonicotinoids are an important class of insecticides whose nitroimino group is reduced in vitro by AOXs. A recent study carried out in the mouse demonstrates the relative contribution of liver AOXs and CYP450 s in the metabolism of the neonicotinoid, imidacloprid, using an in vivo approach (Swenson and Casida 2013). Addition of tungsten or hydralazine in the drinking water of C57BL/6J mice is associated with a selective reduction in the levels of liver AOX activity and no change in the amounts of CYP450 activity. In tungsten- and hydralazine-exposed C57BL/6J mice, CYP450-dependent imidacloprid metabolism is unaffected, whereas the AOX-generated nitrosoguanidine metabolite is substantially decreased. This provides direct in vivo evidence that mouse liver AOX1 and AOX3 play a major role in the reductive metabolism of imidacloprid.
Single nucleotide polymorphisms affecting human AOX1 activity
Given the role played by human AOX1 in xenobiotic metabolism, there is interest in defining the presence of single nucleotide polymorphisms (SNPs) affecting the expression or the catalyitic activity of this enzyme. In fact, the identification of such SNPs should permit the classification of individuals into fast and slow metabolizers, with respect to drugs and toxicants which are recognized as human AOX1 substrates. A major source of inter-individual variability in the levels of AOX1 activity is likely to be related to the presence of SNPs in the coding region of the corresponding gene. Numerous SNPs mapping to the human AOX1 gene are available in the NCBI database (http://www.ncbi.nlm.nih.gov/snp). We determined the allelic frequency of some of these SNPs in a cohort of 180 volunteers representative of the Northern Italian population (Hartmann et al. 2012). In this population, one nonsense polymorphism, one synonymous, and five non-synonymous polymorphisms were identified. The most frequent missense polymorphism involves a G/A substitution which results in the R1297K amino acid exchange. Other relatively frequent SNPs are represented by T/C and G/A substitutions giving rise to the L1271S and N1135S AOX1 variants. The frequency of two further amino acid substitutions (R802C and R921H) is much lower. The recombinant proteins corresponding to the four AOX1 variants generated in our modified E. coli system are catalytically active. Characterization of their enzymatic activity against a number of classic substrates indicates that the R802C and R921H AOX1 variants have reduced catalytic activity. By converse, the N1135S and H1297R amino acid exchanges result in increased catalytic activity. Thus, as observed in certain strain of rats (Adachi et al. 2007; Hartmann et al. 2012), some of the non-synonimous polymorphisms affect the catalytic activity of human AOX1. Individuals characterized by the presence of the R802C or R921H variant are likely to be poor metabolizers, while those endowed with the N1135S or the H1297R are likely to be rapid or extensive metabolizers of drugs known to be bio-transformed by human AOX1. As this type of information is of great clinical relevance in terms of predicting the efficacy and toxicity of such drugs, further studies are necessary to evaluate the influence on enzyme activity exerted by these and other non-synonymous SNPs in the human population worldwide.
Conclusions and future perspectives
The present review article provides an overview of the current knowledge on the structure, evolution and function of mammalian AOXs, a small group of proteins of increasing interest particularly in the medical and toxicological fields, given the ability of these enzymes to metabolize a wide range of drugs and environmental toxicants.
Substantial progress in the definition of the primary and tridimensional structure of mammalian AOXs has been accomplished in the last few years, particularly as a consequence of the very recent crystallization of mouse AOX3 (Coelho et al. 2012; Mahro et al. 2011) and human AOX1 (Coelho et al. 2015). This along with the development of simple and efficient methods for the purification of large quantities of recombinant mammalian AOXs has also resulted in a better understanding of the catalytic mechanisms responsible for the activity of these enzymes. Further insights into the structural determinants guiding the interactions of substrates and inhibitors with the AOX enzymes as well as the release of products from their active site are likely to represent the object of intense efforts in the next future. We expect that some of these efforts will be directed toward the solution of one of the fundamental problems of AOX biology, i.e., the reason for the existence of multiple enzymatic isoforms in certain mammalian species and the presence of a single enzyme in humans. With respect to this, integration of the primary sequence and crystallographic data with the results of site-directed mutagenesis studies and the recent advancements in the field of computational modeling are likely to establish whether different AOX have similar or divergent substrate specificities. These types of approaches are also likely to permit the design and synthesis of selective inhibitors for the various AOX isoforms which will represent useful tools to study the activity of each AOX isoenzyme in the experimental animal. Thus, the recent progress in the elucidation of mammalian AOXs structure is likely to be of interest not only in the specific field of structural biology but also in terms of functional biology, synthetic and medicinal chemistry as well as toxicology.
The strictly conserved structure and sequence of vertebrate AOX genes and the public availability of sequencing dataset for a large number of prokaryotic and eukaryotic genomes has permitted the reconstruction of the primary amino acid structure of numerous AOX proteins (Kurosaki et al. 2013). In addition, the data have been instrumental in the prediction of the complement of active AOX genes, inactive AOX pseudogenes and AOX gene deletions observed in a significant number of vertebrate species. This information is of fundamental relevance from both a basic and an applied perspective. In terms of basic science, it has led to a first picture of AOXs evolutionary history, which indicates that the evolution of vertebrates is characterized by a first phase of asynchronous gene duplications followed by a series of species-specific gene inactivation and gene deletion events. This has resulted in the extant complement of AOX proteins which varies from species to species. In the next few years, it is expected that this picture will become more detailed, as the number of sequenced genomes is likely to increase logarithmically. At the applied level, the available data are already of major interest in the pharmacological and toxicological areas. In fact, they provide information as to the complement of AOX isoenzymes present in animal species widely used for preclinical studies aimed at defining the metabolic and toxicologic profiles of new drug candidates and environmental pollutants. In the realm of drug development, AOX-dependent metabolism of an increasing number of new molecules is becoming a serious problem (Garattini and Terao 2012, 2013). Indeed, the design of new drug candidate avoiding CYP450-dependent metabolism has resulted in an enrichment of chemical structures recognized as substrates by human AOX1 (Pryde et al. 2012). AOX-dependent metabolism cannot be accurately recapitulated in common preclinical in vivo models, like mice and rats, because of the different complement of AOXs present in these animal species relative to humans. The difficulty of scaling clearance from preclinical species to humans is one of the reasons leading to the possible failure of drugs metabolized by human AOX1 in clinical trials (Choughule et al. 2013, 2015; Dalvie et al. 2013; Diamond et al. 2010). The available genomic data predicting the complement of AOX isoforms expressed in popular animal species used as pre-clinical models in drug metabolism studies (Fig. 5) indicate that the best proxies of the human situation are represented by the chimpanzee followed by the guinea pig and the pig. In fact, the liver of these animal species expresses only the orthologue of human AOX1. With respect to this point, however, it should be pointed out both types of animals are predicted to express AOX2 and AOX4 in tissues other than the liver, which may add a confounding factor in the study of drug candidates potentially metabolized by human AOX1. On the basis of these data, we propose that the chimpanzee is likely to be the best surrogate of humans in drug metabolism and pharmacokinetic studies. Clearly, the same considerations hold true for studies aimed at predicting the toxicity of molecules metabolized by human AOX1 in appropriate experimental models in vivo. A potential resolution to the problem of finding simple and cost-effective experimental animals representing predictive proxies of the human situation in the area of xenobiotic metabolism is the development of genetically engineered mice expressing human AOX1 in the context of AOX null mice, a direction we are currently pursuing. In the specific fields of drug development and environmental toxicology, another important issue to be addressed in the next future is the development of simple experimental paradigms to be used for the prediction of human AOX1-dependent metabolism.
Despite the wealth of information generated on the structure and reaction mechanisms in the last few years, there is still a substantial lack of information on the physiological functions of mammalian AOXs. In fact, it is highly unlikely that the primary function of this class of enzymes is xenobiotic metabolism, given the expression of human AOX1 and rodent AOX1, AOX2, AOX3, and AOX4 in multiple tissues, some of which are characterized by very specialized functions. With respect to this, it is expected that further details on the physiological role of AOXs will be gained by the identification of relevant endogenous substrates and products using the direct approaches made possible by the development of current state-of-the-art method for the isolation of significant amounts of catalytically active recombinant and native mammalian AOXs discussed in this review article. Further details on the physiological role of mammalian AOXs will be gained by phenotypic analysis of the AOX knockout mice which are already available or will be generated in the next few years. At present, only knockout mice for AOX2 (Mineko Terao, personal communication) and AOX4 (Terao et al. 2009), two extra-hepatic enzymes, have been generated. Thus, there is an urgent need to extend the range of available knockout mice to the two liver enzymes, AOX1 and AOX3. The data obtained on the AOX2 and AOX4 knockout mice demonstrate that functional inactivation of the two genes is not lethal and does not lead to malformations, supporting the idea that the corresponding enzymes are not involved in embryonal development. This is in line with the data indicating that AOX enzymes start to be active only after birth. A very recent and in-depth phenotypic analysis of the Aox4 knockout animal demonstrates an involvement of AOX4 in the systemic control of circadian rhythms, locomotor activity, and adipogenesis (M. Terao et al. unpublished results). Alterations of circadian rhythms and locomotor activity are in line with the purported involvement, of the Harderian gland, the richest source of AOX4, in the control of the light/dark cycle. The effects exerted by AOX4 on fat deposition and homeostasis are largely unexpected, although our data support the idea that the phenomenon can be explained by actions of the enzyme on thermogenesis and energy balance. In this context, future challenges are represented by the identification of the AOX4-dependent substrates or products, if any, responsible for these systemic effects. In addition, it will be important to establish whether human AOX1 exert a similar action on lipid homeostasis, as supported by recent in vitro data (Sigruener et al. 2007; Weigert et al. 2008). If this is indeed the case, the enzyme may represent a new and viable target for the development of antiobesity drugs.
References
Adachi M, Itoh K, Masubuchi A, Watanabe N, Tanaka Y (2007) Construction and expression of mutant cDNAs responsible for genetic polymorphism in aldehyde oxidase in Donryu strain rats. J Biochem Mol Biol 40(6):1021–1027
Alfaro JF, Joswig-Jones CA, Ouyang W, Nichols J, Crouch GJ, Jones JP (2009) Purification and mechanism of human aldehyde oxidase expressed in Escherichia coli. Drug Metab Dispos 37(12):2393–2398. doi:10.1124/dmd.109.029520
Asai R, Nishino T, Matsumura T, Okamoto K, Igarashi K, Pai EF (2007) Two mutations convert mammalian xanthine oxidoreductase to highly superoxide-productive xanthine oxidase. J Biochem 141(4):525–534. doi:10.1093/jb/mvm054
Barr JT, Choughule K, Jones JP (2014) Enzyme kinetics, inhibition, and regioselectivity of aldehyde oxidase. Methods Mol Biol 1113:167–186. doi:10.1007/978-1-62703-758-7_9
Beedham C, Bruce SE, Critchley DJ, al-Tayib Y, Rance DJ (1987) Species variation in hepatic aldehyde oxidase activity. Eur J Drug Metab Pharmacokinet 12(4):307–310
Beedham C, Padwick DJ, al-Tayib Y, Smith JA (1989) Diurnal variation and melatonin induction of hepatic molybdenum hydroxylase activity in the guinea-pig. Biochem Pharmacol 38(9):1459–1464
Beedham C, Bruce SE, Critchley DJ, Rance DJ (1990) 1-substituted phthalazines as probes of the substrate-binding site of mammalian molybdenum hydroxylases. Biochem Pharmacol 39(7):1213–1221
Beedham C, al-Tayib Y, Smith JA (1992) Role of guinea pig and rabbit hepatic aldehyde oxidase in oxidative in vitro metabolism of cinchona antimalarials. Drug Metab Dispos 20(6):889–895
Bendotti C, Prosperini E, Kurosaki M, Garattini E, Terao M (1997) Selective localization of mouse aldehyde oxidase mRNA in the choroid plexus and motor neurons. NeuroReport 8(9–10):2343–2349
Berger R, Mezey E, Clancy KP et al (1995) Analysis of aldehyde oxidase and xanthine dehydrogenase/oxidase as possible candidate genes for autosomal recessive familial amyotrophic lateral sclerosis. Somat Cell Mol Genet 21(2):121–131
Bittner F, Oreb M, Mendel RR (2001) ABA3 is a molybdenum cofactor sulfurase required for activation of aldehyde oxidase and xanthine dehydrogenase in Arabidopsis thaliana. J Biol Chem 276(44):40381–40384. doi:10.1074/jbc.C100472200
Buzzell GR (1996) The Harderian gland: perspectives. Microsc Res Tech 34(1):2–5. doi:10.1002/(SICI)1097-0029(19960501)34:1<2:AID-JEMT2>3.0.CO;2-W
Carpani G, Racchi M, Ghezzi P, Terao M, Garattini E (1990) Purification and characterization of mouse liver xanthine oxidase. Arch Biochem Biophys 279(2):237–241
Castro GD, Delgado de Layno AM, Costantini MH, Castro JA (2001) Cytosolic xanthine oxidoreductase mediated bioactivation of ethanol to acetaldehyde and free radicals in rat breast tissue. Its potential role in alcohol-promoted mammary cancer. Toxicology 160(1–3):11–18
Cazzaniga G, Terao M, Lo Schiavo P et al (1994) Chromosomal mapping, isolation, and characterization of the mouse xanthine dehydrogenase gene. Genomics 23(2):390–402. doi:10.1006/geno.1994.1515
Cerqueira NM, Coelho C, Bras NF et al (2015) Insights into the structural determinants of substrate specificity and activity in mouse aldehyde oxidases. J Biol Inorg Chem 20(2):209–217. doi:10.1007/s00775-014-1198-2
Chladek J, Martinkova J, Sispera L (1997) An in vitro study on methotrexate hydroxylation in rat and human liver. Physiol Res 46(5):371–379
Chouchana L, Narjoz C, Beaune P, Loriot MA, Roblin X (2012) Review article: the benefits of pharmacogenetics for improving thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol Ther 35(1):15–36. doi:10.1111/j.1365-2036.2011.04905.x
Choughule KV, Barr JT, Jones JP (2013) Evaluation of rhesus monkey and guinea pig hepatic cytosol fractions as models for human aldehyde oxidase. Drug Metab Dispos 41(10):1852–1858. doi:10.1124/dmd.113.052985
Choughule KV, Joswig-Jones CA, Jones JP (2015) Interspecies differences in the metabolism of methotrexate: an insight into the active site differences between human and rabbit aldehyde oxidase. Biochem Pharmacol 96(3):288–295. doi:10.1016/j.bcp.2015.05.010
Coelho C, Mahro M, Trincao J et al (2012) The first mammalian aldehyde oxidase crystal structure: insights into substrate specificity. J Biol Chem 287(48):40690–40702. doi:10.1074/jbc.M112.390419
Coelho C, Foti A, Hartmann T, Santos-Silva T, Leimkuhler S, Romao MJ (2015) Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat Chem Biol 11(10):779–783. doi:10.1038/nchembio.1895
Cohen HJ, Fridovich I (1971a) Hepatic sulfite oxidase. Purification and properties. J Biol Chem 246(2):359–366
Cohen HJ, Fridovich I (1971b) Hepatic sulfite oxidase. The nature and function of the heme prosthetic groups. J Biol Chem 246(2):367–373
Coughlan MP, Rajagopalan KV (1980) The kinetic mechanism of xanthine dehydrogenase and related enzymes. Eur J Biochem 105(1):81–84
Cypher JJ, Tedesco JL, Courtright JB, Kumaran AK (1982) Tissue-specific and substrate-specific detection of aldehyde and pyridoxal oxidase in larval and imaginal tissues of Drosophila melanogaster. Biochem Genet 20(3–4):315–332
Dalvie D, Xiang C, Kang P, Zhou S (2013) Interspecies variation in the metabolism of zoniporide by aldehyde oxidase. Xenobiotica 43(5):399–408. doi:10.3109/00498254.2012.727499
Diamond S, Boer J, Maduskuie TP Jr, Falahatpisheh N, Li Y, Yeleswaram S (2010) Species-specific metabolism of SGX523 by aldehyde oxidase and the toxicological implications. Drug Metab Dispos 38(8):1277–1285. doi:10.1124/dmd.110.032375
Dick RA, Kanne DB, Casida JE (2006) Substrate specificity of rabbit aldehyde oxidase for nitroguanidine and nitromethylene neonicotinoid insecticides. Chem Res Toxicol 19(1):38–43. doi:10.1021/tx050230x
Ding TL, Benet LZ (1979) Comparative bioavailability and pharmacokinetic studies of azathioprine and 6-mercaptopurine in the rhesus monkey. Drug Metab Dispos 7(6):373–377
Falciani F, Ghezzi P, Terao M, Cazzaniga G, Garattini E (1992) Interferons induce xanthine dehydrogenase gene expression in L929 cells. Biochem J 285(Pt 3):1001–1008
Falciani F, Terao M, Goldwurm S et al (1994) Molybdenum(VI) salts convert the xanthine oxidoreductase apoprotein into the active enzyme in mouse L929 fibroblastic cells. Biochem J 298(Pt 1):69–77
Fratelli M, Fisher JN, Paroni G et al (2013) New insights into the molecular mechanisms underlying sensitivity/resistance to the atypical retinoid ST1926 in acute myeloid leukaemia cells: the role of histone H2A.Z, cAMP-dependent protein kinase A and the proteasome. Eur J Cancer 49(6):1491–1500. doi:10.1016/j.ejca.2012.11.013
Fu C, Di L, Han X et al (2013) Aldehyde oxidase 1 (AOX1) in human liver cytosols: quantitative characterization of AOX1 expression level and activity relationship. Drug Metab Dispos 41(10):1797–1804. doi:10.1124/dmd.113.053082
Garattini E, Terao M (2011) Increasing recognition of the importance of aldehyde oxidase in drug development and discovery. Drug Metab Rev 43(3):374–386. doi:10.3109/03602532.2011.560606
Garattini E, Terao M (2012) The role of aldehyde oxidase in drug metabolism. Expert Opin Drug Metab Toxicol 8(4):487–503. doi:10.1517/17425255.2012.663352
Garattini E, Terao M (2013) Aldehyde oxidase and its importance in novel drug discovery: present and future challenges. Expert Opin Drug Discov 8(6):641–654. doi:10.1517/17460441.2013.788497
Garattini E, Fratelli M, Terao M (2008) Mammalian aldehyde oxidases: genetics, evolution and biochemistry. Cell Mol Life Sci 65(7–8):1019–1048. doi:10.1007/s00018-007-7398-y
Garattini E, Fratelli M, Terao M (2009) The mammalian aldehyde oxidase gene family. Hum Genomics 4(2):119–130
Ghaffari T, Nouri M, Saei AA, Rashidi MR (2012) Aldehyde and xanthine oxidase activities in tissues of streptozotocin-induced diabetic rats: effects of vitamin E and selenium supplementation. Biol Trace Elem Res 147(1–3):217–225. doi:10.1007/s12011-011-9291-7
Graessler J, Fischer S (2007) The dual substrate specificity of aldehyde oxidase 1 for retinal and acetaldehyde and its role in ABCA1 mediated efflux. Horm Metab Res 39(11):775–776. doi:10.1055/s-2007-992126
Gruenewald S, Wahl B, Bittner F et al (2008) The fourth molybdenum containing enzyme mARC: cloning and involvement in the activation of N-hydroxylated prodrugs. J Med Chem 51(24):8173–8177. doi:10.1021/jm8010417
Hahnewald R, Leimkuhler S, Vilaseca A, Acquaviva-Bourdain C, Lenz U, Reiss J (2006) A novel MOCS2 mutation reveals coordinated expression of the small and large subunit of molybdopterin synthase. Mol Genet Metab 89(3):210–213. doi:10.1016/j.ymgme.2006.04.008
Hardeland R, Pandi-Perumal SR, Cardinali DP (2006) Melatonin. Int J Biochem Cell Biol 38(3):313–316. doi:10.1016/j.biocel.2005.08.020
Hartmann T, Terao M, Garattini E et al (2012) The impact of single nucleotide polymorphisms on human aldehyde oxidase. Drug Metab Dispos 40(5):856–864. doi:10.1124/dmd.111.043828
Havemeyer A, Lang J, Clement B (2011) The fourth mammalian molybdenum enzyme mARC: current state of research. Drug Metab Rev 43(4):524–539. doi:10.3109/03602532.2011.608682
Heidenreich T, Wollers S, Mendel RR, Bittner F (2005) Characterization of the NifS-like domain of ABA3 from Arabidopsis thaliana provides insight into the mechanism of molybdenum cofactor sulfuration. J Biol Chem 280(6):4213–4218. doi:10.1074/jbc.M411195200
Hille R (1996) The mononuclear molybdenum enzymes. Chem Rev 96(7):2757–2816
Hille R, Sprecher H (1987) On the mechanism of action of xanthine oxidase. Evidence in support of an oxo transfer mechanism in the molybdenum-containing hydroxylases. J Biol Chem 262(23):10914–10917
Holmes RS (1979) Genetics, ontogeny, and testosterone inducibility of aldehyde oxidase isozymes in the mouse: evidence for two genetic loci (Aox-I and Aox-2) closely linked on chromosome 1. Biochem Genet 17(5–6):517–527
Hover BM, Loksztejn A, Ribeiro AA, Yokoyama K (2013) Identification of a cyclic nucleotide as a cryptic intermediate in molybdenum cofactor biosynthesis. J Am Chem Soc 135(18):7019–7032. doi:10.1021/ja401781t
Hover BM, Lilla EA, Yokoyama K (2015a) Mechanistic investigation of cPMP synthase in molybdenum cofactor biosynthesis using an uncleavable substrate analogue. Biochemistry 54(49):7229–7236. doi:10.1021/acs.biochem.5b00857
Hover BM, Tonthat NK, Schumacher MA, Yokoyama K (2015b) Mechanism of pyranopterin ring formation in molybdenum cofactor biosynthesis. Proc Natl Acad Sci USA 112(20):6347–6352. doi:10.1073/pnas.1500697112
Hu R, Xu C, Shen G et al (2006) Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life Sci 79(20):1944–1955. doi:10.1016/j.lfs.2006.06.019
Huang DY, Ichikawa Y (1994) Two different enzymes are primarily responsible for retinoic acid synthesis in rabbit liver cytosol. Biochem Biophys Res Commun 205(2):1278–1283. doi:10.1006/bbrc.1994.2803
Huang DY, Furukawa A, Ichikawa Y (1999) Molecular cloning of retinal oxidase/aldehyde oxidase cDNAs from rabbit and mouse livers and functional expression of recombinant mouse retinal oxidase cDNA in Escherichia coli. Arch Biochem Biophys 364(2):264–272. doi:10.1006/abbi.1999.1129
Huard JM, Youngentob SL, Goldstein BJ, Luskin MB, Schwob JE (1998) Adult olfactory epithelium contains multipotent progenitors that give rise to neurons and non-neural cells. J Comp Neurol 400(4):469–486. doi:10.1002/(SICI)1096-9861(19981102)400:4<469:AID-CNE3>3.0.CO;2-8
Hutzler JM, Obach RS, Dalvie D, Zientek MA (2013) Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin Drug Metab Toxicol 9(2):153–168. doi:10.1517/17425255.2013.738668
Ichida K, Amaya Y, Noda K et al (1993) Cloning of the cDNA encoding human xanthine dehydrogenase (oxidase): structural analysis of the protein and chromosomal location of the gene. Gene 133(2):279–284
Itoh K, Adachi M, Sato J et al (2009) Effects of selenium deficiency on aldehyde oxidase 1 in rats. Biol Pharm Bull 32(2):190–194
Johnson C, Stubley-Beedham C, Stell JG (1984) Elevation of molybdenum hydroxylase levels in rabbit liver after ingestion of phthalazine or its hydroxylated metabolite. Biochem Pharmacol 33(22):3699–3705
Kamli MR, Kim J, Pokharel S, Jan AT, Lee EJ, Choi I (2014) Expressional studies of the aldehyde oxidase (AOX1) gene during myogenic differentiation in C2C12 cells. Biochem Biophys Res Commun 450(4):1291–1296. doi:10.1016/j.bbrc.2014.06.126
Kinsella TJ, Kunugi KA, Vielhuber KA, McCulloch W, Liu SH, Cheng YC (1994) An in vivo comparison of oral 5-iodo-2′-deoxyuridine and 5-iodo-2-pyrimidinone-2′-deoxyribose toxicity, pharmacokinetics, and DNA incorporation in athymic mouse tissues and the human colon cancer xenograft, HCT-116. Cancer Res 54(10):2695–2700
Kinsella TJ, Kunugi KA, Vielhuber KA, Potter DM, Fitzsimmons ME, Collins JM (1998) Preclinical evaluation of 5-iodo-2-pyrimidinone-2′-deoxyribose as a prodrug for 5-iodo-2′-deoxyuridine-mediated radiosensitization in mouse and human tissues. Clin Cancer Res 4(1):99–109
Kinsella TJ, Schupp JE, Davis TW et al (2000) Preclinical study of the systemic toxicity and pharmacokinetics of 5-iodo-2-deoxypyrimidinone-2′-deoxyribose as a radiosensitizing prodrug in two, non-rodent animal species: implications for phase I study design. Clin Cancer Res 6(9):3670–3679
Kitamura S, Tatsumi K (1984a) Involvement of liver aldehyde oxidase in the reduction of nicotinamide N-oxide. Biochem Biophys Res Commun 120(2):602–606
Kitamura S, Tatsumi K (1984b) Reduction of tertiary amine N-oxides by liver preparations: function of aldehyde oxidase as a major N-oxide reductase. Biochem Biophys Res Commun 121(3):749–754
Kitamura S, Sugihara K, Nakatani K et al (1999) Variation of hepatic methotrexate 7-hydroxylase activity in animals and humans. IUBMB Life 48(6):607–611. doi:10.1080/713803569
Kundu TK, Velayutham M, Zweier JL (2012) Aldehyde oxidase functions as a superoxide generating NADH oxidase: an important redox regulated pathway of cellular oxygen radical formation. Biochemistry 51(13):2930–2939. doi:10.1021/bi3000879
Kurosaki M, Demontis S, Barzago MM, Garattini E, Terao M (1999) Molecular cloning of the cDNA coding for mouse aldehyde oxidase: tissue distribution and regulation in vivo by testosterone. Biochem J 341(Pt 1):71–80
Kurosaki M, Terao M, Barzago MM et al (2004) The aldehyde oxidase gene cluster in mice and rats. Aldehyde oxidase homologue 3, a novel member of the molybdo-flavoenzyme family with selective expression in the olfactory mucosa. J Biol Chem 279(48):50482–50498. doi:10.1074/jbc.M408734200
Kurosaki M, Bolis M, Fratelli M et al (2013) Structure and evolution of vertebrate aldehyde oxidases: from gene duplication to gene suppression. Cell Mol Life Sci 70(10):1807–1830. doi:10.1007/s00018-012-1229-5
Lee HF, Mak BS, Chi CS, Tsai CR, Chen CH, Shu SG (2002) A novel mutation in neonatal isolated sulphite oxidase deficiency. Neuropediatrics 33(4):174–179. doi:10.1055/s-2002-34491
Lehrke M, Rump S, Heidenreich T, Wissing J, Mendel RR, Bittner F (2012) Identification of persulfide-binding and disulfide-forming cysteine residues in the NifS-like domain of the molybdenum cofactor sulfurase ABA3 by cysteine-scanning mutagenesis. Biochem J 441(3):823–832. doi:10.1042/BJ20111170
Leimkuhler S, Iobbi-Nivol C (2015) Bacterial molybdoenzymes: old enzymes for new purposes. FEMS Microbiol Rev. doi:10.1093/femsre/fuv043
Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL (2008) Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem 283(26):17855–17863. doi:10.1074/jbc.M801785200
Maeda K, Ohno T, Igarashi S, Yoshimura T, Yamashiro K, Sakai M (2012) Aldehyde oxidase 1 gene is regulated by Nrf2 pathway. Gene 505(2):374–378. doi:10.1016/j.gene.2012.06.010
Mahro M, Coelho C, Trincao J et al (2011) Characterization and crystallization of mouse aldehyde oxidase 3: from mouse liver to Escherichia coli heterologous protein expression. Drug Metab Dispos 39(10):1939–1945. doi:10.1124/dmd.111.040873
Mahro M, Bras NF, Cerqueira NM et al (2013) Identification of crucial amino acids in mouse aldehyde oxidase 3 that determine substrate specificity. PLoS ONE 8(12):e82285. doi:10.1371/journal.pone.0082285
Maia LB, Pereira V, Mira L, Moura JJ (2015) Nitrite reductase activity of rat and human xanthine oxidase, xanthine dehydrogenase, and aldehyde oxidase: evaluation of their contribution to NO formation in vivo. Biochemistry 54(3):685–710. doi:10.1021/bi500987w
Manevski N, Balavenkatraman KK, Bertschi B et al (2014) Aldehyde oxidase activity in fresh human skin. Drug Metab Dispos 42(12):2049–2057. doi:10.1124/dmd.114.060368
Massey V, Edmondson D (1970) On the mechanism of inactivation of xanthine oxidase by cyanide. J Biol Chem 245(24):6595–6598
Matthies A, Nimtz M, Leimkuhler S (2005) Molybdenum cofactor biosynthesis in humans: identification of a persulfide group in the rhodanese-like domain of MOCS3 by mass spectrometry. Biochemistry 44(21):7912–7920. doi:10.1021/bi0503448
Mendel RR (2013) The molybdenum cofactor. J Biol Chem 288(19):13165–13172. doi:10.1074/jbc.R113.455311
Mendel RR, Kruse T (2012) Cell biology of molybdenum in plants and humans. Biochim Biophys Acta 9:1568–1579. doi:10.1016/j.bbamcr.2012.02.007
Mendel RR, Leimkuhler S (2015) The biosynthesis of the molybdenum cofactors. J Biol Inorg Chem 20(2):337–347. doi:10.1007/s00775-014-1173-y
Mira L, Maia L, Barreira L, Manso CF (1995) Evidence for free radical generation due to NADH oxidation by aldehyde oxidase during ethanol metabolism. Arch Biochem Biophys 318(1):53–58. doi:10.1006/abbi.1995.1203
Molotkov A, Fan X, Deltour L et al (2002) Stimulation of retinoic acid production and growth by ubiquitously expressed alcohol dehydrogenase Adh3. Proc Natl Acad Sci USA 99(8):5337–5342. doi:10.1073/pnas.08209329999/8/5337
Moriwaki Y, Yamamoto T, Yamakita J, Takahashi S, Higashino K (1998a) Comparative localization of aldehyde oxidase and xanthine oxidoreductase activity in rat tissues. Histochem J 30(2):69–74
Moriwaki Y, Yamamoto T, Yamakita J, Takahashi S, Tsutsumi Z, Higashino K (1998b) Zonal distribution of allopurinol-oxidizing enzymes in rat liver. Adv Exp Med Biol 431:47–50
Moriwaki Y, Yamamoto T, Takahashi S, Tsutsumi Z, Hada T (2001) Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol Histopathol 16(3):745–753
Neumeier M, Hellerbrand C, Gabele E et al (2006) Adiponectin and its receptors in rodent models of fatty liver disease and liver cirrhosis. World J Gastroenterol 12(34):5490–5494
Niederreither K, Subbarayan V, Dolle P, Chambon P (1999) Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet 21(4):444–448. doi:10.1038/7788
Nishino T (1994) The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J Biochem 116(1):1–6
Nishino T, Okamoto K, Eger BT, Pai EF (2008) Mammalian xanthine oxidoreductase—mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J 275(13):3278–3289. doi:10.1111/j.1742-4658.2008.06489.x
Obach RS, Walsky RL (2005) Drugs that inhibit oxidation reactions catalyzed by aldehyde oxidase do not inhibit the reductive metabolism of ziprasidone to its major metabolite, S-methyldihydroziprasidone: an in vitro study. J Clin Psychopharmacol 25(6):605–608
Obach RS, Prakash C, Kamel AM (2012) Reduction and methylation of ziprasidone by glutathione, aldehyde oxidase, and thiol S-methyltransferase in humans: an in vitro study. Xenobiotica 42(11):1049–1057. doi:10.3109/00498254.2012.683203
Ohkubo M, Sakiyama S, Fujimura S (1983a) Increase of nicotinamide methyltransferase and N1-methyl-nicotinamide oxidase activities in the livers of the rats administered alkylating agents. Cancer Lett 21(2):175–181
Ohkubo M, Sakiyama S, Fujimura S (1983b) Purification and characterization of N1-methylnicotinamide oxidases I and II separated from rat liver. Arch Biochem Biophys 221(2):534–542
Ott G, Havemeyer A, Clement B (2015) The mammalian molybdenum enzymes of mARC. J Biol Inorg Chem 20(2):265–275. doi:10.1007/s00775-014-1216-4
Payne AP (1994) The harderian gland: a tercentennial review. J Anat 185(Pt 1):1–49
Pelikant-Malecka I, Sielicka A, Kaniewska E, Smolenski RT, Slominska EM (2015) Endothelial toxicity of unusual nucleotide metabolites. Pharmacol Rep 67(4):818–822. doi:10.1016/j.pharep.2015.03.020
Pryde DC, Tran TD, Jones P et al (2012) Medicinal chemistry approaches to avoid aldehyde oxidase metabolism. Bioorg Med Chem Lett 22(8):2856–2860. doi:10.1016/j.bmcl.2012.02.069
Rajagopalan KV, Johnson JL (1992) The pterin molybdenum cofactors. J Biol Chem 267(15):10199–10202
Rajapakshe A, Tollin G, Enemark JH (2012) Kinetic and thermodynamic effects of mutations of human sulfite oxidase. Chem Biodivers 9(9):1621–1634. doi:10.1002/cbdv.201200010
Rani Basu L, Mazumdar K, Dutta NK, Karak P, Dastidar SG (2005) Antibacterial property of the antipsychotic agent prochlorperazine, and its synergism with methdilazine. Microbiol Res 160(1):95–100
Reiss J, Cohen N, Dorche C et al (1998) Mutations in a polycistronic nuclear gene associated with molybdenum cofactor deficiency. Nat Genet 20(1):51–53. doi:10.1038/1706
Reiss J, Dorche C, Stallmeyer B, Mendel RR, Cohen N, Zabot MT (1999) Human molybdopterin synthase gene: genomic structure and mutations in molybdenum cofactor deficiency type B. Am J Hum Genet 64(3):706–711. doi:10.1086/302296
Rivera SP, Choi HH, Chapman B et al (2005) Identification of aldehyde oxidase 1 and aldehyde oxidase homologue 1 as dioxin-inducible genes. Toxicology 207(3):401–409. doi:10.1016/j.tox.2004.10.009
Rooseboom M, Commandeur JN, Vermeulen NP (2004) Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev 56(1):53–102. doi:10.1124/pr.56.1.3
Sanoh S, Tayama Y, Sugihara K, Kitamura S, Ohta S (2015) Significance of aldehyde oxidase during drug development: effects on drug metabolism, pharmacokinetics, toxicity, and efficacy. Drug Metab Pharmacokinet 30(1):52–63. doi:10.1016/j.dmpk.2014.10.009
Schumann S, Terao M, Garattini E et al (2009) Site directed mutagenesis of amino acid residues at the active site of mouse aldehyde oxidase AOX1. PLoS ONE 4(4):e5348. doi:10.1371/journal.pone.0005348
Schwartz R, Kjeldgaard NO (1951) The enzymic oxidation of pyridoxal by liver aldehyde oxidase. Biochem J 48(3):333–337
Schwarz G, Mendel RR (2006) Molybdenum cofactor biosynthesis and molybdenum enzymes. Annu Rev Plant Biol 57:623–647. doi:10.1146/annurev.arplant.57.032905.105437
Schwarz G, Mendel RR, Ribbe MW (2009) Molybdenum cofactors, enzymes and pathways. Nature 460(7257):839–847. doi:10.1038/nature08302
Shaw S, Jayatilleke E (1990) The role of aldehyde oxidase in ethanol-induced hepatic lipid peroxidation in the rat. Biochem J 268(3):579–583
Shintani Y, Maruoka S, Gon Y et al (2015) Nuclear factor erythroid 2-related factor 2 (Nrf2) regulates airway epithelial barrier integrity. Allergol Int 64(Suppl):S54–S63. doi:10.1016/j.alit.2015.06.004
Sigruener A, Buechler C, Orso E et al (2007) Human aldehyde oxidase 1 interacts with ATP-binding cassette transporter-1 and modulates its activity in hepatocytes. Horm Metab Res 39(11):781–789. doi:10.1055/s-2007-992129
Stallmeyer B, Drugeon G, Reiss J, Haenni AL, Mendel RR (1999) Human molybdopterin synthase gene: identification of a bicistronic transcript with overlapping reading frames. Am J Hum Genet 64(3):698–705. doi:10.1086/302295
Stanulovic M, Chaykin S (1971) Aldehyde oxidase: catalysis of the oxidation of N 1-methylnicotinamide and pyridoxal. Arch Biochem Biophys 145(1):27–34
Stell JG, Wheelhouse RT, Wright CW (2012) Metabolism of cryptolepine and 2-fluorocryptolepine by aldehyde oxidase. J Pharm Pharmacol 64(2):237–243. doi:10.1111/j.2042-7158.2011.01408.x
Stoddart AM, Levine WG (1992) Azoreductase activity by purified rabbit liver aldehyde oxidase. Biochem Pharmacol 43(10):2227–2235
Stubley C, Stell JG, Mathieson DW (1979) The oxidation of azaheterocycles with mammalian liver aldehyde oxidase. Xenobiotica 9(8):475–484. doi:10.3109/00498257909087261
Sugihara K, Kitamura S, Tatsumi K, Asahara T, Dohi K (1997) Differences in aldehyde oxidase activity in cytosolic preparations of human and monkey liver. Biochem Mol Biol Int 41(6):1153–1160
Swenson TL, Casida JE (2013) Aldehyde oxidase importance in vivo in xenobiotic metabolism: imidacloprid nitroreduction in mice. Toxicol Sci 133(1):22–28. doi:10.1093/toxsci/kft066
Tatsumi K, Kitamura S, Narai N (1986) Reductive metabolism of aromatic nitro compounds including carcinogens by rabbit liver preparations. Cancer Res 46(3):1089–1093
Tayama Y, Miyake K, Sugihara K et al (2007a) Developmental changes of aldehyde oxidase activity in young Japanese children. Clin Pharmacol Ther 81(4):567–572. doi:10.1038/sj.clpt.6100078
Tayama Y, Moriyasu A, Sugihara K, Ohta S, Kitamura S (2007b) Developmental changes of aldehyde oxidase in postnatal rat liver. Drug Metab Pharmacokinet 22(2):119–124
Tayama Y, Sugihara K, Sanoh S et al (2011) Effect of tea beverages on aldehyde oxidase activity. Drug Metab Pharmacokinet 26(1):94–101
Tayama Y, Sugihara K, Sanoh S, Miyake K, Kitamura S, Ohta S (2012) Developmental changes of aldehyde oxidase activity and protein expression in human liver cytosol. Drug Metab Pharmacokinet 27(5):543–547
Terao M, Cazzaniga G, Ghezzi P et al (1992) Molecular cloning of a cDNA coding for mouse liver xanthine dehydrogenase. Regulation of its transcript by interferons in vivo. Biochem J 283(Pt 3):863–870
Terao M, Kurosaki M, Saltini G et al (2000) Cloning of the cDNAs coding for two novel molybdo-flavoproteins showing high similarity with aldehyde oxidase and xanthine oxidoreductase. J Biol Chem 275(39):30690–30700. doi:10.1074/jbc.M005355200
Terao M, Kurosaki M, Marini M et al (2001) Purification of the aldehyde oxidase homolog 1 (AOH1) protein and cloning of the AOH1 and aldehyde oxidase homolog 2 (AOH2) genes. Identification of a novel molybdo-flavoprotein gene cluster on mouse chromosome 1. J Biol Chem 276(49):46347–46363. doi:10.1074/jbc.M105744200
Terao M, Kurosaki M, Barzago MM et al (2006) Avian and canine aldehyde oxidases. Novel insights into the biology and evolution of molybdo-flavoenzymes. J Biol Chem 281(28):19748–19761. doi:10.1074/jbc.M600850200
Terao M, Kurosaki M, Barzago MM et al (2009) Role of the molybdoflavoenzyme aldehyde oxidase homolog 2 in the biosynthesis of retinoic acid: generation and characterization of a knockout mouse. Mol Cell Biol 29(2):357–377. doi:10.1128/MCB.01385-08
Tomita S, Tsujita M, Ichikawa Y (1993) Retinal oxidase is identical to aldehyde oxidase. FEBS Lett 336(2):272–274
Tsujita M, Tomita S, Miura S, Ichikawa Y (1994) Characteristic properties of retinal oxidase (retinoic acid synthase) from rabbit hepatocytes. Biochim Biophys Acta 1204(1):108–116
Ueda O, Kitamura S, Ohashi K, Sugihara K, Ohta S (2003) Xanthine oxidase-catalyzed metabolism of 2-nitrofluorene, a carcinogenic air pollutant, in rat skin. Drug Metab Dispos 31(4):367–372
Ueda O, Sugihara K, Ohta S, Kitamura S (2005) Involvement of molybdenum hydroxylases in reductive metabolism of nitro polycyclic aromatic hydrocarbons in mammalian skin. Drug Metab Dispos 33(9):1312–1318. doi:10.1124/dmd.105.005306
Ventura SM, Dachtler SL (1980) Development and maturation of aldehyde oxidase levels in fetal hepatic tissue of C57BL/6J mice. Enzyme 25(4):213–219
Ventura SM, Dachtler SL (1981) Effects of sex hormones on hepatic aldehyde oxidase activity in C57BL/6J mice. Horm Res 14(4):250–259
Vila R, Kurosaki M, Barzago MM et al (2004) Regulation and biochemistry of mouse molybdo-flavoenzymes. The DBA/2 mouse is selectively deficient in the expression of aldehyde oxidase homologues 1 and 2 and represents a unique source for the purification and characterization of aldehyde oxidase. J Biol Chem 279(10):8668–8683. doi:10.1074/jbc.M308137200
Wahl RC, Rajagopalan KV (1982) Evidence for the inorganic nature of the cyanolyzable sulfur of molybdenum hydroxylases. J Biol Chem 257(3):1354–1359
Weigert J, Neumeier M, Bauer S et al (2008) Small-interference RNA-mediated knock-down of aldehyde oxidase 1 in 3T3-L1 cells impairs adipogenesis and adiponectin release. FEBS Lett 582(19):2965–2972. doi:10.1016/j.febslet.2008.07.034
Wollers S, Heidenreich T, Zarepour M et al (2008) Binding of sulfurated molybdenum cofactor to the C-terminal domain of ABA3 from Arabidopsis thaliana provides insight into the mechanism of molybdenum cofactor sulfuration. J Biol Chem 283(15):9642–9650. doi:10.1074/jbc.M708549200
Yamaguchi Y, Matsumura T, Ichida K, Okamoto K, Nishino T (2007) Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: roles of active site residues in binding and activation of purine substrate. J Biochem 141(4):513–524
Yoshihara S, Tatsumi K (1985) Guinea pig liver aldehyde oxidase as a sulfoxide reductase: its purification and characterization. Arch Biochem Biophys 242(1):213–224
Yoshihara S, Tatsumi K (1997) Purification and characterization of hepatic aldehyde oxidase in male and female mice. Arch Biochem Biophys 338(1):29–34. doi:10.1006/abbi.1996.9774
Zientek MA, Youdim K (2015) Reaction phenotyping: advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab Dispos 43(1):163–181. doi:10.1124/dmd.114.058750
Ziouzenkova O, Orasanu G, Sharlach M et al (2007) Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med 13(6):695–702. doi:10.1038/nm1587
Acknowledgments
The financial support granted by the Fondazione Italo Monzino and the Associazione Italiana per la Ricerca contro il Cancro (AIRC) to Enrico Garattini was fundamental for the extension of this review article. This work was also financially supported by the Deutsche Forschungsgemeinschaft (DFG) grant LE1171/8-1 to Silke Leimkühler and by. Fundação para a Ciência e Tecnologia through projects UID/Multi/04378/2013, PTDC/BBB-BEP/1185/2014, EXCL/QEQ-COM/0394/2012, PTDC/BIA-PRO/118377/2010 (M.J.R., T.S.-S., C.C.), SFRH/BPD/84581/2012 (C.C.) and DAAD-441.00 (M.J.R., T.S.-S., S.L.). We also thank the I02 staff of the Diamond Light Source (DLS), the X06DA-PXIII staff from the Swiss Light Source (SLS) and the staff from ID14-1, ID29-1, and ID23-1 from the European Synchrotron Radiation Facility (ESRF). We would also like to acknowledge the help of Mr. Felice Deceglie and Mr. Alessandro Soave for the artwork.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Terao, M., Romão, M.J., Leimkühler, S. et al. Structure and function of mammalian aldehyde oxidases. Arch Toxicol 90, 753–780 (2016). https://doi.org/10.1007/s00204-016-1683-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-016-1683-1