
Abstract
The incidence, prevalence, and hospitalization rates associated with cardiovascular diseases (CVDs) are projected to increase substantially in the world. Understanding of the biological and pathophysiological mechanisms of survival can help the researchers to develop new management modalities. Numerous experimental studies have demonstrated that mid-chain HETEs are strongly involved in the pathogenesis of the CVDs. Mid-chain HETEs are biologically active eicosanoids that result from the metabolism of arachidonic acid (AA) by both lipoxygenase and CYP1B1 (lipoxygenase-like reaction). Therefore, identifying the localizations and expressions of the lipoxygenase and CYP1B1 and their associated AA metabolites in the cardiovascular system is of major importance in understanding their pathological roles. Generally, the expression of these enzymes is shown to be induced during several CVDs, including hypertension and cardiac hypertrophy. The induction of these enzymes is associated with the generation of mid-chain HETEs and subsequently causation of cardiovascular events. Of interest, inhibiting the formation of mid-chain HETEs has been reported to confer a protection against different cardiac hypertrophy and hypertension models such as angiotensin II, Goldblatt, spontaneously hypertensive rat and deoxycorticosterone acetate (DOCA)-salt-induced models. Although the exact mechanisms of mid-chain HETEs-mediated cardiovascular dysfunction are not fully understood, the present review proposes several mechanisms which include activating G-protein-coupled receptor, protein kinase C, mitogen-activated protein kinases, and nuclear factor kappa B. This review provides a clear understanding of the role of mid-chain HETEs in the pathogenesis of cardiovascular diseases and their importance as novel targets in the treatment for hypertension and cardiac hypertrophy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
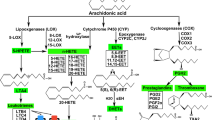
Mounting evidence is shedding light on the role of arachidonic acid (AA) metabolites in the pathogenesis of cardiovascular disease (CVD) (Roman 2002). AA is released following activation of phospholipase A2 and subsequent metabolism by cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P-450 (CYP) pathways. These enzymes insert oxygen at different positions in AA to generate a major family of biologically active mediators called eicosanoids (Capdevila et al. 1981). CYP, cysteinato-heme mixed function mono-oxygenases enzymes, oxidizes AA into epoxyeicosatrienoic acids (EETs) and hydroxyeicosatetraenoic acids (HETEs) which are known to play an important role in the maintenance of cardiovascular health (Zordoky and El-Kadi 2010). CYP ω-hydroxylases, namely CYP4 family, metabolize AA into its cardiotoxic form 20-HETE (Anwar-Mohamed et al. 2013; Elshenawy et al. 2013; Fava et al. 2012; Gross et al. 2005; Schwartzman et al. 1996; Wu and Schwartzman 2011; Yousif et al. 2009; Zordoky et al. 2008, 2010), whereas CYP epoxygenases, mainly CYP2B, CYP2C, and CYP2J subfamilies, metabolize AA into four regioisomers of cardioprotective EETs, 14,15-EET, 11,12-EET, 8,9-EET, and 5,6-EET metabolites (Roman 2002). EETs are further metabolized by soluble epoxide hydrolase (sEH) into their corresponding degradation products dihydroxyeicosatrienoic acids (DHETs) (Imig et al. 2002). Mid-chain hydroxyeicosatetraenoic acids (mid-chain HETEs), typified by 5-, 12-, and 15-HETE, are biologically active eicosanoids that result from the metabolism of AA by both LOX- and CYP-catalyzed bis-allylic oxidation reaction (LOX-like reaction) (Fig. 1). In this review, we will focus on lipoxygenase products of AA, namely mid-chain HETEs, and we will discuss the role of 5-, 12-, and 15-HETEs in the pathogenesis of hypertension and cardiac hypertrophy.

Arachidonic acid metabolism pathway. Arachidonic acid is released following the activation of phospholipase A2 and subsequent metabolism by cyclooxygenase (COX), lipoxygenase (LOX) and cytochrome P-450 (CYP) pathways. These enzymes insert oxygen at different positions in AA to generate a major family of biologically active mediators called eicosanoids
Biosynthesis of mid-chain HETEs
LOX
Regulation of LOX enzymes
LOXs, non-heme iron dioxygenase enzymes, constitute a family of lipid-peroxidizing enzymes that insert molecular oxygen into free and esterified polyunsaturated fatty acids. The LOX enzymes are named according to the specific carbon atoms of AA that are oxidized (Chen et al. 1994). For instance, the 12-LOX oxygenates AA at C-12 and catalyzes the formation of 12-hydroxyperoxyeicosatetraenoic acid (12-HPETE), which then is subsequently converted into 12-hydroxyeicosaenoic acid (12-HETE) by glutathione peroxidase. The platelet-type 12-lipoxygenase was the first mammalian LOX to be cloned as a functionally distinct isoform and is expressed in leukocytes and epidermal cells (Yamamoto 1992). Interestingly, though some LOXs form exclusively one metabolite from AA, others are categorized as dual-specificity LOX [12-LOX (leukocyte type), 15-LOX-1] because they form both 12-HETE and 15-HETE metabolites at the same time (Yamamoto 1992). 15-LOX-1 has been shown to catalyze the metabolism of linoleic acid to synthesize hydroxy octadecadienoic acids. A second 15-LOX gene has been discovered in human in 1997 (Brash et al. 1997). Based on the amino acid sequence, it seems that the murine homolog of 15-LOX-2 has primarily 8-LOX activity, while the rat homolog has not been characterized to date (Jisaka et al. 2000). Unlike other LOXs, 5-LOX requires the presence of 5-LOX activating protein (FLAP) for productive 5-HETE synthesis in vivo. FLAP is a member of the Membrane-Associated Proteins in Eicosanoid and Glutathione metabolism (MAPEG) superfamily, localizing to the nuclear envelope. FLAP has been shown to be existed as a trimer, creating a binding pocket that allows AA to laterally diffuse into the protein complex from the membrane (Ferguson et al. 2007). The cytosolic loops of FLAP interact with the 5-LOX catalytic domain and transfer AA into the 5-LOX active site.
LOX expressions in cardiovascular system
12/15-LOX was originally isolated from porcine leukocytes (Yokoyama et al. 1986), but its tissue distribution is now known to be relatively wide, including the adrenal gland, the brain, and the kidneys (Gu et al. 1994; Katoh et al. 1994; Watanabe et al. 1993). Significant amounts of 12/15-LOX mRNA were also detected in rat spleen, aorta, lung, and leukocytes (Hada et al. 1994). In endothelial cells, the basal level of 12-LOX is required for serum-stimulated endothelial cell proliferation and for minimally modified low-density lipoprotein-induced monocyte binding to endothelial cells (Honda et al. 1999; Tang et al. 1995). Although the expression of 12/15-LOX in heart tissue is relatively low compared with the blood vessels, the induced 12/15-LOX activities in heart were four times greater than in reticulocytes, previously the richest known source of the enzyme (Bailey et al. 1995). Furthermore, the 5-LOX mRNA content was significantly greater in the heart compared to the brain in mice (Dzitoyeva et al. 2009). The induction of LOX has been shown to play a major role in the pathogenesis of cardiovascular diseases (CVDs) including hypertension and atherosclerosis. Furthermore, a nonsynonymous polymorphism in 12-LOX was shown to be associated with essential hypertension and urinary 12-HETE (Quintana et al. 2006). 5-LOX polymorphism has been reported to be related to the vulnerability of the carotid atherosclerosis plaques (Jin et al. 2010). The 5-LOX protein was abundantly expressed in arterial walls of patients afflicted with various lesion stages of atherosclerosis of the aorta and of coronary and carotid arteries (Spanbroek et al. 2003).
CYP1B1
Regulation of CYP1B1 enzyme
CYP1B1, CYP-catalyzed bis-allylic oxidation (LOX-like reaction), also metabolizes AA to produce mid-chain HETEs (Choudhary et al. 2004). CYP1B1 is a monooxygenase enzyme that is involved in a number of cellular functions such as metabolism of xenobiotics (Walisser et al. 2005). CYP1B1 gene was cloned in 1994 from tetrachloro-dibenzo-1/2-dioxin-treated human keratinocyte cells (Sutter et al. 1994). CYP1B1 is a tumor-related form of CYPs which is constitutively expressed in extrahepatic tissues and is markedly overexpressed in a wide variety of primary tumors (McFadyen et al. 2001a). The presence of CYP1B1 in tumor tissues may be of importance in the modulation of these tumors by anticancer drugs (McFadyen et al. 2001b; Murray et al. 2001). In this regard, the high expression level of CYP1B1 in tumor tissues, with lack of expression in normal tissues, was found to be partially regulated through proteasomal degradation of the enzyme (Bandiera et al. 2005). CYP1B1 has been shown to be responsible for the bioactivation of a variety of environmental carcinogens such as polycyclic aromatic hydrocarbons (PAHs) to epoxide and diol epoxide intermediates (Shimada and Fujii-Kuriyama 2004). The biochemical and carcinogenic effects of PAHs are primarily initiated by binding to and activation of a cytosolic ligand-activated transcription factor, aryl hydrocarbon receptor (AhR). Mechanistically, upon binding with its ligands, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), AhR dissociates from its inhibitory proteins (Denison et al. 1986) allowing it to translocate to the nucleus, where it heterodimerizes with a nuclear transcription factor protein called the AhR nuclear translocator (ARNT) (Whitelaw et al. 1994). The heterodimeric AhR-ARNT complex then binds to specific DNA recognition sequences, GCGTG, within the xenobiotic-responsive element (XRE) located in the promoter region of CYP1B1 gene (Korashy and El-Kadi 2006; Nebert et al. 2004). Beside a transcriptional mechanism, CYP1B1 expression has been shown to be controlled by posttranslational mechanisms (Murray et al. 2001). Several studies demonstrated that the constitutive and inducible expressions of CYP1B1 mRNA do not correlate with the expression of AhR mRNA. In addition, the constitutive Cyp1b1 mRNA and protein were expressed in ARNT-deficient murine hepatoma cells as compared to wild-type (WT) cells (Eltom et al. 1999). These results suggested that other mechanisms possibly contributed to the regulation of CYP1B1, including non-AhR-mediated pathways and/or posttranscriptional mechanisms.
CYP1B1 expression in cardiovascular system
Though most CYPs are expressed in the liver, extrahepatic expression of these genes was reported in the kidney, lung, and brain (Malik et al. 2012). Among extrahepatic sites, CYP1B1 has been reported to be constitutively expressed in adult human heart at mRNA level and in the human fetal ventricular cardiomyocyte, RL-14 cell line, at mRNA and protein levels (Choudhary et al. 2005; Maayah et al. 2015). Furthermore, Cyp1b1 mRNA is predominantly expressed at a significant level in the heart of both AhR-WT and AhR-null adult mice heart representing about 13 % of the total cardiac CYPs (Choudhary et al. 2003). In female NMRI mice heart, ethoxy resurofin O-deethylase (EROD) activity, the functional marker of Cyp1b1, was 8- and 180-fold lower than the lung and hepatic EROD activity, respectively (Granberg et al. 2000). In blood vessel, CYP1B1 has been shown to be constitutively expressed in vascular smooth muscle cells, retinal endothelial cells, and coronary artery smooth muscle cells (Conway et al. 2009; Dubey et al. 2003; Tang et al. 2009). Although CYP1B1 is expressed in normal tissues and is constitutively active, the induction of CYP1B1 has been shown to play a major role in the pathogenesis of CVDs including ischemic heart diseases, myocardial infarction, hypertension, atherosclerosis, cardiac hypertrophy, and heart failure (Korashy and El-Kadi 2006; Malik et al. 2012). Furthermore, CYP1B1 polymorphism was shown to play a role in the pathogenesis of heart diseases. In this regard, hazard ratio for heart disease among never smokers was 1.9 (95 % confidence interval: 1.2–3.2) for CYP1B1*3 GG (19 %) versus CC (32 %) according to the Copenhagen City Heart Study (Kaur-Knudsen et al. 2009) (Table 1).
Metabolism of mid-chain HETEs
5-HETE
5-HETE is metabolized by acyltransferase-dependent acylation into cellular phospholipids and glycerides (Arai et al. 1997; O’Flaherty et al. 1986; Stenson and Parker 1979). Microsome-bound nicotinamide adenine dinucleotide phosphate (NAD+)-dependent dehydrogenase (5-hydroxyicosanoid dehydrogenase [5-HEDH]) converts 5-HETE into its 5-keto analog, 5-oxo-6E,8Z,11Z,14Z-eicosatetraenoate (5-oxo-ETE, 5-oxoETE); (Powell et al. 1992). CYP4F2 and CYP4F3 oxidize 5-HETE to 5,20-dihydroxyETE (5,20-diHETE) (Kikuta et al. 1998, 2000; O’Flaherty et al. 1986). 12-lipoxygenase metabolizes 5-HETE to 5,12-diHETE, whereas cyclooxygenase-2 further metabolizes 5-HETE into its corresponding degradation products, 5-(S),15(R)-diHETE, and 5-(S),11(R)-diHETE (Borgeat et al. 1981; Mulugeta et al. 2010; Tejera et al. 2012).
12-HETE
12-HETE is metabolized to 12-oxo-ETE by microsomal NAD+-dependent 12-hydroxyeicosanoid dehydrogenase in porcine polymophonuclear leukocytes (Powell and Rokach 2015). 12-Oxo-ETE is further metabolized in porcine neutrophils by the NADH-dependent cytosolic enzyme, 12-oxoeicosanoid Δ10-reductase, to 12-oxo-6,8,14-eicosatrienoic acid (12-oxo-ETrE, i.e., 10,11-dihydro-12-oxo-ETE) (Powell and Rokach 2015). CYP4F2 and CYP4F3 oxidize 12-HETE to 12,20-dihydroxyETE (12,20-diHETE) (Kikuta et al. 1998, 2000; Marcus et al. 1984). Tetranor-12-HETE is the major β-oxidation product resulting from peroxisomal metabolism of 12-HETE in numerous tissues including vascular smooth muscle cells (Lacape et al. 1992). 12-HETE released from platelets is converted into 5, 12-dihydroxy-(E,Z,E,Z)-6,8,10,14-eicosatetraenoic acid [5, 12-DHETE] by the 5-lipoxygenase in Ca2+-ionophorestimulated neutrophils (Marcus et al. 1982).
15-HETE
15-HETE is oxidized to its keto analog, 15-oxo-ETE, by NAD+-dependent 15-hydroxyprostaglandin dehydrogenase, 15-oxo-ETE, similar to 15-HETE, in which its product can be converted to 13-cysteinyl-glycyl-glutamyl and then 13-cysteinyl-glycine products (Bergholte et al. 1987; Hammond et al. 2012). 5-LOX oxidizes 15-HETE to form its 5,6-trans epoxide derivative which may then rearrange to the lipoxins (LX), LXA4 and LXB4 or to 5,15-dihydroperoxy-6E,8Z,11Z,13E-eicosatetraenoate (5,15-diHETE) (Serhan 2005). 15-HETE may be also acylated into membrane phospholipids, particularly phosphatidylinositol and phosphatidylethanolamine (Brezinski and Serhan 1990; Brinckmann et al. 1998; Maskrey et al. 2007; Thomas et al. 2010). The phosphatidylethanolamine-bound 15-HETE may be then metabolized to phosphatidylethanolamine-bound 15-oxo-ETE (Hammond et al. 2012).
Mid-chain HETEs and their role in cardiovascular diseases
The role of mid-chain HETEs in the vasculature
Prior commencing the role of mid-chain HETEs in the pathogenesis of hypertension, it is imperative to discuss the vasoactive functions of these metabolites. Increased formation of mid-chain HETE metabolites by the biological agents involved in cardiovascular dysfunction has been reported in vascular smooth muscle cells, endothelial cells, and monocytes (Conrad et al. 1992; Natarajan et al. 1993, 1996; Patricia et al. 1999). Mid-chain HETEs have direct effects like chemotaxis, changes in vascular tone and production of vascular endothelial growth factor, a potent angiogenic agent (Honda et al. 1999; Nakao et al. 1982; Stern et al. 1989; Tang et al. 1995). Mid-chain HETEs have been shown to exhibit direct mitogenic effects and increase the levels of the key extracellular matrix protein fibronectin in vascular smooth muscle cells (Natarajan et al. 1994). Furthermore, they also mediate the hypertrophic effect of angiotensin II and have a direct hypertrophic effect on vascular smooth muscle cells (Reddy et al. 2002; Zhang et al. 2014).
The role of 5- and 15-HETEs as a vasoactive monohydroxyeicosatetraenoic acid has been investigated on the pulmonary artery of isolated perfused lung (Burhop et al. 1988). It has been shown that 5- and 15-HETEs were able to induce pulmonary vasoconstriction, lung vascular permeability, and edema (Burhop et al. 1988). 15-HETE mediates hypoxia-induced pulmonary vascular medial thickening, intimal endothelial cells migration, and angiogenesis (Ma et al. 2011; Shen et al. 2013). Mechanistically, 15-HETE controlled the cell cycle progression from the G0/G1 phase to the G2/M+S phase and enhanced the microtubule formation in cell nucleus (Ma et al. 2011). The effect of 15-HETE was mediated via Rho-kinase pathway (ROCK) (Ma et al. 2010). The prominence of ROCK in chronic hypoxic pulmonary hypertension is emphasized because of its potential role in maintaining vasoconstriction and the vascular wall cell proliferation (Kroll et al. 2009).
The vasoactive effects of 12-HETE have been investigated on isolated perfused renal arcuate arteries of the dog using videomicroscopy (Ma et al. 1991). 12-HETE was found to act as a vasoconstrictor in small renal arteries since it reduces vascular diameter by 63 um (from 306 um), which was 37 % of the maximal vasoconstrictor response to norepinephrine (Ma et al. 1991; Yiu et al. 2003). Mechanistically, the vasoconstrictor response induced by 12-HETE was associated with depolarization of vascular smooth muscles. This is supported by a previous finding demonstrated that 12-HETE is an inhibitor of the Na+-K+-ATPase in the corneal epithelium (Masferrer et al. 1990; Schwartzman et al. 1987) and the kidney (Masferrer et al. 1990). Furthermore, the incubation of renal arteries obtained from ischemic kidneys with nordihydroguaiaretic acid (NDGA), the LOX inhibitor, showed no effect on the formation of 12-HETE, suggesting that the 12-HETE formed by renal arteries may be produced by the LOX pathway-independent mechanism (Ma et al. 1991). In agreement with this suggestion, it has been demonstrated that inhibitors of CYP attenuate the myogenic response of dog renal arcuate arteries (Kauser et al. 1991). The L-type calcium channel may be also involved in 12-HETE-mediated vasoconstriction in renal blood vessels. In that, the vasoconstrictive effect of 12-HETE was abolished during L-type calcium channel inhibition (Yiu et al. 2003). Renal myocyte Ca2+ response following exposure to 12-HETE was greatly reduced in the absence of extracellular Ca2+ or calcium channel blockade implying it as an important mechanism responsible for the afferent arteriolar vasoconstriction stimulated by 12-HETE (Yiu et al. 2003).
The role of mid-chain HETEs in the pathogenesis of hypertension
Hypertension is a powerful risk factor for heart disease in which the force of the blood against arterial walls is high enough to cause adverse heart events, such as cardiac hypertrophy, acute myocardial infraction, stroke, and coronary artery disease (Chaturvedi 2004; DiNicolantonio et al. 2015; Vakili et al. 2001). Accumulating data provide convincing evidence that mid-chain HETEs are involved in the development of hypertension. In that, the generation of mid-chain HETEs was shown to be increased in patients with essential hypertension (Dolegowska et al. 2009; Gonzalez-Nunez et al. 2001). This generation suggests a role for these metabolites in the pathogenesis of essential hypertension.
Experimentally, mice lacking macrophage 12/15-LOX has been reported to be resistant to toward both N(G)-nitro-l-arginine-methyl ester (L-NAME)- and deoxycorticosterone acetate/high-salt-induced hypertension (Kriska et al. 2012). Furthermore, 12-HETE participates in angiotensin II-induced hypertension by the modulation of angiotensin II-induced aldosterone secretion. In this regard, BW755c, a non-selective LOX blocker, inhibited the angiotensin II-stimulated level of aldosterone in a dose-dependent manner (Nadler et al. 1987). The specific role of 12-HETE is supported by the following findings, first; angiotensin II induces 12-HETE production in adrenal glomerulosa cells; second, the inability of BW755c to block the binding of angiotensin II with its receptor, suggesting an AT-I receptor-independent mechanism; third, the addition of 12-HETE and 12-HPETE restores the angiotensin II stimulatory effects during LOX inhibition (Nadler et al. 1987). The above observations suggest that the 12-HETE formation may be an obligatory step in angiotensin II control of aldosterone secretion. Moreover, it raises a question of whether or not 12-HETE is a mediator of angiotensin II effect in vascular tissue paralleling to its effect in the adrenal cortex. This hypothesis was confirmed by a previous study investigated the potential role of 12-HETE in the vasculature in an angiotensin II-dependent model of hypertension. In that, the two-kidney, one-clip (2 K, lC) Goldblatt hypertensive rat was used as a model since it is primarily dependent on the renin–angiotensin system (DeForrest et al. 1982; Freeman et al. 1977). Acute and chronic administration of phenidone, a non-selective LOX inhibitor, prevents the development of hypertension in this model (Nozawa et al. 1990). The antihypertensive effect of phenidone was accompanied by a suppression of 12-HETE formations produced by aortic segments in the 2K, 1C rats (Nozawa et al. 1990). In agreement with the above results, it has been shown that 12-HETE potentiates the angiotensin II-induced pressor response (Takai et al. 2001). In addition, renal microvascular 12-HETE formation has been reported to be increased in response to angiotensin II-induced renal vasoconstriction (Yiu et al. 2003). Inhibiting the formation of 12-HETE markedly attenuated the in vitro contractile response to angiotensin II of femoral artery rings parallel with lowering the pressor effect in vivo (Stern et al. 1989). The previous studies suggested that the formation of 12-HETE in vascular tissue may mediate, at least in part, the vasoconstrictor actions of angiotensin II.
The mechanism by which the inhibition of 12-HETE formation attenuates the rise in blood pressure produced by angiotensin II has been investigated by studying changes in cytosolic calcium in cultured rat vascular smooth muscle cells using the fluorescent dye fura-2 (Saito et al. 1992). In that, baicalein and 5,8,11-eicosatriynoic acid, 12-LOX inhibitors, repressed angiotensin II-induced increases in cytosolic calcium in both normal and calcium-poor buffer (Saito et al. 1992). The addition of 12-HETE alone to the cells had no acute effect on the intracellular calcium concentration. However, the addition of 12-HETE restored the initial calcium response to angiotensin II in vascular smooth muscle cells pretreated with LOX inhibitors. 12-HETE, by increasing angiotensin II-induced cytosolic calcium in vascular tissue, may enhance pressor-induced vascular reactivity (Sasaki et al. 1997). Furthermore, 5,8,11-eicosatriynoic acid, 12-HETE formation inhibitor, repressed vasopressin and endothelin-stimulated induction of the intracellular calcium (Saito et al. 1992). Taken together, 12-HETE may participate in the contractile action of angiotensin II through modulation of the intracellular calcium in vascular smooth muscle cells.
Spontaneously hypertensive rat often with the Wistar-Kyoto rat as the normotensive control is the most commonly used model of hypertension, with over 5000 PubMed references in the last 10 years. The importance of this model comes from the following factors; first, it is clinically relevant; second, it has uniform polygenetic disposition and excitatory factors; and third, it lacks inter-individual variation (Lindpaintner et al. 1992). Accordingly, the modulation in the formation of mid-chain HETEs using this model would reflect, at least in part, their importance in the pathogenesis of essential hypertension.
The association between the formation of 12-HETE and intra-arterial blood pressure in spontaneously hypertensive rats and Wistar-Kyoto rats has been investigated using both a cross-sectional analysis and an acute pharmacological intervention (Sasaki et al. 1997; Stern et al. 1996). 12-HETE production was substantially induced in spontaneously hypertensive rats compared with Wistar-Kyoto rats. An overall linear correlation between 12-HETE and systolic pressure suggested a positive relationship between systolic arterial pressure and the formation of 12-HETE (Stern et al. 1996). This is consistent with the finding that specific 12-LOX inhibitors, cinnamyl-3,4-dihydroxycyanocinnamate and 5,8,11-eicosatriynoic acid, significantly provoked a noticeable hypotensive effect in spontaneously hypertensive rats but not in Wistar-Kyoto rats (Sasaki et al. 1997; Stern et al. 1996). This reduction in arterial pressure was accompanied by a clear inhibition in 12-HETE formation in both serum and aortic smooth muscle, suggesting an important role of 12-HETE in the pathogenesis of elevated arterial blood pressure in spontaneously hypertensive rats.
Both 5- and 15-HETEs have also been reported to be implicated in the development of hypertension. In that respect, the formation of 5- and 15-HETEs was markedly increased in female spontaneously hypertensive rats (Koeners et al. 2011). Treatment of female spontaneously hypertensive rat with 12-(3-adamantan-1-yl-ureido)-dodecanoic acid during the perinatal phase was accompanied by a marked decrease in the formation of 5- and 15-HETEs, suggesting an important role of these mono-HETEs in the pathogenesis of hypertension (Koeners et al. 2011). The importance of 15-HETE as a potential mediator of hypertension is highlighted by its high formation level in placentae from pregnancies complicated by pregnancy-induced hypertension compared with gestation-matched controls (Mitchell and Koenig 1991). Mechanistically, 15-HETE and its hydroperoxy precursor mediated their effect through the inhibition prostacyclin biosynthesis which may contribute to the pathological sequelae of pregnancy-induced hypertension (Mitchell and Koenig 1991).
The role of mid-chain HETEs in the pathogenesis of cardiac hypertrophy
Cardiac hypertrophy is a major risk factor for heart diseases which frequently occur following acute events, such as myocardial infraction, or accompanying chronic insults such as hypertension (Vakili et al. 2001). At early stage of pathological cardiac hypertrophy, the heart walls thicken in an attempt to compensate for the increased stress (Carreno et al. 2006). However, prolonged hypertrophy deteriorates heart functions and eventually leads to heart failure. Despite the substantial progress of heart research during the past two decades, the molecular pathogenesis of cardiac hypertrophy is still ambiguous.
Several lines of evidence are supporting the role of mid-chain HETEs in the development of cardiac hypertrophy. In that, the formation of mid-chain HETEs was shown to be increased during pressure overload-induced cardiac hypertrophy (El-Sherbeni and El-Kadi 2014). The importance of descending aortic constriction (DAC) as a model of cardiac hypertrophy is pronounced since it is more clinically relevant as the cardiac hypertrophy is developed over relatively longer period of time (Patten and Hall-Porter 2009). Of particular interest in this study, the generation of mid-chain HETEs was accompanied by a high expression level of CYP1B1 protein. The role of CYP1B1 in the formation of mid-chain HETEs was confirmed by the ability of the recombinant CYP1B1 enzyme to catalyze the formation of mid-chain HETEs (Choudhary et al. 2004; El-Sherbeni and El-Kadi 2014). In addition to DAC model, we have also demonstrated a high formation level of mid-chain HETEs in rats treated with angiotensin II. Interestingly, pretreatment of rats with tetramethoxy stilbene (TMS), a selective CYP1B1 inhibitor, significantly inhibited angiotensin II-induced cardiac hypertrophy (data not published yet). Of importance, the protection against cardiac hypertrophy associated with a marked decrease in the formation of mid-chain HETEs not only suggest a crucial role of mid-chain HETEs in cardiac hypertrophy but also confirmed CYP1B1 as an important generator of mid-chain HETEs.
The overexpression of 12-LOX in cardiac fibroblast cells was used as a model to investigate the hypertrophic effect of 12-HETE (Wen et al. 2003). The importance of cardiac fibroblast cells is confirmed by the fact that the growth of fibroblast cell and its concomitant deposition of extracellular matrix proteins are one of the characterizations of cardiac fibrosis (Lal et al. 2014). The previous detrimental effects account for the abnormal myocardial stiffness and ultimate ventricular dysfunction that is seen in many forms of pathogenic cardiac hypertrophy (Souders et al. 2009). The expressed 12-LOX enzyme was functionally intact after transfection since overexpressed 12-LOX cardiac fibroblasts showed a higher level of 12-HETE in comparison with control cells. Overexpression of 12-LOX induced cell [(3)H]leucine and [(3)H]thymidine incorporation, cell protein content, fibronectin content, collagen protein expression, and enlargement of cell size compared with that of mock-transfected cells (Wen et al. 2001). 12-LOX overexpression leads to morphologic evidence of cellular hypertrophy in rat cardiac fibroblasts. This is supported by cell morphologic examination using hematoxylin and eosin (H&E) staining. This demonstrated that long axis of nuclei and the mean number of nucleoli of 12-LOX-transfected cells were significantly higher than mock-transfected cells (Wen et al. 2001, 2003).
15-HETE has been shown to increase the sensitivity of the isoproterenol-mediated β-adrenergic response in cardiomyocytes and has been proposed to be implicated in heart failure by induction of cardiac fibrosis (Kayama et al. 2009; Levick et al. 2007; Wallukat et al. 1994; Zhang et al. 2014). Furthermore, it has been shown that norepinephrine induced its hypertrophic effect through the induction of 12- and 15-HETEs (Parmentier et al. 2001). Mechanistically, 15-HETE is markedly incorporated into the cellular phosphatidylinositol pool. The 15-HETE-containing phosphatidylinositols may then be converted to 15-HETE-substituted diacylglycerol. This diacylglycerol species may in turn modulate a protein kinase C (PKC) (Wallukat et al. 1994). 15-HETE also induced adventitia fibrosis and fibroblasts phenotypic alterations depended on signaling of the transforming growth factor-β1 (Zhang et al. 2014). The above observation comes in agreement with a previous finding illustrated that baicalein, 12/15LOX inhibitor, attenuated myocardial fibrosis in spontaneously hypertensive rats (Kong et al. 2011). Moreover, 12/15 LOX inhibitors, baicalein and wogonin, have been reported to suppress collagen deposition in response to angiotensin II (Kong et al. 2010).
5-HETE has been shown to participate in the pathogenesis of angiotensin II-induced hypertrophy (Revermann et al. 2011). In that, LP105, 5-LOX blocker, inhibited angiotensin II-induced hypertrophy in ApoE−/− mice. This was manifested by the ability of LP105 to show a lower heart rate, a trend toward reduced heart-to-body weight ratio and it significantly prevented the increase in aortic weight and diameter mediated by angiotensin II (Revermann et al. 2011). In agreement with the previous result, selenium was reported to reduce diabetic cardiac hypertrophy through down-regulation of 5-LOX and its corresponding metabolite, 5-HETE (Dhanya et al. 2014).
Although the previous studies have illustrated the potential hypertrophic effect of mid-chain HETEs in the cardiovascular system, none of them have utilized human cardiomyocytes to study the cardiac hypertrophic effect of mid-chain HETEs. Therefore, we have investigated recently, for the first time, the role of mid-chain HETEs to induce cellular hypertrophy using human ventricular cardiomyocytes, RL-14 cell line (Maayah and El-Kadi 2015). The ability of mid-chain HETEs to induce cellular hypertrophy was evidenced first by the induction of the cardiac hypertrophy markers brain natriuretic peptide (BNP), atrial natriuretic peptide (ANP), α-myocin heavy chain (α-MHC), β-myocin heavy chain (β-MHC) in time- and concentration-dependent manners. The second evidence for the induction of cellular hypertrophy was the capability of mid-chain HETEs to increase the cell surface area of human ventricular cardiomyocytes in comparison with control cells. Our previous study provided the first evidence of the ability of mid-chain HETEs to induce cellular hypertrophy in the human ventricular cardiomyocytes (Maayah and El-Kadi 2015).
The role of mid-chain HETEs in the pathogenesis of heart failure and cardiomyopathy
The role of mid-chain HETEs is not restricted on cardiac hypertrophy but also involved in the development of cardiac dysfunction and heart failure. In this regard, it has been demonstrated that 12- and 15-HETEs were markedly up-regulated in heart failure (Kayama et al. 2009). The role of 12- and 15-HETEs in the pathogenesis of heart failure was investigated in transgenic mice that overexpressed 12/15-LOX in cardiomyocytes. The overexpression of 12/15-LOX and its corresponding 12- and 15-HETE metabolites was able to induce systolic dysfunction, infiltration of macrophages, up-regulation of monocyte chemoattractant protein 1, and cardiac fibrosis (Kayama et al. 2009). In HL-1 mouse cardiac myocytes, 12-HETE increases intramitochondrial calcium and mitochondrial NO, and induces apoptosis (Nazarewicz et al. 2007). Furthermore, treatment for cardiac fibroblasts and endothelial cells with 12-HETE significantly induced the expression of monocyte chemoattractant protein 1. On the other hand, disruption of 12/15-LOX significantly inhibited cardiac monocyte chemoattractant protein 1 expression, macrophage infiltration, and restoring systolic dysfunction induced by chronic pressure overload, suggesting an important role of 12- and 15-HETEs in the development of heart failure (Kayama et al. 2009).
Mid-chain HETEs were also involved in the pathogenesis of heart failure induced by doxorubicin. In this regard, we have demonstrated that doxorubicin treatment caused cardiac dysfunction and fibrosis in vitro in the human ventricular cardiomyocytes, RL-14 cell line, and in vivo in rats. This was associated with proportional increase in the formation of mid-chain HETEs. The direct involvement of mid-chain HETEs in the doxorubicin-induced cardiac dysfunction was supported by the ability of TMS, a CYP1B1 inhibitor, to attenuate doxorubicin-induced cardiotoxicity as well as it inhibited the formation of mid-chain HETEs. The above study has confirmed the role of mid-chain HETEs in the development of cardiac dysfunction and heart failure.
12- and 15-HETEs have been also shown to be implicated in the development of diabetic cardiomyopathy. Treatment of mice with streptozotocin, a well-known diabetes inducer, up-regulated the expression of 5-LOX and 12/15-LOX and its corresponding AA metabolites, 12- and 15-HETEs; it also induced cardiac dysfunction and fibrosis (Kumar et al. 2013; Suzuki et al. 2015). Interestingly, disruption of 12/15-LOX significantly inhibited the induction of TNF-α, nuclear factor kappa B (NF-κB), reactive oxygen species and eventually attenuated streptozotocin-induced cardiac dysfunction and fibrosis (Suzuki et al. 2015). In a manner similar to what was observed in vivo, neonatal cultured cardiomyocytes incubated with high glucose conditions illustrated a high expression of 12/15-LOX as well as TNF-α, NF-κB, and collagen markers which were inhibited by treatment of the 12/15-LOX inhibitor implying the role of 12- and 15-HETEs in the development of diabetic cardiomyopathy (Suzuki et al. 2015). Tables 2 and 3 summarize the effect and the role of mid-chain HETEs in the development of CVDs.
Molecular mechanism of mid-chain HETEs action
The process of cardiac hypertrophy and myopathy is complex and involves multiple cross-regulated signaling pathways (Frey and Olson 2003) that culminate in massive alterations in myocardial architecture (Fard et al. 2000). Understanding the mechanism by which mid-chain HETEs are involved in cardiac hypertrophy and myopathy could be a critical issue in cardiac homeostasis. However, these molecular mechanisms have not been fully elucidated, and no precise cellular receptors for mid-chain HETEs have yet been identified. Importantly, some potential pathways, such as PKC, mitogen-activated protein kinases (MAPKs), and NF-κB, by which these AA metabolites may stimulate cellular growth and hypertrophy have been explored. PKC, MAPKs, and NF-kB are pivotal to this process as central mediators of cardiac remodeling in response to injury and/or cardiac wall stress. Therefore, it is necessary to discuss the role of mid-chain HETEs on each pathway.
MAPKs
MAPKs are serine/threonine-specific protein kinases that are involved in the regulation of various cellular responses such as gene expression, mitosis, differentiation, proliferation, and survival/apoptosis (Kayama et al. 2009). MAPKs were found to exert control over those genes that stimulate protein synthesis and initiate hypertrophy (Thorburn et al. 1994). When hypertrophy stimulus is initiated at cell membrane, activated MAPKs move through large pores on the nuclear membrane, translocating into the nucleus, and activate transcription factors involved in cardiac hypertrophy (Pearson et al. 2001). MAPK signaling cascade consists of extracellular-regulated kinases (ERK), c-Jun NH2-terminal kinases (JNKs), and p38 MAPK (Sopontammarak et al. 2005). Previous studies analyzing MAPK activities in cardiac hypertrophy and myopathy have demonstrated differential effects; in that, persistent activation of p38 and JNK can promote apoptosis, resulting in cardiac dilation and dysfunction (Pearson et al. 2001), whereas ERK1/2 has been proposed to regulate smooth muscle contraction and to promote cellular hypertrophy (Modesti et al. 2008; Pearson et al. 2001; Sopontammarak et al. 2005). With regard to mid-chain HETEs, we have demonstrated recently that the activation of ERK1/2 signaling pathway positively regulates the induction of the cellular hypertrophy in response to mid-chain HETEs (Maayah et al. 2015). This was supported by two interesting findings; first, the ability of mid-chain HETEs to induce the phosphorylated ERK1/2; second, blocking of the phosphorylated ERK1/2 using the ERK1/2 inhibitor, U0126, significantly attenuated mid-chain HETE-induced cellular hypertrophy (Maayah et al. 2015). Our results come in agreement with a previous finding illustrated that mid-chain HETEs induced cell growth in cancer and cardiac fibroblast cells through ERK1/2 signaling pathway (Cabral et al. 2013; Garcia-Verdugo et al. 2012; Guo et al. 2011a; Kang et al. 2013; Lu et al. 2006; O’Flaherty et al. 2002; Song et al. 2015; Szekeres et al. 2000). Norepinephrine has been reported to stimulate cytosolic phospholipase A(2)-dependent phospholipase D(2) through mid-chain HETEs via ERK pathway by a mechanism involving tyrosine phosphorylation of phospholipase D(2) in rabbit vascular smooth muscle (Parmentier et al. 2001). Furthermore, it has been shown that angiotensin II induced cellular hypertrophy in H9c2 cells through ERK1/2 but not p38 or JNK (Zong et al. 2013). Of particular interest in this study, baicalein, 12/15 LOX inhibitor, blocked the cellular hypertrophic effect of angiotensin II through ERK1/2 signaling pathway (Zong et al. 2013). The effect of mid-chain HETEs is not restricted only on ERK1/2 but also involve the activation of p38 pathway. In that, 12-HETE has been shown to induce hypertrophy in cardiac fibroblast through p38 signaling pathway (Wen et al. 2003). Furthermore, treatment of porcine vascular smooth muscle cells with 12-HETE led to hypertrophy through the activation of Ras and p38 MAPK (Reddy et al. 2002). Inhibition of p38 using, SB202190, significantly blocked the hypertrophy induced by 12-HETE in both cardiac fibroblast and porcine vascular smooth muscle cells, suggesting an important role of p38 in 12-HETE induced cellular hypertrophy (Reddy et al. 2002; Wen et al. 2003). Angiotensin II also induced protein synthesis and hypertrophy in rat vascular smooth muscle through mid-chain HETEs/p38 signaling pathways (Yaghini et al. 2007). Moreover, 12- and 15-HETEs have been reported to augment AT-1 receptor and angiotensin II signaling through ERK and p38 signaling pathways (Xu et al. 2008).
NF-κB
Cardiac hypertrophy and myopathy are also regulated by several transcription factors such as NF-κB, myocyte enhancer factor 2 (MEF2), and homeobox transcription factors Csx/Nkx 2-5 (Akazawa and Komuro 2003). Among these transcription factors, NF-κB plays a wide range of physiological and pathophysiological functions, such as B cell proliferation, cell cycle control, carcinogenesis and cardiac hypertrophy, and myopathy (Grabellus et al. 2002). Biochemical analysis has established that the major form of NF-κB consists of two distinct polypeptides of 50 and 65 kDa, termed p50 and p65. Upon activation by inflammatory mediators and hypertrophy agonist, NF-κB binds to its responsive element sequences, κB, to initiate target gene transcription that is involved in cardiac hypertrophy (Leychenko et al. 2011). NF-κB has been shown to be activated in the failing human heart (Grabellus et al. 2002). Genetic NF-κB inhibition attenuates angiotensin II-induced hypertrophy, suggesting an important role of NF-κB in cardiac hypertrophy (Esposito et al. 2002). Furthermore, it has been demonstrated that blockade of NF-κB ameliorates myocardial hypertrophy in response to aortic banding and chronic infusion of angiotensin II, suggesting an important role of NF-κB as a signaling pathway in the regulation of cardiac hypertrophy (Kawano et al. 2005). Recently, we have demonstrated that mid-chain HETEs were able to induce the binding activity of NF-κB to their responsive elements (Maayah et al. 2015). The direct evidence for the involvement of NF-κB in the mid-chain HETEs-mediated induction of cellular hypertrophy was supported by the observation that blocking of NF-κB using pyrrolidinedithiocarbamate (PDTC) significantly resulted in restoration of the mRNA expression of the hypertrophy markers to their normal levels implying that the activation of NF-κB is required for the induction of cardiac hypertrophy (Maayah et al. 2015). Our results are consistent with previous findings reported that mid-chain HETEs induced cell growth and angiogenesis through NF-κB signaling pathway (Kandouz et al. 2003; Prato et al. 2010; Stoltz et al. 1996; Vonach et al. 2011). Viral vector-mediated 12/15-LOX overexpression in vascular smooth muscle cells stimulated the expression of NF-κB (Dwarakanath et al. 2008). Of particular interest in this study, mid-chain HETEs induced NF-κB through MAPK-dependent mechanism (Dwarakanath et al. 2008; Guo et al. 2011b). Inhibition of 12/15 LOX, baicalein, attenuates angiotensin II-induced cardiac hypertrophy and fibrosis through the inhibition of ERK1/2 and NF-κB signaling pathways in mice (Wang et al. 2015).
PKC and Gq-protein coupled receptor (GPCR)
The PKC is a family of multifunctional isoenzymes expressed in different tissues which plays a pivotal role in apoptosis, migration, adhesion, tumorigenesis, cardiac hypertrophy, angiogenesis, platelet function, and inflammation (Newton 2001). PKC was named PKM when it was discovered by Inoue et al. in 1977 since it was hypothesized that magnesium ion is essential for its activation (Inoue et al. 1977). However, after understanding the crucial role of calcium and the phospholipid for its activation, the protein was renamed as PKC. PKC has three isoforms, α, β, and γ, which differ in their composition of the 50 amino acids at the C-terminal end. They are expressed in various organs of the body, and this specificity is associated with their physiological functions. Of importance, PKCα is found in almost all the organs especially in the heart (Wetsel et al. 1992). PKCα functions distinctly from PKCβ and PKCγ in regulating cardiac contractility and heart failure in which transgenic mice with greater PKCα activity showed decreased cardiac contractility, ventricular dilation, and secondary hypertrophy, suggesting that increased PKCα signaling is detrimental to the heart (Braz et al. 2004; Hahn et al. 2003; Liu et al. 2009). Inhibition of PKCα protects against cardiac hypertrophy induced by angiotensin II (Yan et al. 2010). PKCα is regulated by GPCR in which activation of GPCR activates the effector enzyme phospholipase C (PLC). PLC cleaves phosphor inositol bisphosphate (PIP2) in the membrane to yield diacylglycerol (DAG) and inositol trisphosphate. DAG remains in the membrane and activates PKC (Hahn et al. 2003).
It has been demonstrated that 12-HETE activates PKCα through GPCR-mediated hydrolysis of inositol phospholipids (Liu et al. 1995). Several studies also suggest that 12-HETE activates the PKCα/ERK1/2 axis via an unidentified plasma membrane GPCR (Szekeres et al. 2000). Mechanistically, mid-chain HETEs mainly 12-HETE treatment specifically induced GTPγS coupling in membrane fractions of GPCR31-transfected cells (Guo et al. 2011b). Furthermore, 12-HETE stimulated ERK1/2 and NF-κB activation in GPR31-transfected cells. In contrast, there was no detectable ERK1/2 or NF-κB activation in 12-HETE-treated mock-transfected cells (Guo et al. 2011b). Furthermore, it has been shown that 12-HETE stimulates PKCα/NF-κB axis in freshly isolated aortic endothelial cells (Bolick et al. 2005). 5- and 15-HETEs have been shown to induce their pathological responses through PKCα/MAPK signaling pathway (Awasthi et al. 2001; Guo et al. 2009; Rao et al. 1994). In addition, 15-HETE specifically stimulates a signal transduction cascade leading to a supersensitivity of the cells toward β-adrenergic agonists, which involves the phosphatidylinositol cycle and a PKC (Wallukat et al. 1994). Figure 2 summarizes the molecular mechanism of action of mid-chain HETEs.

Molecular mechanism of mid-chain HETEs. Mid-chain HETEs bind to G-protein-coupled receptor (GPCR), induce GTPγS coupling, and activate protein kinase C (PKC). The activated PKC phosphorylates MAPK signaling cascade, extracellular-regulated kinases (ERK), c-Jun NH2-terminal kinases (JNKs), and p38. Phosphorylate MAPKs then stimulate NF-κB binding to its responsive element sequences, κB, to initiate target gene transcription that is involved in cardiac hypertrophy
Mid-chain HETEs as a promising drug targets
As suggested in the discussions above, inhibiting the formation of mid-chain HETEs can be achieved by suppressing both LOXs and CYP1B1 enzymes. Unlike receptor antagonism, inhibition of these enzymes results directly in reducing the production of fatty acid metabolites with concomitant damping of the associated inflammatory and hypertrophy activities that contribute to the pathogenesis of cardiovascular diseases.
Baicalein, a low molecular weight 5,6,7-trihydroxyflavone isolated from Scutellaria baicalensis Georgy roots, is a key component of chinese herbal medicine Scutellaria species, commonly used to treat bacterial and viral infections and cardiovascular diseases in China (Li-Weber 2009). Early studies conducted on rat platelets showed that baicalein exerts potent inhibitory activity against 12/15-LOX in addition to CYP1B1 (Chan et al. 2002; Deschamps et al. 2006). Baicalein exerts protective effects against I/R injury, hypertension, and cardiac dysfunction in mouse or rat models (Li-Weber 2009). Baicalein significantly attenuated angiotensin II-induced elevation of blood pressure, cardiac hypertrophy, and fibrosis. These beneficial effects were associated with inhibition of inflammation, oxidative stress, and multiple signaling pathways ERK1/2 and NF-κB (Wang et al. 2015). In addition to baicalein, the flavonoid luteolin, 3′,4′,5,7-tetrahydroxyflavone, and naturally occurring furocoumarins, imperatorin, display a wide range of pharmacological properties including anti-inflammatory and antioxidant activities (Abad et al. 2001; Guo et al. 2012; Lopez-Lazaro 2009). Luteolin and imperatorin exert an inhibitory effect against both LOXs and CYP1B1 at nanomolar concentration (Abad et al. 2001; Kim et al. 2005; Mammen et al. 2005; Sadik et al. 2003). Recent study has demonstrated that luteolin protects against the progression of diabetes mellitus-induced cardiac dysfunction by the attenuation of myocardial oxidative stress (Wang et al. 2012). Imperatorin can attenuate cardiac hypertrophy both in vivo and in vitro and halt the process leading from hypertrophy to heart failure (Zhang et al. 2012). However, naturally occurring furocoumarin and flavonoid compounds are known to have poor bioavailability in that they are rapidly metabolized and excreted which limit their uses clinically.
Zileuton [Leutrol, N-(1-benzo(b)-thien-2yl) ethyl-N-hydroxyurea] is a specific 5-LOX inhibitor that was developed by Abbott (Carter et al. 1991). Zileuton apparently inhibits 5-LOX via iron chelation but is lacking of 12- and 15-LOX inhibitory activity. Furthermore, it has an inhibitory activity against CYP1 family (Wang and Zhou 2009). Of interest, the 5-HETE formation inhibitor, zileuton, has been shown to protect cardiomyocytes from H2O2-induced cytotoxicity which suggests its possible application as a potent therapeutic agent for the prevention of ischemia and heart failure (Kwak et al. 2010).
Mid-chain HETEs probably produced by the CYP pathway might be involved in the mitogenesis and the regulation of cellular growth. This is supported by the finding that CYP inhibitors such as SKF-525A persuade a cell cycle delay and inhibit cellular hypertrophy, whereas LOX inhibitors such as NDGA have failed to produce such effect (Nieves and Moreno 2006). The inhibition of cellular growth in response to SKF-525A was associated with CYP inhibition and the subsequent impairment of mid-chain HETEs synthesis. Interestingly, exogenous addition of mid-chain HETEs reversed the effects of SKF-525A confirming an important role of CYP in the regulation of mid-chain HETEs (Nieves and Moreno 2006).
TMS, a selective CYP1B1 inhibitor, and Cyp1b1 gene disruption have been shown to reduce the formation of mid-chain HETEs induced by angiotensin II (Jennings et al. 2012). Of particular interest in this study, the levels of 12/15 LOX, Cyp4a, and Cyp4f protein were not changed in the Cyp1b1−/− mice, suggesting a CYP1B1-specific production of mid-chain HETEs. TMS and Cyp1b1 gene disruption also exert protective effects against angiotensin II-induced hypertension and associated cardiac hypertrophy, fibrosis, and inflammation (Jennings et al. 2010). Furthermore, they reversed deoxycorticosterone-salt-induced hypertension and cardiac and vascular hypertrophy and minimized renal dysfunction through the inhibition of reactive oxygen species (ROS) and MAPKs (Sahan-Firat et al. 2010). TMS displays antihypertensive effect and inhibits its associated cardiovascular events in spontaneously hypertensive rats, primarily by inhibiting ROS, pro-inflammatory cytokines, catecholamines, and MAPKs (Jennings et al. 2014b).
Although CYP1B1 inhibition has attenuated angiotensin II-induced hypertension and associated pathophysiological changes in male mice and rats, CYP1B1 plays a critical role in maintaining the reduced hypertensive effect of angiotensin II and its associated pathophysiological changes in female mice and rats, most likely through the generation of 2-methoxyestradiol metabolite (Jennings et al. 2014a). This is supported by a recent finding demonstrated that angiotensin II caused cardiovascular remodeling and endothelial dysfunction and increased vascular reactivity and oxidative stress in Cyp1b1(−/−) but not in Cyp1b1(+/+) female mice (Jennings et al. 2014a). Furthermore, the induction of cardiovascular changes by angiotensin II was associated with a dramatic decrease in the formation of 2-methoxyestradiol in Cyp1b1−/− mice, suggesting a 2-methoxyestradiol-dependent mechanism.
Of particular interest, 2-methoxyestrogen has been shown to exert feedback inhibition of CYP1B1 and possesses cardioprotective activity by inhibiting vascular smooth muscle cell growth in arteries (Dawling et al. 2003). Recently, FDA has approved 2-methoxyestradiol sustained-release injection indicated for the treatment for pulmonary arterial hypertension and ovarian carcinoma. In endothelial cell cultures, 2-methoxyestradiol was shown to significantly reduce endothelin-1 levels and increase prostacyclin production. In various animal models, these effects have translated into a significant reduction in vascular remodeling and right ventricular hypertrophy, resulting in reduced disease severity and improved survival (Tofovic et al. 2010). Furthermore, 2-methoxyestradiol attenuates hypertension and coronary vascular remodeling in spontaneously hypertensive rats and deoxycorticosterone-induced hypertension (Bonacasa et al. 2008; Yuan et al. 2013). However, whether 2-methoxyestradiol would inhibit the formation of mid-chain HETEs and subsequently protect against left ventricular hypertrophy has never been examined before and needs further investigations.
Conclusion
In conclusion, mid-chain HETEs induced by CYP1B1 enzyme could serve as a novel target for the development of therapeutic agents for the treatment for hypertension and cardiac hypertrophy.
References
Abad MJ, de las Heras B, Silvan AM et al (2001) Effects of furocoumarins from Cachrys trifida on some macrophage functions. J Pharm Pharmacol 53(8):1163–1168
Akazawa H, Komuro I (2003) Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res 92(10):1079–1088. doi:10.1161/01.RES.0000072977.86706.23
Anwar-Mohamed A, El-Sherbeni A, Kim SH et al (2013) Acute arsenic treatment alters cytochrome P450 expression and arachidonic acid metabolism in lung, liver and kidney of C57Bl/6 mice. Xenobiotica 43(8):719–729. doi:10.3109/00498254.2012.754113
Arai M, Imai H, Metori A, Nakagawa Y (1997) Preferential esterification of endogenously formed 5-hydroxyeicosatetraenoic acid to phospholipids in activated polymorphonuclear leukocytes. Eur J Biochem FEBS 244(2):513–519
Awasthi S, Vivekananda J, Awasthi V, Smith D, King RJ (2001) CTP:phosphocholine cytidylyltransferase inhibition by ceramide via PKC-alpha, p38 MAPK, cPLA2, and 5-lipoxygenase. Am J Physiol Lung Cell Mol Physiol 281(1):L108–L118
Bailey JM, Makheja AN, Lee R, Simon TH (1995) Systemic activation of 15-lipoxygenase in heart, lung, and vascular tissues by hypercholesterolemia: relationship to lipoprotein oxidation and atherogenesis. Atherosclerosis 113(2):247–258
Bandiera S, Weidlich S, Harth V, Broede P, Ko Y, Friedberg T (2005) Proteasomal degradation of human CYP1B1: effect of the Asn453Ser polymorphism on the post-translational regulation of CYP1B1 expression. Mol Pharmacol 67(2):435–443
Bergholte JM, Soberman RJ, Hayes R, Murphy RC, Okita RT (1987) Oxidation of 15-hydroxyeicosatetraenoic acid and other hydroxy fatty acids by lung prostaglandin dehydrogenase. Arch Biochem Biophys 257(2):444–450
Bolick DT, Orr AW, Whetzel A et al (2005) 12/15-lipoxygenase regulates intercellular adhesion molecule-1 expression and monocyte adhesion to endothelium through activation of RhoA and nuclear factor-κB. Arterioscler Thromb Vasc Biol 25(11):2301–2307. doi:10.1161/01.ATV.0000186181.19909.a6
Bonacasa B, Sanchez ML, Rodriguez F et al (2008) 2-Methoxyestradiol attenuates hypertension and coronary vascular remodeling in spontaneously hypertensive rats. Maturitas 61(4):310–316. doi:10.1016/j.maturitas.2008.09.028
Borgeat P, Picard S, Vallerand P, Sirois P (1981) Transformation of arachidonic acid in leukocytes. Isolation and structural analysis of a novel dihydroxy derivative. Prostaglandins Med 6(6):557–570
Brash AR, Boeglin WE, Chang MS (1997) Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci USA 94(12):6148–6152
Braz JC, Gregory K, Pathak A et al (2004) PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 10(3):248–254. doi:10.1038/nm1000
Brezinski ME, Serhan CN (1990) Selective incorporation of (15S)-hydroxyeicosatetraenoic acid in phosphatidylinositol of human neutrophils: agonist-induced deacylation and transformation of stored hydroxyeicosanoids. Proc Natl Acad Sci USA 87(16):6248–6252
Brinckmann R, Schnurr K, Heydeck D, Rosenbach T, Kolde G, Kuhn H (1998) Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 91(1):64–74
Burhop KE, Selig WM, Malik AB (1988) Monohydroxyeicosatetraenoic acids (5-HETE and 15-HETE) induce pulmonary vasoconstriction and edema. Circ Res 62(4):687–698
Cabral M, Martin-Venegas R, Moreno JJ (2013) Role of arachidonic acid metabolites on the control of non-differentiated intestinal epithelial cell growth. Int J Biochem Cell Biol 45(8):1620–1628. doi:10.1016/j.biocel.2013.05.009
Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW (1981) Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci USA 78(9):5362–5366
Carreno JE, Apablaza F, Ocaranza MP, Jalil JE (2006) Cardiac hypertrophy: molecular and cellular events. Rev Esp Cardiol 59(5):473–486
Carter GW, Young PR, Albert DH et al (1991) 5-lipoxygenase inhibitory activity of zileuton. J Pharmacol Exp Ther 256(3):929–937
Chan HY, Chen ZY, Tsang DS, Leung LK (2002) Baicalein inhibits DMBA-DNA adduct formation by modulating CYP1A1 and CYP1B1 activities. Biomed Pharmacother 56(6):269–275
Chaturvedi S (2004) The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure (JNC 7): is it really practical? Natl Med J India 17(4):227
Chen XS, Kurre U, Jenkins NA, Copeland NG, Funk CD (1994) cDNA cloning, expression, mutagenesis of C-terminal isoleucine, genomic structure, and chromosomal localizations of murine 12-lipoxygenases. J Biol Chem 269(19):13979–13987
Choudhary D, Jansson I, Schenkman JB, Sarfarazi M, Stoilov I (2003) Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch Biochem Biophys 414(1):91–100
Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB (2004) Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab Dispos 32(8):840–847
Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB (2005) Expression patterns of mouse and human CYP orthologs (families 1-4) during development and in different adult tissues. Arch Biochem Biophys 436(1):50–61. doi:10.1016/j.abb.2005.02.001
Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E (1992) Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci USA 89(1):217–221
Conway DE, Sakurai Y, Weiss D et al (2009) Expression of CYP1A1 and CYP1B1 in human endothelial cells: regulation by fluid shear stress. Cardiovasc Res 81(4):669–677. doi:10.1093/cvr/cvn360
Dawling S, Roodi N, Parl FF (2003) Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res 63(12):3127–3132
DeForrest JM, Knappenberger RC, Antonaccio MJ, Ferrone RA, Creekmore JS (1982) Angiotensin II is a necessary component for the development of hypertension in the two kidney, one clip rat. Am J Cardiol 49(6):1515–1517
Denison MS, Vella LM, Okey AB (1986) Structure and function of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Species difference in molecular properties of the receptors from mouse and rat hepatic cytosols. J Biol Chem 261(9):3987–3995
Deschamps JD, Kenyon VA, Holman TR (2006) Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg Med Chem 14(12):4295–4301. doi:10.1016/j.bmc.2006.01.057
Dhanya BL, Swathy RP, Indira M (2014) Selenium downregulates oxidative stress-induced activation of leukotriene pathway in experimental rats with diabetic cardiac hypertrophy. Biol Trace Elem Res 161(1):107–115. doi:10.1007/s12011-014-0076-7
DiNicolantonio JJ, Fares H, Niazi AK et al (2015) β-Blockers in hypertension, diabetes, heart failure and acute myocardial infarction: a review of the literature. Open Heart 2(1):e000230. doi:10.1136/openhrt-2014-000230
Dolegowska B, Blogowski W, Kedzierska K et al (2009) Platelets arachidonic acid metabolism in patients with essential hypertension. Platelets 20(4):242–249. doi:10.1080/09537100902849836
Dubey RK, Gillespie DG, Zacharia LC, Barchiesi F, Imthurn B, Jackson EK (2003) CYP450- and COMT-derived estradiol metabolites inhibit activity of human coronary artery SMCs. Hypertension 41(3 Pt 2):807–813. doi:10.1161/01.HYP.0000048862.28501.72
Dwarakanath RS, Sahar S, Lanting L et al (2008) Viral vector-mediated 12/15-lipoxygenase overexpression in vascular smooth muscle cells enhances inflammatory gene expression and migration. J Vasc Res 45(2):132–142. doi:10.1159/000109966
Dzitoyeva S, Imbesi M, Ng LW, Manev H (2009) 5-Lipoxygenase DNA methylation and mRNA content in the brain and heart of young and old mice. Neural Plast 2009:209596. doi:10.1155/2009/209596
Elshenawy OH, Anwar-Mohamed A, El-Kadi AO (2013) 20-Hydroxyeicosatetraenoic acid is a potential therapeutic target in cardiovascular diseases. Curr Drug Metab 14(6):706–719
El-Sherbeni AA, El-Kadi AO (2014) Alterations in cytochrome P450-derived arachidonic acid metabolism during pressure overload-induced cardiac hypertrophy. Biochem Pharmacol 87(3):456–466. doi:10.1016/j.bcp.2013.11.015
Eltom SE, Zhang L, Jefcoate CR (1999) Regulation of cytochrome P-450 (CYP) 1B1 in mouse Hepa-1 variant cell lines: a possible role for aryl hydrocarbon receptor nuclear translocator (ARNT) as a suppressor of CYP1B1 gene expression. Mol Pharmacol 55(3):594–604
Esposito G, Rapacciuolo A, Naga Prasad SV et al (2002) Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 105(1):85–92
Fard A, Wang CY, Takuma S et al (2000) Noninvasive assessment and necropsy validation of changes in left ventricular mass in ascending aortic banded mice. J Am Soc Echocardiogr 13(6):582–587
Fava C, Ricci M, Melander O, Minuz P (2012) Hypertension, cardiovascular risk and polymorphisms in genes controlling the cytochrome P450 pathway of arachidonic acid: a sex-specific relation? Prostaglandins Other Lipid Mediat 98(3–4):75–85. doi:10.1016/j.prostaglandins.2011.11.007
Ferguson AD, McKeever BM, Xu S et al (2007) Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science 317(5837):510–512. doi:10.1126/science.1144346
Freeman RH, Davis JO, Watkins BE, Lohmeier TE (1977) Mechanisms involved in two-kidney renal hypertension induced by constriction of one renal artery. Circ Res 40(5 Suppl 1):I29–I35
Frey N, Olson EN (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65:45–79
Garcia-Verdugo I, BenMohamed F, Tattermusch S et al (2012) A role for 12R-lipoxygenase in MUC5AC expression by respiratory epithelial cells. Eur Respir J 40(3):714–723. doi:10.1183/09031936.00023111
Gonzalez-Nunez D, Claria J, Rivera F, Poch E (2001) Increased levels of 12(S)-HETE in patients with essential hypertension. Hypertension 37(2):334–338
Grabellus F, Levkau B, Sokoll A et al (2002) Reversible activation of nuclear factor-κB in human end-stage heart failure after left ventricular mechanical support. Cardiovasc Res 53(1):124–130
Granberg AL, Brunstrom B, Brandt I (2000) Cytochrome P450-dependent binding of 7,12-dimethylbenz[a]anthracene (DMBA) and benzo[a]pyrene (B[a]P) in murine heart, lung, and liver endothelial cells. Arch Toxicol 74(10):593–601
Gross GJ, Falck JR, Gross ER, Isbell M, Moore J, Nithipatikom K (2005) Cytochrome P450 and arachidonic acid metabolites: role in myocardial ischemia/reperfusion injury revisited. Cardiovasc Res 68(1):18–25. doi:10.1016/j.cardiores.2005.06.007
Gu JL, Natarajan R, Ben-Ezra J et al (1994) Evidence that a leukocyte type of 12-lipoxygenase is expressed and regulated by angiotensin II in human adrenal glomerulosa cells. Endocrinology 134(1):70–77. doi:10.1210/endo.134.1.8275971
Guo L, Tang X, Chu X et al (2009) Role of protein kinase C in 15-HETE-induced hypoxic pulmonary vasoconstriction. Prostaglandins Leukot Essent Fatty Acids 80(2–3):115–123. doi:10.1016/j.plefa.2008.11.007
Guo AM, Liu X, Al-Wahab Z et al (2011a) Role of 12-lipoxygenase in regulation of ovarian cancer cell proliferation and survival. Cancer Chemother Pharmacol 68(5):1273–1283. doi:10.1007/s00280-011-1595-y
Guo Y, Zhang W, Giroux C et al (2011b) Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem 286(39):33832–33840. doi:10.1074/jbc.M110.216564
Guo W, Sun J, Jiang L et al (2012) Imperatorin attenuates LPS-induced inflammation by suppressing NF-κB and MAPKs activation in RAW 264.7 macrophages. Inflammation 35(6):1764–1772. doi:10.1007/s10753-012-9495-9
Hada T, Hagiya H, Suzuki H et al (1994) Arachidonate 12-lipoxygenase of rat pineal glands: catalytic properties and primary structure deduced from its cDNA. Biochim Biophys Acta 1211(2):221–228
Hahn HS, Marreez Y, Odley A et al (2003) Protein kinase Cα negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res 93(11):1111–1119. doi:10.1161/01.RES.0000105087.79373.17
Hammond VJ, Morgan AH, Lauder S et al (2012) Novel keto-phospholipids are generated by monocytes and macrophages, detected in cystic fibrosis, and activate peroxisome proliferator-activated receptor-gamma. J Biol Chem 287(50):41651–41666. doi:10.1074/jbc.M112.405407
Honda HM, Leitinger N, Frankel M et al (1999) Induction of monocyte binding to endothelial cells by MM-LDL: role of lipoxygenase metabolites. Arterioscler Thromb Vasc Biol 19(3):680–686
Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD (2002) Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39(2 Pt 2):690–694
Inoue M, Kishimoto A, Takai Y, Nishizuka Y (1977) Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. J Biol Chem 252(21):7610–7616
Jennings BL, Sahan-Firat S, Estes AM et al (2010) Cytochrome P450 1B1 contributes to angiotensin II-induced hypertension and associated pathophysiology. Hypertension 56(4):667–674. doi:10.1161/HYPERTENSIONAHA.110.154518
Jennings BL, Anderson LJ, Estes AM et al (2012) Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension 59(2):348–354. doi:10.1161/HYPERTENSIONAHA.111.183301
Jennings BL, George LW, Pingili AK et al (2014a) Estrogen metabolism by cytochrome P450 1B1 modulates the hypertensive effect of angiotensin II in female mice. Hypertension 64(1):134–140. doi:10.1161/HYPERTENSIONAHA.114.03275
Jennings BL, Montanez DE, May ME Jr et al (2014b) Cytochrome P450 1B1 contributes to increased blood pressure and cardiovascular and renal dysfunction in spontaneously hypertensive rats. Cardiovasc Drugs Ther 28(2):145–161. doi:10.1007/s10557-014-6510-4
Jin X, He Y, Zhu M et al (2010) The relationship between the polymorphism of SG13S114 A/T in ALOX5AP gene and the vulnerability of carotid atherosclerosis in Chinese Han population. Int J Clin Exp Med 3(1):28–32
Jisaka M, Kim RB, Boeglin WE, Brash AR (2000) Identification of amino acid determinants of the positional specificity of mouse 8S-lipoxygenase and human 15S-lipoxygenase-2. J Biol Chem 275(2):1287–1293
Kandouz M, Nie D, Pidgeon GP, Krishnamoorthy S, Maddipati KR, Honn KV (2003) Platelet-type 12-lipoxygenase activates NF-κB in prostate cancer cells. Prostaglandins Other Lipid Mediat 71(3–4):189–204
Kang KH, Ling TY, Liou HH et al (2013) Enhancement role of host 12/15-lipoxygenase in melanoma progression. Eur J Cancer 49(12):2747–2759. doi:10.1016/j.ejca.2013.03.030
Katoh T, Lakkis FG, Makita N, Badr KF (1994) Co-regulated expression of glomerular 12/15-lipoxygenase and interleukin-4 mRNAs in rat nephrotoxic nephritis. Kidney Int 46(2):341–349
Kaur-Knudsen D, Nordestgaard BG, Tybjaerg-Hansen A, Bojesen SE (2009) CYP1B1 genotype and risk of cardiovascular disease, pulmonary disease, and cancer in 50,000 individuals. Pharmacogenet Genomics 19(9):685–694. doi:10.1097/FPC.0b013e32833042cb
Kauser K, Clark JE, Masters BS et al (1991) Inhibitors of cytochrome P-450 attenuate the myogenic response of dog renal arcuate arteries. Circ Res 68(4):1154–1163
Kawano S, Kubota T, Monden Y et al (2005) Blockade of NF-κB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res 67(4):689–698. doi:10.1016/j.cardiores.2005.04.030
Kayama Y, Minamino T, Toko H et al (2009) Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J Exp Med 206(7):1565–1574. doi:10.1084/jem.20082596
Kikuta Y, Kusunose E, Sumimoto H et al (1998) Purification and characterization of recombinant human neutrophil leukotriene B4 omega-hydroxylase (cytochrome P450 4F3). Arch Biochem Biophys 355(2):201–205. doi:10.1006/abbi.1998.0724
Kikuta Y, Kusunose E, Kusunose M (2000) Characterization of human liver leukotriene B(4) omega-hydroxylase P450 (CYP4F2). J Biochem 127(6):1047–1052
Kim HJ, Lee SB, Park SK, Kim HM, Park YI, Dong MS (2005) Effects of hydroxyl group numbers on the B-ring of 5,7-dihydroxyflavones on the differential inhibition of human CYP 1A and CYP1B1 enzymes. Arch Pharmacal Res 28(10):1114–1121
Koeners MP, Wesseling S, Ulu A et al (2011) Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am J Physiol Endocrinol Metab 300(4):E691–E698. doi:10.1152/ajpendo.00710.2010
Kong EK, Huang Y, Sanderson JE, Chan KB, Yu S, Yu CM (2010) Baicalein and Wogonin inhibit collagen deposition in SHR and WKY cardiac fibroblast cultures. BMB Rep 43(4):297–303
Kong EK, Yu S, Sanderson JE, Chen KB, Huang Y, Yu CM (2011) A novel anti-fibrotic agent, baicalein, for the treatment of myocardial fibrosis in spontaneously hypertensive rats. Eur J Pharmacol 658(2–3):175–181. doi:10.1016/j.ejphar.2011.02.033
Korashy HM, El-Kadi AO (2006) The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab Rev 38(3):411–450. doi:10.1080/03602530600632063
Kriska T, Cepura C, Magier D, Siangjong L, Gauthier KM, Campbell WB (2012) Mice lacking macrophage 12/15-lipoxygenase are resistant to experimental hypertension. Am J Physiol Heart Circ Physiol 302(11):H2428–H2438. doi:10.1152/ajpheart.01120.2011
Kroll J, Epting D, Kern K et al (2009) Inhibition of Rho-dependent kinases ROCK I/II activates VEGF-driven retinal neovascularization and sprouting angiogenesis. Am J Physiol Heart Circ Physiol 296(3):H893–H899. doi:10.1152/ajpheart.01038.2008
Kumar S, Prasad S, Sitasawad SL (2013) Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocin-induced diabetic rats. PLoS One 8(7):e67009. doi:10.1371/journal.pone.0067009
Kwak HJ, Park KM, Choi HE, Lim HJ, Park JH, Park HY (2010) The cardioprotective effects of zileuton, a 5-lipoxygenase inhibitor, are mediated by COX-2 via activation of PKC delta. Cell Signal 22(1):80–87. doi:10.1016/j.cellsig.2009.09.014
Lacape G, Daret D, Crockett R, Rigaud M, Larrue J (1992) Dual metabolic pathways of 12-HETE in rat aortic smooth muscle cells. Prostaglandins 44(3):167–176
Lal H, Ahmad F, Zhou J et al (2014) Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 130(5):419–430. doi:10.1161/CIRCULATIONAHA.113.008364
Levick SP, Loch DC, Taylor SM, Janicki JS (2007) Arachidonic acid metabolism as a potential mediator of cardiac fibrosis associated with inflammation. J Immunol 178(2):641–646
Leychenko A, Konorev E, Jijiwa M, Matter ML (2011) Stretch-induced hypertrophy activates NFkB-mediated VEGF secretion in adult cardiomyocytes. PLoS One 6(12):e29055. doi:10.1371/journal.pone.0029055
Lindpaintner K, Kreutz R, Ganten D (1992) Genetic variation in hypertensive and ‘control’ strains. What are we controlling for anyway? Hypertension 19(5):428–430
Liu B, Khan WA, Hannun YA et al (1995) 12(S)-hydroxyeicosatetraenoic acid and 13(S)-hydroxyoctadecadienoic acid regulation of protein kinase C-alpha in melanoma cells: role of receptor-mediated hydrolysis of inositol phospholipids. Proc Natl Acad Sci USA 92(20):9323–9327
Liu Q, Chen X, Macdonnell SM et al (2009) Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res 105(2):194–200. doi:10.1161/CIRCRESAHA.109.195313
Li-Weber M (2009) New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat Rev 35(1):57–68. doi:10.1016/j.ctrv.2008.09.005
Lopez-Lazaro M (2009) Distribution and biological activities of the flavonoid luteolin. Mini Rev Med Chem 9(1):31–59
Lu C, Liu Y, Tang X, Ye H, Zhu D (2006) Role of 15-hydroxyeicosatetraenoic acid in phosphorylation of ERK1/2 and caldesmon in pulmonary arterial smooth muscle cells. Can J Physiol Pharmacol 84(10):1061–1069. doi:10.1139/y06-057
Ma YH, Harder DR, Clark JE, Roman RJ (1991) Effects of 12-HETE on isolated dog renal arcuate arteries. Am J Physiol 261(2 Pt 2):H451–H456
Ma J, Liang S, Wang Z et al (2010) ROCK pathway participates in the processes that 15-hydroxyeicosatetraenoic acid (15-HETE) mediated the pulmonary vascular remodeling induced by hypoxia in rat. J Cell Physiol 222(1):82–94. doi:10.1002/jcp.21923
Ma C, Li Y, Ma J et al (2011) Key role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension 58(4):679–688. doi:10.1161/HYPERTENSIONAHA.111.171561
Maayah ZH, El-Kadi AO (2015) 5-, 12- and 15-hydroxyeicosatetraenoic acids induce cellular hypertrophy in the human ventricular cardiomyocyte, RL-14 cell line, through MAPK- and NF-κB-dependent mechanism. Arch Toxicol. doi:10.1007/s00204-014-1419-z
Maayah ZH, Elshenawy OH, Althurwi HN, Abdelhamid G, El-Kadi AO (2015) Human fetal ventricular cardiomyocyte, RL-14 cell line, is a promising model to study drug metabolizing enzymes and their associated arachidonic acid metabolites. J Pharmacol Toxicol Methods 71:33–41. doi:10.1016/j.vascn.2014.11.005
Malik KU, Jennings BL, Yaghini FA et al (2012) Contribution of cytochrome P450 1B1 to hypertension and associated pathophysiology: a novel target for antihypertensive agents. Prostaglandins Other Lipid Mediat 98(3–4):69–74. doi:10.1016/j.prostaglandins.2011.12.003
Mammen JS, Kleiner HE, DiGiovanni J, Sutter TR, Strickland PT (2005) Coumarins are competitive inhibitors of cytochrome P450 1B1, with equal potency for allelic variants. Pharmacogenet Genomics 15(3):183–188
Marcus AJ, Broekman MJ, Safier LB et al (1982) Formation of leukotrienes and other hydroxy acids during platelet-neutrophil interactions in vitro. Biochem Biophys Res Commun 109(1):130–137
Marcus AJ, Safier LB, Ullman HL et al (1984) 12S,20-dihydroxyicosatetraenoic acid: a new icosanoid synthesized by neutrophils from 12S-hydroxyicosatetraenoic acid produced by thrombin- or collagen-stimulated platelets. Proc Natl Acad Sci USA 81(3):903–907
Masferrer JL, Rios AP, Schwartzman ML (1990) Inhibition of renal, cardiac and corneal (Na(+)–K+)ATPase by 12(R)-hydroxyeicosatetraenoic acid. Biochem Pharmacol 39(12):1971–1974
Maskrey BH, Bermudez-Fajardo A, Morgan AH et al (2007) Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem 282(28):20151–20163. doi:10.1074/jbc.M611776200
McFadyen MC, Cruickshank ME, Miller ID et al (2001a) Cytochrome P450 CYP1B1 over-expression in primary and metastatic ovarian cancer. Br J Cancer 85(2):242–246
McFadyen MC, McLeod HL, Jackson FC, Melvin WT, Doehmer J, Murray GI (2001b) Cytochrome P450 CYP1B1 protein expression: a novel mechanism of anticancer drug resistance. Biochem Pharmacol 62(2):207–212
Mitchell MD, Koenig JM (1991) Increased production of 15-hydroxyeicosatetraenoic acid by placentae from pregnancies complicated by pregnancy-induced hypertension. Prostaglandins Leukot Essent Fatty Acids 43(1):61–62
Modesti PA, Serneri GG, Gamberi T et al (2008) Impaired angiotensin II—extracellular signal-regulated kinase signaling in failing human ventricular myocytes. J Hypertens 26(10):2030–2039. doi:10.1097/HJH.0b013e328308de68
Mulugeta S, Suzuki T, Hernandez NT, Griesser M, Boeglin WE, Schneider C (2010) Identification and absolute configuration of dihydroxy-arachidonic acids formed by oxygenation of 5S-HETE by native and aspirin-acetylated COX-2. J Lipid Res 51(3):575–585. doi:10.1194/jlr.M001719
Murray GI, Melvin WT, Greenlee WF, Burke MD (2001) Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu Rev Pharmacol Toxicol 41:297–316
Nadler JL, Natarajan R, Stern N (1987) Specific action of the lipoxygenase pathway in mediating angiotensin II-induced aldosterone synthesis in isolated adrenal glomerulosa cells. J Clin Investig 80(6):1763–1769. doi:10.1172/JCI113269
Nakao J, Ooyama T, Ito H, Chang WC, Murota S (1982) Comparative effect of lipoxygenase products of arachidonic acid on rat aortic smooth muscle cell migration. Atherosclerosis 44(3):339–342
Natarajan R, Gu JL, Rossi J et al (1993) Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci USA 90(11):4947–4951
Natarajan R, Gonzales N, Lanting L, Nadler J (1994) Role of the lipoxygenase pathway in angiotensin II-induced vascular smooth muscle cell hypertrophy. Hypertension 23(1 Suppl):I142–I147
Natarajan R, Bai W, Rangarajan V et al (1996) Platelet-derived growth factor BB mediated regulation of 12-lipoxygenase in porcine aortic smooth muscle cells. J Cell Physiol 169(2):391–400. doi:10.1002/(SICI)1097-4652(199611)169:2<391:AID-JCP19>3.0.CO;2-C
Nazarewicz RR, Zenebe WJ, Parihar A et al (2007) 12(S)-hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch Biochem Biophys 468(1):114–120. doi:10.1016/j.abb.2007.09.018
Nebert DW, Dalton TP, Okey AB, Gonzalez FJ (2004) Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem 279(23):23847–23850
Newton AC (2001) Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101(8):2353–2364
Nieves D, Moreno JJ (2006) Hydroxyeicosatetraenoic acids released through the cytochrome P-450 pathway regulate 3T6 fibroblast growth. J Lipid Res 47(12):2681–2689. doi:10.1194/jlr.M600212-JLR200
Nozawa K, Tuck ML, Golub M, Eggena P, Nadler JL, Stern N (1990) Inhibition of lipoxygenase pathway reduces blood pressure in renovascular hypertensive rats. Am J Physiol 259(6 Pt 2):H1774–H1780
O’Flaherty JT, Wykle RL, Redman J, Samuel M, Thomas M (1986) Metabolism of 5-hydroxyicosatetraenoate by human neutrophils: production of a novel omega-oxidized derivative. J Immunol 137(10):3277–3283
O’Flaherty JT, Rogers LC, Chadwell BA et al (2002) 5(S)-hydroxy-6,8,11,14-E, Z, Z, Z-eicosatetraenoate stimulates PC3 cell signaling and growth by a receptor-dependent mechanism. Cancer Res 62(23):6817–6819
Parmentier JH, Muthalif MM, Saeed AE, Malik KU (2001) Phospholipase D activation by norepinephrine is mediated by 12(s)-, 15(s)-, and 20-hydroxyeicosatetraenoic acids generated by stimulation of cytosolic phospholipase a2. tyrosine phosphorylation of phospholipase d2 in response to norepinephrine. J Biol Chem 276(19):15704–15711. doi:10.1074/jbc.M011473200
Patricia MK, Kim JA, Harper CM et al (1999) Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler Thromb Vasc Biol 19(11):2615–2622
Patten RD, Hall-Porter MR (2009) Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail 2(2):138–144. doi:10.1161/CIRCHEARTFAILURE.108.839761
Pearson G, Robinson F, Beers Gibson T et al (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22(2):153–183. doi:10.1210/edrv.22.2.0428
Powell WS, Rokach J (2015) Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta 1851(4):340–355. doi:10.1016/j.bbalip.2014.10.008
Powell WS, Gravelle F, Gravel S (1992) Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. J Biol Chem 267(27):19233–19241
Prato M, Gallo V, Giribaldi G, Aldieri E, Arese P (2010) Role of the NF-κB transcription pathway in the haemozoin- and 15-HETE-mediated activation of matrix metalloproteinase-9 in human adherent monocytes. Cell Microbiol 12(12):1780–1791. doi:10.1111/j.1462-5822.2010.01508.x
Quintana LF, Guzman B, Collado S, Claria J, Poch E (2006) A coding polymorphism in the 12-lipoxygenase gene is associated to essential hypertension and urinary 12(S)-HETE. Kidney Int 69(3):526–530. doi:10.1038/sj.ki.5000147
Rao GN, Baas AS, Glasgow WC, Eling TE, Runge MS, Alexander RW (1994) Activation of mitogen-activated protein kinases by arachidonic acid and its metabolites in vascular smooth muscle cells. J Biol Chem 269(51):32586–32591
Reddy MA, Thimmalapura PR, Lanting L, Nadler JL, Fatima S, Natarajan R (2002) The oxidized lipid and lipoxygenase product 12(S)-hydroxyeicosatetraenoic acid induces hypertrophy and fibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMP response element-binding protein activation. Mediation of angiotensin II effects. J Biol Chem 277(12):9920–9928. doi:10.1074/jbc.M111305200
Revermann M, Mieth A, Popescu L et al (2011) A pirinixic acid derivative (LP105) inhibits murine 5-lipoxygenase activity and attenuates vascular remodelling in a murine model of aortic aneurysm. Br J Pharmacol 163(8):1721–1732. doi:10.1111/j.1476-5381.2011.01321.x
Roman RJ (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82(1):131–185. doi:10.1152/physrev.00021.2001
Sadik CD, Sies H, Schewe T (2003) Inhibition of 15-lipoxygenases by flavonoids: structure-activity relations and mode of action. Biochem Pharmacol 65(5):773–781
Sahan-Firat S, Jennings BL, Yaghini FA et al (2010) 2,3′,4,5′-Tetramethoxystilbene prevents deoxycorticosterone-salt-induced hypertension: contribution of cytochrome P-450 1B1. Am J Physiol Heart Circ Physiol 299(6):H1891–H1901. doi:10.1152/ajpheart.00655.2010
Saito F, Hori MT, Ideguchi Y et al (1992) 12-Lipoxygenase products modulate calcium signals in vascular smooth muscle cells. Hypertension 20(2):138–143
Sasaki M, Hori MT, Hino T, Golub MS, Tuck ML (1997) Elevated 12-lipoxygenase activity in the spontaneously hypertensive rat. Am J Hypertens 10(4 Pt 1):371–378
Schwartzman ML, Balazy M, Masferrer J, Abraham NG, McGiff JC, Murphy RC (1987) 12(R)-hydroxyicosatetraenoic acid: a cytochrome-P450-dependent arachidonate metabolite that inhibits Na+, K+-ATPase in the cornea. Proc Natl Acad Sci USA 84(22):8125–8129
Schwartzman ML, da Silva JL, Lin F, Nishimura M, Abraham NG (1996) Cytochrome P450 4A expression and arachidonic acid omega-hydroxylation in the kidney of the spontaneously hypertensive rat. Nephron 73(4):652–663
Serhan CN (2005) Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 73(3–4):141–162. doi:10.1016/j.plefa.2005.05.002
Shen T, Ma J, Zhang L et al (2013) Positive feedback-loop of telomerase reverse transcriptase and 15-lipoxygenase-2 promotes pulmonary hypertension. PLoS One 8(12):e83132. doi:10.1371/journal.pone.0083132
Shimada T, Fujii-Kuriyama Y (2004) Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci 95(1):1–6
Song YS, Kim MS, Lee DH, Oh DK, Yoon DY (2015) 15-Hydroxyeicosatetraenoic acid inhibits phorbol-12-myristate-13-Acetate-induced MUC5AC expression in NCI-H292 respiratory epithelial cells. J Microbiol Biotechnol 25(5):589–597
Sopontammarak S, Aliharoob A, Ocampo C, Arcilla RA, Gupta MP, Gupta M (2005) Mitogen-activated protein kinases (p38 and c-Jun NH2-terminal kinase) are differentially regulated during cardiac volume and pressure overload hypertrophy. Cell Biochem Biophys 43(1):61–76. doi:10.1385/CBB:43:1:061
Souders CA, Bowers SL, Baudino TA (2009) Cardiac fibroblast: the renaissance cell. Circ Res 105(12):1164–1176. doi:10.1161/CIRCRESAHA.109.209809
Spanbroek R, Grabner R, Lotzer K et al (2003) Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc Natl Acad Sci USA 100(3):1238–1243. doi:10.1073/pnas.242716099
Stenson WF, Parker CW (1979) Metabolism of arachidonic acid in ionophore-stimulated neutrophils. Esterification of a hydroxylated metabolite into phospholipids. J Clin Investig 64(5):1457–1465. doi:10.1172/JCI109604
Stern N, Golub M, Nozawa K et al (1989) Selective inhibition of angiotensin II-mediated vasoconstriction by lipoxygenase blockade. Am J Physiol 257(2 Pt 2):H434–H443
Stern N, Kisch ES, Knoll E (1996) Platelet lipoxygenase in spontaneously hypertensive rats. Hypertension 27(5):1149–1152
Stoltz RA, Abraham NG, Laniado-Schwartzman M (1996) The role of NF-κB in the angiogenic response of coronary microvessel endothelial cells. Proc Natl Acad Sci USA 93(7):2832–2837
Sutter TR, Tang YM, Hayes CL et al (1994) Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J Biol Chem 269(18):13092–13099
Suzuki H, Kayama Y, Sakamoto M et al (2015) Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes 64(2):618–630. doi:10.2337/db13-1896
Szekeres CK, Tang K, Trikha M, Honn KV (2000) Eicosanoid activation of extracellular signal-regulated kinase1/2 in human epidermoid carcinoma cells. J Biol Chem 275(49):38831–38841. doi:10.1074/jbc.M002673200
Takai S, Jin D, Kirimura K, Fujimoto Y, Miyazaki M (2001) 12-Hydroxyeicosatetraenoic acid potentiates angiotensin II-induced pressor response in rats. Eur J Pharmacol 418(1–2):R1–R2
Tang DG, Renaud C, Stojakovic S, Diglio CA, Porter A, Honn KV (1995) 12(S)-HETE is a mitogenic factor for microvascular endothelial cells: its potential role in angiogenesis. Biochem Biophys Res Commun 211(2):462–468. doi:10.1006/bbrc.1995.1836
Tang Y, Scheef EA, Wang S et al (2009) CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood 113(3):744–754. doi:10.1182/blood-2008-03-145219
Tejera N, Boeglin WE, Suzuki T, Schneider C (2012) COX-2-dependent and -independent biosynthesis of dihydroxy-arachidonic acids in activated human leukocytes. J Lipid Res 53(1):87–94. doi:10.1194/jlr.M017822
Thomas CP, Morgan LT, Maskrey BH et al (2010) Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J Biol Chem 285(10):6891–6903. doi:10.1074/jbc.M109.078428
Thorburn J, McMahon M, Thorburn A (1994) Raf-1 kinase activity is necessary and sufficient for gene expression changes but not sufficient for cellular morphology changes associated with cardiac myocyte hypertrophy. J Biol Chem 269(48):30580–30586
Tofovic SP, Jones TJ, Bilan VP, Jackson EK, Petrusevska G (2010) Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J Cardiovasc Pharmacol 56(5):475–483. doi:10.1097/FJC.0b013e3181f215e7
Vakili BA, Okin PM, Devereux RB (2001) Prognostic implications of left ventricular hypertrophy. Am Heart J 141(3):334–341. doi:10.1067/mhj.2001.113218
Vonach C, Viola K, Giessrigl B et al (2011) NF-κB mediates the 12(S)-HETE-induced endothelial to mesenchymal transition of lymphendothelial cells during the intravasation of breast carcinoma cells. Br J Cancer 105(2):263–271. doi:10.1038/bjc.2011.194
Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA (2005) Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci USA 102(49):17858–17863
Wallukat G, Morwinski R, Kuhn H (1994) Modulation of the beta-adrenergic response of cardiomyocytes by specific lipoxygenase products involves their incorporation into phosphatidylinositol and activation of protein kinase C. J Biol Chem 269(46):29055–29060
Wang B, Zhou SF (2009) Synthetic and natural compounds that interact with human cytochrome P450 1A2 and implications in drug development. Curr Med Chem 16(31):4066–4218
Wang G, Li W, Lu X, Bao P, Zhao X (2012) Luteolin ameliorates cardiac failure in type I diabetic cardiomyopathy. J Diabetes Complicat 26(4):259–265. doi:10.1016/j.jdiacomp.2012.04.007
Wang AW, Song L, Miao J et al (2015) Baicalein attenuates angiotensin II-induced cardiac remodeling via inhibition of AKT/mTOR, ERK1/2, NF-κB, and calcineurin signaling pathways in mice. Am J Hypertens 28(4):518–526. doi:10.1093/ajh/hpu194
Watanabe T, Medina JF, Haeggstrom JZ, Radmark O, Samuelsson B (1993) Molecular cloning of a 12-lipoxygenase cDNA from rat brain. Eur J Biochem FEBS 212(2):605–612
Wen Y, Gu J, Liu Y, Wang PH, Sun Y, Nadler JL (2001) Overexpression of 12-lipoxygenase causes cardiac fibroblast cell growth. Circ Res 88(1):70–76
Wen Y, Gu J, Peng X, Zhang G, Nadler J (2003) Overexpression of 12-lipoxygenase and cardiac fibroblast hypertrophy. Trends Cardiovasc Med 13(4):129–136
Wetsel WC, Khan WA, Merchenthaler I et al (1992) Tissue and cellular distribution of the extended family of protein kinase C isoenzymes. J Cell Biol 117(1):121–133
Whitelaw ML, Gustafsson JA, Poellinger L (1994) Identification of transactivation and repression functions of the dioxin receptor and its basic helix-loop-helix/PAS partner factor Arnt: inducible versus constitutive modes of regulation. Mol Cell Biol 14(12):8343–8355
Wu CC, Schwartzman ML (2011) The role of 20-HETE in androgen-mediated hypertension. Prostaglandins Other Lipid Mediat 96(1–4):45–53. doi:10.1016/j.prostaglandins.2011.06.006
Xu ZG, Yuan H, Lanting L et al (2008) Products of 12/15-lipoxygenase upregulate the angiotensin II receptor. J Am Soci Nephrol 19(3):559–569. doi:10.1681/ASN.2007080939
Yaghini FA, Li F, Malik KU (2007) Expression and mechanism of spleen tyrosine kinase activation by angiotensin II and its implication in protein synthesis in rat vascular smooth muscle cells. J Biol Chem 282(23):16878–16890. doi:10.1074/jbc.M610494200
Yamamoto S (1992) Mammalian lipoxygenases: molecular structures and functions. Biochim Biophys Acta 1128(2–3):117–131
Yan L, Huang H, Tang QZ et al (2010) Breviscapine protects against cardiac hypertrophy through blocking PKC-α-dependent signaling. J Cell Biochem 109(6):1158–1171. doi:10.1002/jcb.22495
Yiu SS, Zhao X, Inscho EW, Imig JD (2003) 12-Hydroxyeicosatetraenoic acid participates in angiotensin II afferent arteriolar vasoconstriction by activating L-type calcium channels. J Lipid Res 44(12):2391–2399. doi:10.1194/jlr.M300183-JLR200
Yokoyama C, Shinjo F, Yoshimoto T, Yamamoto S, Oates JA, Brash AR (1986) Arachidonate 12-lipoxygenase purified from porcine leukocytes by immunoaffinity chromatography and its reactivity with hydroperoxyeicosatetraenoic acids. J Biol Chem 261(35):16714–16721
Yousif MH, Benter IF, Roman RJ (2009) Cytochrome P450 metabolites of arachidonic acid play a role in the enhanced cardiac dysfunction in diabetic rats following ischaemic reperfusion injury. Auton Autacoid Pharmacol 29(1–2):33–41. doi:10.1111/j.1474-8673.2009.00429.x
Yuan W, Yu Y, Li J et al (2013) Estrogen metabolite 2-methoxyestradiol prevents hypertension in deoxycorticosterone acetate-salt rats. Cardiovasc Drugs Ther 27(1):17–22. doi:10.1007/s10557-012-6428-7
Zhang Y, Cao Y, Duan H, Wang H, He L (2012) Imperatorin prevents cardiac hypertrophy and the transition to heart failure via NO-dependent mechanisms in mice. Fitoterapia 83(1):60–66. doi:10.1016/j.fitote.2011.09.011
Zhang L, Li Y, Chen M et al (2014) 15-LO/15-HETE mediated vascular adventitia fibrosis via p38 MAPK-dependent TGF-beta. J Cell Physiol 229(2):245–257. doi:10.1002/jcp.24443
Zong J, Zhang DP, Zhou H et al (2013) Baicalein protects against cardiac hypertrophy through blocking MEK-ERK1/2 signaling. J Cell Biochem 114(5):1058–1065. doi:10.1002/jcb.24445
Zordoky BN, El-Kadi AO (2010) Effect of cytochrome P450 polymorphism on arachidonic acid metabolism and their impact on cardiovascular diseases. Pharmacol Ther 125(3):446–463. doi:10.1016/j.pharmthera.2009.12.002
Zordoky BN, Aboutabl ME, El-Kadi AO (2008) Modulation of cytochrome P450 gene expression and arachidonic acid metabolism during isoproterenol-induced cardiac hypertrophy in rats. Drug Metab Dispos 36(11):2277–2286. doi:10.1124/dmd.108.023077
Zordoky BN, Anwar-Mohamed A, Aboutabl ME, El-Kadi AO (2010) Acute doxorubicin cardiotoxicity alters cardiac cytochrome P450 expression and arachidonic acid metabolism in rats. Toxicol Appl Pharmacol 242(1):38–46. doi:10.1016/j.taap.2009.09.012
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research [Grant 106665] to A.O.S.E. Z.H.M. is the recipient Izaak Walton Killam Memorial Scholarship and Alberta Innovates-Health solution Graduate Student Scholarship. The authors are grateful to Samya Elkhatali for her helpful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest.
Rights and permissions
About this article

Cite this article
Maayah, Z.H., El-Kadi, A.O.S. The role of mid-chain hydroxyeicosatetraenoic acids in the pathogenesis of hypertension and cardiac hypertrophy. Arch Toxicol 90, 119–136 (2016). https://doi.org/10.1007/s00204-015-1620-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-015-1620-8