
Abstract
Purpose
We investigated the contribution of cytochrome P450 (CYP) 1B1 to hypertension and its pathogenesis by examining the effect of its selective inhibitor, 2,4,3′,5′-tetramethoxystilbene (TMS), in spontaneously hypertensive rats (SHR).
Methods
Blood pressure (BP) was measured bi-weekly. Starting at 8 weeks, TMS (600 μg/kg, i.p.) or its vehicle was injected daily. At 14 weeks, samples were collected for measurement.
Results
TMS reversed increased BP in SHR (207 ± 7 vs. 129 ± 2 mmHg) without altering BP in Wistar-Kyoto rats. Increased CYP1B1 activity in SHR was inhibited by TMS (RLU: aorta, 5.4 ± 0.7 vs. 3.7 ± 0.7; heart, 6.0 ± 0.8 vs. 3.4 ± 0.4; kidney, 411 ± 45 vs. 246 ± 10). Increased vascular reactivity, cardiovascular hypertrophy, endothelial and renal dysfunction, cardiac and renal fibrosis in SHR were minimized by TMS. Increased production of reactive oxygen species and NADPH oxidase activity in SHR, were diminished by TMS. In SHR, TMS reduced increased plasma levels of nitrite/nitrate (46.4 ± 5.0 vs. 28.1 ± 4.1 μM), hydrogen-peroxide (36.0 ± 3.7 vs. 14.1 ± 3.8 μM), and thiobarbituric acid reactive substances (6.9 ± 1.0 vs. 3.4 ± 1.5 μM). Increased plasma levels of pro-inflammatory cytokines and catecholamines, and cardiac activity of extracellular signal-regulated kinase, p38 mitogen-activated protein kinase, c-Src tyrosine kinase, and protein kinase B in SHR were also inhibited by TMS.
Conclusions
These data suggests that increased oxidative stress generated by CYP1B1 contributes to hypertension, increased cytokine production and sympathetic activity, and associated pathophysiological changes in SHR. CYP1B1 could be a novel target for developing drugs to treat hypertension and its pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytochrome P450 (CYP) 1B1 is a heme-thiolate monooxygenase expressed in non-hepatic tissues including the cardiovascular system, and is involved in NADPH-dependent metabolism of various substrates including fatty acids, steroids, and xenobiotics [1]. CYP1B1 has been implicated in some experimental models of systemic [2–4] and pulmonary [5] arterial hypertension. CYP1B1 also mediates angiotensin (Ang) II-induced vascular smooth muscle cell (VSMC) migration, proliferation and hypertrophy via generation of reactive oxygen species (ROS) through metabolism of arachidonic acid (AA), released by activation of cytosolic phospholipase A2 (cPLA2) [6]. ROS, activation of immune cells, and production of proinflammatory cytokines promote the development of hypertension and associated pathophysiology in various experimental models of hypertension, including SHR [7, 8]. ROS also stimulate sympathetic activity [9], which leads to activation of the immune system [8]. We have shown that ROS generated by NADPH oxidase in a CYP1B1-dependent manner contribute to the development of Ang II- and DOCA-salt-induced hypertension and associated pathophysiological changes in cardiovascular and renal systems [2–4, 10, 11]. These observations raise the possibility that ROS generated via CYP1B1 might contribute to hypertension, increased sympathetic activity and cytokine production, and associated cardiovascular and renal pathophysiology consequent to activation of one or more signaling molecules in SHR, an experimental model of human essential hypertension. To test this hypothesis, we have investigated the effect of 2,4,3′,5′-tetramethoxystilbene (TMS), a selective inhibitor of CYP1B1 activity [12], on hypertension and its pathogenesis in SHR. Our results show that TMS lowers BP and prevents associated cardiovascular and renal changes in SHR, primarily by inhibiting ROS generated via CYP1B1; decreasing circulating levels of proinflammatory cytokines and catecholamines; and decreasing activation of ERK1/2, p38 MAPK, c-Src and Akt.
Materials and Methods
Materials
TMS and trans-resveratrol were purchased from Cayman Chemical (Ann Arbor, MI). DHE was from Invitrogen™ (Carlsbad, CA), the CYP1B1 antibody was from BD Biosciences (Franklin Lakes, NJ), and antibodies against α-smooth muscle-specific actin, ERK1/2, p38 MAPK, c-Src, and Akt were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The primary phospho ERK1/2, phospho p38 MAPK, phospho c-Src, and phospho Akt antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). All other chemicals were purchased from Sigma (St. Louis, MO).
Animals and blood pressure measurements
All experiments were performed according to the protocols approved by the University of Tennessee Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. 36 three-weeks-old male SHR and 36 age-matched WKY (Harlan Laboratories, Indianapolis, IN) were used throughout this research. Each strain of rats was split into two groups; one group was injected daily with TMS (600 μg/kg) and the other with vehicle (DMSO; 100 μl) beginning at 8 weeks of age. Systolic BP and mean arterial pressure (MAP) were measured twice a week from 4 weeks of age; we used a noninvasive tail cuff method (Kent Scientific, Torrington, CT; model XBP 1000). Prior to the beginning of each experiment, rats were acclimated to the BP measuring device for 1 week.
CYP1B1 activity assay
CYP1B1 activity was determined using the P450-Glo™ Assay Kit (Promega, Madison, WI) as previously described [2, 3]. This assay has been shown to be specific for CYP1B1, as in our previous studies in mice lacking CYP1B1 (Cyp1b1 −/−), limited CYP1B1 activity was found in tissues from Cyp1b1 −/− mice, compared to their wild type counterparts [4, 11]. At the completion of the experiments, animals were anesthetized with ketamine (60 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.), the left ventricle was punctured, and blood was flushed out by perfusion with cold saline (3 min). The aorta, heart, and kidney were dissected free, cleaned of surrounding tissue, snap-frozen in liquid N2, and stored at −80 °C until use. Tissue samples were homogenized in ice-cold 100 mM potassium phosphate buffer (pH 7.4) with a TissueLyser II (QIAGEN, Valencia, CA) (2 × 3 min). Following homogenization, samples were centrifuged at 10,000 g for 20 min at 4 °C; the supernatant was collected and stored at −80 °C until further use. Protein content in the samples was determined by the Bradford method, and 500 μg of protein was added to a reaction mixture containing 20 μM luciferin-CEE substrate and 100 mM potassium phosphate buffer (pH 7.4) and incubated at 37 °C for 10 min. 100 μM of NADPH (final concentration) was added, and the solution was further incubated at 37 °C for 45 min. Finally, a 1:1 volume of luciferin detection reagent was added to the samples, and they were mixed for 10 s after which they were incubated at room temperature for 20 min. Luminescence was measured with a luminometer (Turner Designs, Sunny Vale, CA; model TD-20/20). Potassium phosphate buffer was used as a blank and subtracted from each reading; activity was expressed as relative luminescence units (RLU).
To determine the IC50 of TMS and resveratrol on CYP1B1 activity, cardiac lysates from vehicle-treated WKY and SHR were prepared as described above, but, prior to adding luciferin-CEE, lysates were incubated with various concentrations of TMS (1 pM - 100 nM) and resveratrol (1 nM - 1 mM) for 1 h at 37 °C. After this incubation period, samples were processed as described above and the IC50 calculated.
Western blot analysis
Animals were anesthetized and the aorta, heart, and kidney removed as described above. Tissue samples were homogenized in lysis buffer, and protein content was determined by the Bradford method. Approximately 10 μg of protein was loaded and resolved on 8 % SDS-polyacrylamide gels and processed for Western blot analysis as previously described [2, 13]. Blots were probed with different primary and corresponding secondary antibodies, and intensity of the bands was measured with ImageJ 1.42 software (http://rsb.info.nih.gov/nih-image; National Institutes of Health). Protein expression of CYP1B1 was calculated as a ratio of its expression to that of α-actin, and activities of signaling molecules (ERK1/2, p38 MAPK, c-Src, and Akt) were calculated as a ratio of phosphorylated protein to their corresponding non-phosphorylated levels.
Measurement of vascular function and structure
Vascular reactivity
Following anesthesia (described above), the aorta, and mesenteric and renal arteries were quickly dissected free, cleaned of surrounding tissue, and approximately 2-mm rings were mounted in a wire myograph system (Danish Myo Technology, Aarhus, Denmark; model 610 M). Vessels were continuously bathed in Krebs buffer (composition in mM: 118 NaCl, 4.7 KCl, 25 NaHCO3, 1.2 MgSO4, 1.2 KH2PO4, 11.1 glucose, 2.5 CaCl2.2H2O) at 37 °C, which was gassed with 95 % O2 and 5 % CO2 to maintain the pH at 7.4. An initial tension of 9 mN (aorta) and 5 mN (mesenteric and renal arteries) was placed on the vessels and they were allowed to equilibrate for approximately 30 min. To confirm the viability of the vessels, they were initially tested for constriction to 60 mM KCl and then washed three times with fresh Krebs buffer. Cumulative concentration response curves to phenylephrine (PE) and endothelin-1 (ET-1) were obtained and responses measured as force of contraction (mN).
Endothelium-dependent and -independent vasodilation
Endothelial function was examined by constricting the vessels with the concentration of PE that evoked a maximal response followed by adding increasing concentrations of acetylcholine (ACh). Changes in the response of vessels to ACh were measured and presented as a percentage of the PE-induced constriction. Endothelium-independent vasodilation was studied by constricting the vessels with the concentration of PE that evoked a maximal response followed by adding increasing concentrations of sodium nitroprusside (SNP). Changes in the response of vessels to SNP were measured and presented as a percentage of the PE-induced constriction.
Media: lumen ratio
Following anesthesia (described above), the vessels were dissected free, cleaned of surrounding tissue, placed in O.C.T. compound, and frozen at −80 °C. 10 μm sections were cut using a cryostat and stained with standard hematoxylin and eosin. Sections were viewed with an Olympus® inverted system microscope (Olympus America, Inc., Melville, NY; model BX41), photographed using a SPOT™ Insight™ digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI; model Insight 2MP Firewire), and images were analyzed using ImageJ 1.42.
Measurement of renal hemodynamics
Renal blood flow
Renal blood flow (RBF) was measured in rats using a 1.0 V Transonic renal flow probe which was attached to a TS420 perivascular flow meter (Transonic Systems, Ithaca, NY) as described previously [14]. Briefly, rats were anesthetized as described above and placed on a 37 °C heated surgery table. The left kidney was exposed from a subcostal flank incision and the renal artery was isolated from the renal vein. After equilibrating for approximately 15 min, RBF was recorded for 10 min and the average RBF over this time period was calculated and expressed in ml/min.
Renal vascular resistance
Renal vascular resistance was calculated as MAP/RBF.
Metabolic study for analysis of renal function
To assess renal function, individual rats were housed in metabolic cages for a period of 24 h at the completion of the experiment, allowing for the measurement of food and water consumption and the separation of urine from fecal material and food waste. Urine was collected in tubes that contained a small volume of mineral oil to prevent evaporation. Following calculation of volume, urine was aliquoted and stored at −80 °C until further analysis. Urine was analyzed for osmolality using a Vapro® vapor pressure osmometer (Wescor, South Logan, UT; model 5520), protein content by the standard Bradford method, and Na+ concentration, using a flame photometer (Instrumentation Laboratory, Inc., Lexington, MA; model 443). Creatinine clearance was calculated and used as an estimate of glomerular filtration rate (GFR) by measuring creatinine concentration in serum and urine samples (30 μl) with a QuantiChrom™ creatinine assay kit (BioAssay Systems, Hayward, CA). For serum collection, approximately 1.5 ml blood was withdrawn directly from the abdominal aorta following anesthesia of animals (described above). The blood was transferred to an Eppendorf tube and allowed to clot for approximately 30 min at room temperature, after which it was centrifuged 1,500 g for 15 min at 4 °C. The serum (approximately 300–400 μl) was collected and stored at −80 °C until further analysis. Urine was also analyzed for levels of hydrogen peroxide, thiobarbituric acid reactive substances (TBARS), and nitrate/nitrite, described below.
Measurement of heart weight: body weight ratio
At the completion of the experiments and prior to sacrifice, animals were weighed. Immediately following sacrifice, hearts were removed and their wet weight determined. The ratio of heart weight (mg) to body weight (g) was calculated and used as a measure of cardiac hypertrophy.
Immunohistochemical analysis
At the completion of the experiments, animals were anesthetized as described above, the carotid artery was cannulated, and the animals were perfused with saline (3 min). The heart and kidneys were dissected free and placed in Optimal Cutting Temperature (O.C.T.) compound (Sakura Finetek USA, Inc., Torrance, CA) and frozen at −80 °C. Cardiac sections (5 μm) were cut using a cryostat (Leica Microsystems, Bannockburn, IL; model CM1850) and processed for α-smooth muscle actin (myofibroblasts) as described previously [15] or stained with Masson’s trichrome for collagen, according to the manufacturer’s instructions (Sigma). Renal sections (10 μm) were cut and processed for α-smooth muscle actin or stained with Masson’s trichrome (collagen deposition) as described previously [16]. Tissue sections were viewed as described above.
Measurement of ROS production
To measure ROS production, aortic, cardiac, and renal sections were exposed to DHE, following the previously described and validated method [17]. Fresh, unfixed tissue samples were placed in O.C.T. compound and frozen at −80 °C. 30 μm sections were cut using a cryostat and placed on a glass slide. Sections were incubated in PBS for 30 min at 37 °C, and then DHE, 2 μM for vascular [17] and cardiac [18] sections, or 5 μM for renal sections [19], was topically applied. Cover slips were applied, and sections were further incubated at 37 °C in a light-protected, humidified chamber for 30 min. Sections were then rinsed in PBS, and fluorescence was detected using a 585-nm filter on an Olympus® inverted system microscope (Olympus America, Inc.; model IX50). Images were photographed using an Olympus® digital camera (Olympus America, Inc., model DP71) and analyzed using ImageJ 1.42. For analysis, fluorescence intensity of each group of animals was calculated by measuring 4 separate fields of view across 2 independent sections from each animal (n = 5 in each group). The average intensity of the 2 sections was used as the total fluorescence intensity for each animal.
Measurement of NADPH oxidase activity
NADPH oxidase activity was determined in tissue homogenates by measuring lucigenin (N,N′-dimethyl-9,9′-biacridinium dinitrate)-enhanced chemiluminescence, as described previously, with some modifications [2, 3]. Following anesthesia (described above), the aorta, heart, and kidneys were isolated, cleaned of surrounding tissue, snap-frozen in liquid N2, and stored at −80 °C until use. Tissue samples were homogenized and sonicated in lysis buffer containing protease inhibitors (20 mM phosphate buffer, 1 mM EGTA, 10 μg/ml aprotinin, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 0.5 mM phenylmethylsulphonylfluoride, and 150 mM sucrose). Samples were then centrifuged at 3,000 g for 10 min at 4 °C, and supernatants were kept on ice until use. Protein content in the samples was determined by the Bradford method, and equal amounts of protein were combined 1:1 with a reaction mixture containing 5 μM lucigenin (final concentration) and 100 μM NADPH (final concentration). Luminescence was measured every minute for 10 min with a luminometer. Lysis buffer was used as a blank and subtracted from each reading and activity expressed as arbitrary units.
Measurement of plasma and urinary levels of oxidative stress markers
Plasma
At the completion of the experiments, animals were anesthetized as described above, and approximately 4–5 ml of blood was withdrawn directly from the abdominal aorta and transferred to K+-EDTA tubes (BD Vacutainer®; BD Biosciences). Blood was centrifuged at 1,500 g for 15 min at 4 °C, and the plasma (approximately 1.5-2.0 ml) was collected and stored in 200 μl aliquots at −80 °C until further analysis.
Hydrogen peroxide
Plasma and urine levels of H2O2 were measured using an Amplex® red hydrogen peroxide/peroxidase assay kit (Invitrogen™) according to the manufacturer’s instructions, using 50 μl of sample.
Thiobarbituric acid reactive substances
Plasma and urine levels of TBARS, an indicator of lipid peroxidation, were measured using a TBARS assay kit (Cayman Chemical) according to the manufacturer’s instructions, using 100 μl of sample.
Nitrate/nitrite
Plasma and urine levels of nitrate/nitrite, an indicator of total nitric oxide bioavailability, were measured using a nitrate/nitrite colorimetric assay kit (Cayman Chemical) according to the manufacturer’s instructions, using 40 μl of plasma sample and 80 μl of urine sample.
Measurement of plasma levels of cytokines
Levels of various cytokines were measured in 40 μl of plasma using a rat cytokine 10-plex panel (Invitrogen™) according to the manufacturer’s instructions. Samples were analyzed using a Bio-Plex® 200 system (Bio-Rad, Hercules, CA).
Determination of plasma levels of catecholamines
Plasma levels of epinephrine and norepinephrine were kindly measured using HPLC by Dr. Raymond Johnson of the CMN/KC neurochemistry core laboratory at Vanderbilt University, using 50 μl of sample. Catecholamines were first extracted from plasma by mixing with dihydroxybenzylamine (internal standard), Tris buffer (pH 8.5), and 100 mg Al2O3. This solution was allowed to mix for 30 min, after which, the buffer was aspirated off, and the Al2O3 was washed with distilled water. The wash was again thoroughly aspirated off, and the catecholamines were desorbed from the Al2O3 with 200 μl of 0.1 N acetic acid. 75 μl of sample was injected into a biogenic amine HPLC chromatograph (Waters Corporation, Milford, MA) [20].
Statistical analysis
Data were analyzed by one-way analysis of variance followed by Neuman-Keuls post-hoc test or Student’s t-test. The values of a minimum of three different experiments are expressed as the mean ± SEM. P values < 0.05 were considered statistically significant.
Results
TMS reduced BP in SHR
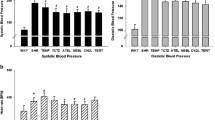
To determine the contribution of CYP1B1 in development of hypertension in SHR, we examined the effect of the selective CYP1B1 inhibitor, TMS [12], on BP in SHR and WKY rats. Systolic BP steadily increased in SHR from 4 weeks of age (Fig. 1). Starting from 8 weeks of age, daily injections of TMS reduced systolic BP in SHR to levels observed at the beginning of the experiment (207 ± 7 vs. 129 ± 2 mmHg; Fig. 1). Systolic BP was not altered in WKY injected with TMS or its vehicle (129 ± 7 vs. 127 ± 4 mmHg; Fig. 1).

TMS reversed increased BP in SHR. WKY and SHR were given daily i.p. injections of vehicle (DMSO, 100 μl) or the CYP1B1 inhibitor, TMS (600 μg/kg), starting from 8 weeks of age. Systolic blood pressure (SBP) was measured by tail cuff twice a week beginning from 4 weeks of age. * P < 0.05 vs. corresponding WKY; † P < 0.05 vs. corresponding vehicle (n = 6 for all experiments; data are expressed as mean ± SEM)
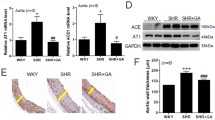
TMS reduced CYP1B1 activity but not protein expression in WKY and SHR
SHR had increased aortic, cardiac, and renal CYP1B1 activity compared to WKY (Fig. 2a, c, e, respectively). TMS treatment decreased CYP1B1 activity in WKY and SHR (Fig. 2a, c, e, respectively). TMS treatment did not alter protein expression of CYP1B1 in aorta, heart, or kidney from WKY and SHR (Fig. 2b, d, f, respectively). These observations suggest that the increase in BP in SHR is dependent on CYP1B1 activity.

TMS reduced increased CYP1B1 activity in SHR, which is not associated with changes in protein expression. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. At the completion of the experiment, aorta a, heart c, and kidney e were collected for measurement of CYP1B1 activity with the P450-Glo™ assay as described in Methods. CYP1B1 activity is expressed as relative luminescence units (RLU). CYP1B1 protein expression was measured by western blot in aorta b, heart d, and kidney f from WKY and SHR given daily i.p. injections of vehicle (DMSO) or TMS, using approximately 10 μg of protein for loading as described in Methods. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 3–6 for all experiments; data are expressed as mean ± SEM)
TMS reduced increased vascular reactivity, vascular smooth muscle hypertrophy, and renal vascular resistance in SHR
The increase in vascular reactivity to vasoconstrictor agents, and vascular hypertrophy is associated with hypertension in several experimental models including SHR [2, 10, 21–23]. Therefore, we investigated the contribution of CYP1B1 to increased vascular reactivity associated with hypertension in SHR. The responses to PE and ET-1 of the aorta (Fig. 3a, b, respectively), mesenteric artery (Fig. 3c, d, respectively), and renal artery (Fig. 3e, f, respectively) from SHR were increased; these increases were reduced in TMS-treated aorta, mesenteric and renal artery from SHR (Fig. 3a–f). The increase in vascular reactivity, which correlated with an increase in media: lumen ratio, an indicator of vascular smooth muscle hypertrophy, was also reduced by TMS treatment (Table 1).

TMS reduced increased vascular reactivity in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. Vascular reactivity was measured as described in Methods. Responses of the aorta (a, b), mesenteric artery (c, d), and renal artery (e, f) from WKY and SHR treated with vehicle or TMS to increasing concentrations of phenylephrine (PE) and endothelin-1 (ET-1), respectively are shown. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 5–6 for all experiments; data are expressed as mean ± SEM)
Renal blood flow (RBF) was decreased in vehicle-treated SHR, and this decrease was not altered by TMS treatment (Table 2). In contrast, renal vascular resistance (RVR) was increased in vehicle-treated SHR; this increase was reduced by TMS treatment (Table 2). These data indicate that increased vascular reactivity to PE and ET-1, vascular hypertrophy, and increased RVR associated with hypertension in SHR are also dependent on CYP1B1 activity.
TMS improved endothelial function in SHR
Endothelial function, as indicated by relaxation to vasodilator agents, is impaired in human hypertension [24], and is either impaired or unaltered in some experimental models of hypertension [25]. To determine if endothelial function in SHR is dependent on CYP1B1 activity, we examined the effect of TMS on ACh-induced relaxation of blood vessels from SHR preconstricted with PE. Vehicle-treated SHR displayed endothelial dysfunction in the aorta (Fig. 4a), mesenteric artery (Fig. 4c), and renal artery (Fig. 4e), as demonstrated by a decreased relaxation response to increasing concentrations of ACh; these responses were improved by TMS treatment (52.7 ± 7.9, 49.1 ± 7.8, and 53.2 ± 5.2 vs. 80.4 ± 8.5, 85.2 ± 7.2, and 79.5 ± 9.5 % with TMS, in aorta, mesenteric and renal artery, respectively; Fig. 4a, c, e, respectively). Endothelium-independent relaxations to SNP were not different in the aorta (Fig. 4b), mesenteric artery (Fig. 4d), or renal artery (Fig. 4f) from any of the treatment groups. These results indicate that CYP1B1 contributes to impaired endothelial function in SHR.

TMS improved endothelial function in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. Endothelial function was measured as described in Methods. Responses of the aorta (a, b), mesenteric artery (c, d), and renal artery (e, f) from WKY and SHR treated with vehicle or TMS to increasing concentrations of acetylcholine (ACh) and sodium nitroprusside (SNP) are shown. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 5–6 for all experiments; data are expressed as mean ± SEM)
TMS improves renal dysfunction in SHR
The kidney is known to play an important role in the development of hypertension and increased BP promotes renal dysfunction and end organ damage [26–28]. Therefore, to determine the contribution of CYP1B1 to renal function and cardiac and renal damage we examined the effect of TMS in SHR and WKY. Vehicle- and TMS-treated SHR had a lower food intake than their corresponding WKY counterparts, but there was no difference in either water intake or urine output between groups (Table 3). Vehicle-treated SHR displayed a decrease in GFR, as indicated by increased serum creatinine (0.48 ± 0.03 vs. 0.84 ± 0.11 mg/dl, Table 3), and an associated decrease in creatinine clearance (0.83 ± 0.15 vs. 0.55 ± 0.12 ml/min, Table 3). In addition, vehicle-treated SHR had decreased urinary excretion of Na+, decreased urine osmolality, and increased proteinuria (Table 3). Treatment with TMS improved these changes in SHR and was without effect in WKY (Table 3), suggesting that CYP1B1 is involved in renal dysfunction associated with hypertension in SHR.
TMS reduced cardiac hypertrophy, and cardiac and renal fibrosis in SHR
SHR had increased heart: body weight ratio (HW: BW), an indicator of cardiac hypertrophy, compared to WKY (Table 4). In WKY, TMS alone had no effect on HW: BW, but minimized the increase in SHR (Table 4). Hearts from vehicle-treated SHR, but not WKY, also displayed fibrosis as demonstrated by α-smooth muscle actin-positive myofibroblasts and collagen deposition in the myocardium, as demonstrated by Masson’s trichrome staining; these were reduced by TMS (Fig. 5a, b, respectively). In addition to the heart, kidneys from vehicle-treated SHR also displayed interstitial fibrosis, as demonstrated by increased α-smooth muscle actin staining (Fig. 5c). Masson’s trichrome staining revealed substantial collagen deposition in vehicle-treated SHR (Fig. 5d); these changes were reduced by TMS treatment (Fig. 5c, d, respectively). These observations support our hypothesis that CYP1B1 contributes to the cardiac and renal damage associated with hypertension in SHR.

TMS decreased cardiac and renal fibrosis in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. At the completion of the experiments, cardiac and renal tissue was removed and processed for immunohistochemistry as described in Methods. α-Smooth muscle actin (α-SMA)-positive myofibroblasts are present in the myocardium of vehicle-treated SHR (a arrows); these myofibroblasts are absent in vehicle-treated WKY and TMS-treated rats. b Masson’s trichrome staining revealed increased collagen deposition (intense blue staining) in the perivascular space of vehicle-treated SHR; however, this staining was reduced in TMS-treated SHR and absent from vehicle- and TMS-treated WKY. Renal fibrosis was observed in vehicle-treated SHR, as demonstrated by increased α-SMA-positive myofibroblasts in the interstitial space and around the glomeruli (c arrows); these myofibroblasts are absent in vehicle-treated WKY and TMS-treated rats. Masson’s trichrome staining revealed increased collagen deposition (intense blue staining) in vehicle-treated SHR (d), which was reduced in vehicle-treated WKY and TMS treated rats. Photomicrographs are representative images of at least three animals from each experimental group. Magnification for all images is × 100, and scale bar represents 200 μm
TMS reduced increased vascular, cardiac, and renal superoxide production and NADPH oxidase activity in SHR
ROS generated via activation of NADPH oxidase has been implicated in the endothelial dysfunction and the cardiac and renal pathophysiological changes and has been implicated in the development of hypertension in various models including SHR [29–31]. These observations and our previous demonstration that ROS generated via activation of CYP1B contributes to Ang II- and DOCA-salt induced hypertension [2–4] led us to determine if ROS production and NADPH oxidase activity associated with hypertension in SHR is dependent on CYP1B1 activity. Vehicle-treated SHR displayed increased superoxide production in the aorta (Fig. 6a), mesenteric (Fig. 6c), and renal (Fig. 6d) arteries, as indicated by increased 2-hydroxyethidium (2-OHE) fluorescence; TMS treatment reduced this increase (Fig. 6a, c, d, respectively). The increase in superoxide production observed in the aorta of vehicle-treated SHR was associated with a 1.8 fold increase in NADPH oxidase activity; TMS treatment reduced this increase (Fig. 6b). It should be noted that due to insufficient tissue quantity, we could not measure NADPH oxidase activity in the mesenteric or renal arteries.

TMS reduced vascular oxidative stress and NADPH oxidase activity in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. Superoxide production, as determined by 2-hydroxyethidium (2-OHE) fluorescence intensity, was measured in the aorta (a), and mesenteric (c) and renal (d) arteries as described in Methods. b NADPH oxidase activity was measured in aortic homogenates with a lucigenin-based luminescence assay as described in Methods. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 5–6 for all experiments; data are expressed as mean ± SEM). Magnification: A = x20; C, D = x100
Vehicle-treated SHR also displayed increased superoxide production in the heart and kidney (Fig. 7a, c, respectively), which was reduced by TMS treatment. Correlating with the increased superoxide production, vehicle-treated SHR were also found to have a 2.7 fold and 6.2 fold increase in cardiac and renal NADPH oxidase activity, respectively; TMS treatment reduced this increase (Fig. 7b, d). These data suggest that increased NADPH oxidase activity and production of ROS via CYP1B1 in the cardiovascular and renal systems could contribute to the endothelial and renal dysfunction, cardiovascular and renal damage associated with hypertension in SHR. Since increased oxidative stress also results in an increase in generation of H2O2, TBARS and alterations in NO production, we determined the effect of TMS on these parameters in SHR and WKY.

TMS reduced cardiac and renal oxidative stress and NADPH oxidase activity in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. Superoxide production, as determined by 2-OHE fluorescence intensity, was measured in the heart (a) and kidney (c) as described in Methods. In the kidney of vehicle-treated SHR, superoxide production was identified predominately in the glomerulus (c arrows), which was reduced by TMS. NADPH oxidase activity was measured in cardiac (b) and renal (d) homogenates with a lucigenin-based luminescence assay as described in Methods. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 5–6 for all experiments; data are expressed as mean ± SEM). Magnification for all images is × 100
TMS reduced increased plasma and urine levels of H2O2, thiobarbituric acid reactive substances, and NOx in SHR
Plasma and urine levels of H2O2, a product of the scavenging action of superoxide dismutase (Fig. 8a, d respectively), and TBARS (Fig. 8b, e respectively), a product of lipid peroxidation, were increased in vehicle-treated SHR. Vehicle-treated SHR also had increased plasma and urine levels of NOx (Fig. 8c, f, respectively), possibly in an attempt to lower BP by modulating blood vessel diameter, and cardiac and renal function. All of these increases were absent in SHR treated with TMS (Fig. 8a–f), supporting the view that ROS generated via CYP1B1 contribute to hypertension and its pathogenesis in SHR.

TMS reduced increased plasma and urine levels of markers of oxidative stress in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. At the completion of the experiment, plasma was isolated and urine collected, and levels of hydrogen peroxide (H2O2; a, d), thiobarbituric acid reactive substances (TBARS; b, e), nitrite/nitrate (NOx; c, f), were measured using commercially available kits as described in Methods. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 6–10 for all experiments; data are expressed as mean ± SEM)
TMS reduced increased plasma levels of proinflammatory cytokines and catecholamines in SHR
ROS increase activity of immune cells and production of proinflammatory cytokines, which have been implicated in the development of hypertension and associated pathophysiological changes in various experimental models of hypertension, including SHR [7, 8]. Moreover, ROS also increase activity of the sympathetic nervous system [9], which stimulates activation of the immune system [8]. Therefore, we investigated the role of CYP1B1 in activation of immune cells and sympathetic activity in SHR and WKY. We used a rat cytokine 10-plex panel, which allowed for simultaneous measurement of multiple cytokines from a single sample. Vehicle-treated SHR had increased plasma levels of IL-1β, IL-2, IL-6, and IL-12 (Table 5). TMS treatment brought the levels of these cytokines down to levels observed in WKY (Table 5). Levels of IL-6 were reduced by TMS when compared to vehicle-treated SHR, but still above the level observed in TMS-treated WKY; the reason for this is currently unclear. Plasma levels of IL-4 and TNF-α were not different between any of the treatment groups (Table 5). We were unable to detect quantifiable levels of IL-1α, IL-10, granulocyte-macrophage colony-stimulating factor, or interferon-γ in our assay.
Vehicle-treated SHR had higher plasma levels of norepinephrine and epinephrine than WKY (Table 6). TMS treatment lowered levels of norepinephrine and epinephrine in SHR (Table 6). These observations indicate that increased levels of IL-1β, IL-2, IL-6, and IL-12, and sympathetic activity, as indicated by increased plasma levels of norepinephrine and epinephrine, are mediated by increased ROS production dependent on CYP1B1 activity.
TMS reduced increased cardiac activity of ERK1/2, p38 MAPK, c-Src, and Akt in SHR
ROS is known to activate one or more signaling molecules including ERK1/2, p38 MAPK, c-Src, and Akt that have been implicated in cardiovascular hypertrophy [32]. Therefore, we determined the contribution of CYP1B1 in the activation of these signaling molecules in the cardiac tissue of SHR and WKY. Cardiac activities of ERK1/2, p38 MAPK, c-Src, and Akt, as measured by their phosphorylation, were increased by 1.4 fold, 1.3 fold, 1.7 fold, and 1.9 fold, respectively in SHR; TMS reduced the increase in activity of these kinases in SHR, but had no effect on their activity in WKY (Fig. 9a–d, respectively). These data suggest that the cardiac hypertrophy in SHR is most likely mediated by one or more of these signaling molecules by a mechanism dependent on CYP1B1 activity.

TMS reduced increased cardiac activities of various signal transduction molecules in SHR. Animals were given daily i.p. injections of DMSO (vehicle) or the CYP1B1 inhibitor, TMS, from 8 weeks of age. Activities of ERK1/2 (a), p38 MAPK (b), c-Src (c) and Akt (d), as determined by phosphorylation of these kinases, were measured in cardiac homogenates by western blot as described in Methods. * P < 0.05 vs. WKY; † P < 0.05 vs. corresponding vehicle (n = 3 for all experiments; data are expressed as mean ± SEM)
Discussion
This study demonstrated that, in SHR, CYP1B1 contributes to hypertension and associated cardiovascular and renal pathophysiological changes by increased oxidative stress; increased levels of catecholamines and cytokines; and in the heart by activation of ERK1/2, p38 MAPK, c-Src, and Akt. At 8 weeks of age SHR were hypertensive; after which, daily administration of TMS, an inhibitor of CYP1B1 activity, gradually decreased BP to a level that was not different from that observed in WKY. This decrease in BP was associated with reduced cardiovascular hypertrophy, cardiac and renal fibrosis, renal dysfunction, RVR, response of the aorta, and mesenteric and renal arteries to PE and ET-1, and improvement of endothelial dysfunction, as indicated by enhanced relaxation to ACh. These findings suggest that the cardiovascular and renal pathophysiological changes associated with increased BP in SHR most likely depend on CYP1B1 activity. Supporting this view was our demonstration that CYP1B1 activity increased in aorta, heart, and kidney of SHR but was inhibited by TMS. Because CYP1B1 protein level was not altered in these tissues of SHR, with or without TMS treatment, the increase in CYP1B1 activity, which could be due to a biochemical modification of this enzyme or increased levels of NADPH/P450 reductase, remains to be investigated. Since the assay kit we used to measure CYP1B1 activity also measures CYP1A1 activity, it could also participate in the actions of TMS. However, this is unlikely because CYP1A1 is not expressed in significant amounts in the aorta, heart, and kidney of rat [3]. Moreover, TMS exhibits 50 and 500 fold more selectivity for CYP1B1 than CYP1A1 and CYP1A2, respectively [12]. Furthermore, the CYP1B1 assay kit used in this study does not detect any activity in the heart or kidney of Cyp1b1 −/− mice [4, 11]. TMS also does not inhibit the activity of 12/15 lipoxygenase or CYP4A, as indicated by lack of effect of TMS on the conversion of AA into 12-hydroxeicosatetraenoic (12-HETE) or 20-HETE, respectively in VSMCs of rat [6].
CYP1B1 is constitutively active and requires a substrate(s) to produce its effect. Recently, we reported that the effect of Ang II to stimulate VSMC growth was mediated by AA released from tissue lipids by cPLA2 and its metabolism by CYP1B1 and generation of ROS [6]. In the present study, TMS reduced CYP1B1 activity in WKY, but did not alter BP. Therefore, the increase in BP and associated cardiovascular pathophysiological changes that were prevented by TMS in SHR are most likely due to increased availability of AA and/or other fatty acids to CYP1B1 and generation of ROS by increased activity of neurohumoral systems such as the renin-angiotensin system in SHR, but not WKY [33]. Supporting this view was our demonstration that TMS treatment also attenuated NADPH oxidase activity and ROS generation in the aorta, heart, and kidney, and decreased plasma and urine levels of H2O2 in SHR, but not in WKY. Moreover, plasma and urine levels of another marker of oxidative stress, TBARS, which are formed by lipid peroxidation, were also increased in SHR, but not in WKY, and inhibited by TMS treatment in SHR.
Increased oxidative stress causes inactivation of NO; a compensatory increase in cardiac, renal, and vascular expression of endothelial and inducible NO synthases, increased urinary and plasma levels of NOx [34, 35], and endothelial dysfunction despite increased vascular NO [36] have been reported in SHR. In our study in SHR, the increase in plasma and urinary levels of NOx were attenuated by TMS, suggesting that ROS generated by CYP1B1 leads to a further production of ROS from NO. These observations, and reports that antioxidant therapy reduce BP and cardiovascular hypertrophy, improve vascular dysfunction in SHR [35–38], and inhibit NADPH oxidase and increase superoxide dismutase activity in stroke-prone SHR [39], suggest that the effect of TMS to normalize BP and minimize associated cardiovascular hypertrophy and cardiac fibrosis, and vascular reactivity and endothelial dysfunction may include inhibition of ROS generated by CYP1B1.
In this study, we cannot exclude the possibility that improvement in one or more of the pathophysiological changes is the result of decreased BP by TMS, although decreased BP in SHR by hydralazine did not affect end-organ damage [40]. Hydralazine, which is known to lower BP by a direct relaxation of vascular smooth muscle cells at concentrations up to 100 μM, did not alter CYP1B1 activity in cardiac tissue of Sprague Dawley rats, measured in vitro, whereas TMS at a concentration of 1 μM inhibited cardiac CYP1B1 activity by 90 % (our unpublished data). TMS is a derivative of resveratrol that is known to exert antioxidant and cardiovascular protective effects [41], and inhibit the activity of CYP1B1 by decreasing its expression induced by a procarcinogen, 2,3,7,8-tetrachlorodibenzo-p-dioxin [42]. Therefore, it is possible that TMS could exert its effects in SHR by its metabolism to resveratrol. However, this is unlikely because resveratrol suppresses ROS production and improves endothelial dysfunction, but has either no effect or decreases BP in SHR only at doses 25 to 50 times higher than that of TMS [43–46], which in our study completely reversed the increase in BP in SHR. Moreover, CYP1B1 activity, but not its expression, was increased in SHR, and CYP1B1 activity, but not its expression, was inhibited by TMS. In addition, exogenous TMS incubated with cardiac tissue from WKY and SHR was 1000 times more potent in inhibiting CYP1B1 activity than was resveratrol (IC50 for TMS, 5 nM vs. resveratrol, 5 μM).
ROS also stimulate sympathetic activity [9], and increased sympathetic nerve activity in SHR has been implicated in the development of hypertension [47, 48]. Therefore, it is possible that TMS might also lower BP and minimize associated pathophysiological changes in the cardiovascular system by inhibiting sympathetic nerve activity by acting directly and/or by reducing ROS generated via CYP1B1 in the brain. Supporting this view is our finding that plasma levels of both norepinephrine and epinephrine that were markedly increased in SHR were reduced by TMS.
The sympathetic nervous system also promotes activation of the immune system and dysregulation of the immune system has been proposed to contribute to the pathogenesis of hypertension in SHR [8]. Also, interaction of the innate and/or adaptive immune system(s), ROS, and inflammation has been implicated in several experimental models of hypertension [7]. Our finding that increased plasma levels of the proinflammatory cytokines IL-1β and IL-6 were minimized by treatment with TMS in SHR suggest that the antihypertensive and associated cardiovascular protective actions of this agent could be due to an anti-inflammatory effect as a result of decreased ROS production via CYP1B1. However, we cannot exclude some of its effects that are independent of CYP1B1 because TMS that did not alter BP in WKY also reduced the plasma levels of IL-6. In the present study we also found an increase in plasma levels of IL-2 in SHR that was prevented by TMS. Currently, the effect of IL-2 on BP in SHR is controversial [49–51]; as such, further work is required to determine the exact role of IL-2 on BP in SHR. Interestingly, a marked increase in plasma levels of IL-12 that is known to stimulate differentiation of naïve CD4+ T cells into T-helper-1-cells that generate interferon-γ [52] was also observed without any detectable levels of interferon-γ in SHR. Whether IL-12 exerts anti- or pro-hypertensive effects in SHR remains to be determined.
The mechanism by which increased ROS generated via CYP1B1 exert their cardiovascular and proinflammatory effects and cause hypertension in SHR could be due to activation of one or more signaling molecules including ERK1/2, p38 MAPK, c-Src, and/or Akt, which have been implicated in the action of Ang II and other vasoactive agents and growth factors, on cardiovascular hypertrophy, and renal dyshomeostasis, inflammation and fibrosis in hypertension [30, 32, 53]. ROS and these signaling molecules activate various transcription factors that are involved in cardiovascular hypertrophy, inflammation and fibrosis [32, 53]. The demonstration that activity of these signaling molecules, as indicated by their phosphorylation, was increased in the heart of SHR, but not WKY, and was inhibited by TMS, suggests that they mediate the pathophysiological effects of ROS generated via CYP1B1 in SHR. CYP1B1 can metabolize various substrates including steroids, retinoids and fatty acids [54]. Previously, we have shown that metabolism of AA by CYP1B1 results in generation of ROS that by activating ERK1/2 and p38 MAPK stimulate VSMC migration, proliferation, and hypertrophy [6], and cardiovascular hypertrophy, renal dysfunction and fibrosis [2, 10, 11]. ROS are believed to activate the mitogen-activated kinases and also tyrosine kinases by their oxidative modification and inactivation of protein phosphatases [31, 53, 55]. Therefore, the mechanism by which inhibition of CYP1B1 by TMS results in decreased activity of ERK1/2, p38 MAPK, c-Src, and Akt could be the consequence of decreased ROS production from AA and/or other fatty acids generated by increased activity of neurohumoral systems in SHR.
In conclusion, the findings of the present study, based on the use of a selective inhibitor of CYP1B1, TMS, demonstrate that in SHR, CYP1B1 contributes to hypertension and associated pathophysiology including cardiovascular hypertrophy; cardiac and renal fibrosis; renal dysfunction; increased RVR; increased vascular reactivity; endothelial dysfunction; increased oxidative stress; inflammation; and activation of ERK1/2, p38 MAPK, c-Src, and Akt. Moreover, CYP1B1 could serve as a novel target for developing agents like TMS to treat essential hypertension and other cardiovascular diseases. Further studies utilizing selective CYP1B1 gene disruption in SHR and WKY are required to confirm the observed effects of TMS and to establish the contribution of CYP1B1 to hypertension in SHR.
References
Nebert DW, Dalton TP. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006;6:947–60.
Jennings BL, Sahan-Firat S, Estes AM, et al. Cytochrome P450 1B1 contributes to angiotensin II–induced hypertension and associated pathophysiology. Hypertension. 2010;56:667–74.
Sahan-Firat S, Jennings BL, Yaghini FA, et al. 2,3′,4,5′-Tetramethoxystilbene prevents deoxycorticosterone-salt-induced hypertension: contribution of cytochrome P-450 1B1. Am J Physiol Heart Circ Physiol. 2010;299:H1891–901.
Jennings BL, Estes AM, Anderson LJ, et al. Cytochrome P450 1B1 gene disruption minimizes deoxycorticosterone acetate-salt–induced hypertension and associated cardiac dysfunction and renal damage in mice. Hypertension. 2012;60:1510–6.
White K, Johansen AK, Nilsen M, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–98.
Yaghini FA, Song CY, Lavrentyev EN, et al. Angiotensin II–induced vascular smooth muscle cell migration and growth are mediated by cytochrome P450 1B1–dependent superoxide generation. Hypertension. 2010;55:1461–7.
Harrison DG, Guzik TJ, Lob HE, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–40.
Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res. 2012;111:1190–7.
Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol. 2004;287:H695–703.
Jennings BL, Anderson LJ, Estes AM, et al. Involvement of cytochrome P-450 1B1 in renal dysfunction, injury, and inflammation associated with angiotensin II-induced hypertension in rats. Am J Physiol Renal Physiol. 2012;302:F408–20.
Jennings BL, Anderson LJ, Estes AM, et al. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension. 2012;59:348–54.
Chun Y-J, Kim S, Kim D, Lee S-K, Guengerich FP. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 2001;61:8164–70.
Yaghini FA, Li F, Malik KU. Expression and mechanism of spleen tyrosine kinase activation by angiotensin II and its implication in protein synthesis in rat vascular smooth muscle cells. J Biol Chem. 2007;282:16878–90.
Dobrowolski L, Walkowska A, Kompanowska-Jezierske E, Kuczeriszka M, Sadowski A. Effects of ATP on rat renal haemodynamics and excretion: Role of sodium intake, nitric oxide and cytochrome P450. Acta Phsyiol. 2007;189:77–85.
Zhao W, Ahokas RA, Weber KT, Sun Y. ANG II-induced cardiac molecular and cellular events: role of aldosterone. Am J Phsyiol Heart Circ Physiol. 2006;291:H336–43.
Suganami T, Mori K, Tanaka I, et al. Role of prostaglandin E receptor EP1 subtype in the development of renal injury in genetically hypertensive rats. Hypertension. 2003;42:1183–90.
Miller Jr FJ, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–305.
Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A. 2006;103:7432–7.
Fan YY, Kohno M, Nakano D, et al. Cilnidipine suppresses podocyte injury and proteinuria in metabolic syndrome rats: possible involvement of N-type calcium channel in podocyte. J Hypertens. 2010;28:1034–43.
Mefford IN. Biomedical uses of high-performance liquid chromatography with electrochemical detection. Methods Biochem Anal. 1985;31:221–58.
Warshaw DM, Mulvany MJ, Halpern W. Mechanical and morphological properties of arterial resistance vessels in young and old spontaneously hypertensive rats. Circ Res. 1979;45:250–9.
Collis MG, DeMey C, Vanhoutte PM. Renal vascular reactivity in the young spontaneously hypertensive rat. Hypertension. 1980;2:45–52.
Berecek KH, Stocker M, Gross F. Changes in renal vascular reactivity at various stages of deoxycorticosterone hypertension in rats. Circ Res. 1980;46:619–24.
Panza JA, Quyyumi AA, Brush Jr JE, Epstein SE. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. New Eng J Med. 1990;323:22–7.
Vapaatalo H, Mervaala E, Nurminen M-L. Role of endothelium and nitric oxide in experimental hypertension. Physiol Res. 2000;49:1–10.
Rettig R, Folberth C, Stauss H, Kopf D, Waldherr R, Unger T. Role of the kidney in primary hypertension: a renal transplantation study. Am J Physiol Renal Physiol. 1990;258:F606–11.
Hultström M. Development of structural kidney damage in spontaneously hypertensive rats. J Hypertens. 2012;30:1087–91.
Hall JE, Granger JP, de Carmo JM, et al. Hypertension: physiology and pathophysiology. Compr Physiol. 2012;2:2393–442.
Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension. Diabetes Care. 2008;31:S170–80.
Nistala R, Whaley-Connell A, Sowers JR. Redox control of renal function and hypertension. Antioxid Redox Signal. 2008;10:2047–89.
Datla SR, Griendling KK. Reactive oxygen species, NADPH oxidases, and hypertension. Hypertension. 2010;56:325–30.
Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97.
Bolterman RJ, Manriquez MC, Ruiz MCO, Juncos LA, Romero JC. Effects of captopril on the renin angiotensin system, oxidative stress, and endothelin in normal and hypertensive rats. Hypertension. 2005;46:943–7.
Vaziri ND, Ni Z, Oveisi F. Upregulation of renal and vascular nitric oxide synthase in young spontaneously hypertensive rats. Hypertension. 1998;31:1248–54.
Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension. 2000;36:957–64.
Ülker S, McMaster D, McKeown PP, Bayraktutan U. Impaired activities of antioxidant enzymes elicit endothelial dysfunction in spontaneous hypertensive rats despite enhanced vascular nitric oxide generation. Cardiovasc Res. 2003;59:488–500.
Akpaffiong MJ, Taylor AA. Antihypertensive and vasodilator actions of antioxidants in spontaneously hypertensive rats. Am J Hypertens. 1998;11:1450–60.
Monroy-Ruiz J, Sevilla M-Á, Carrón R, Montero M-J. Astaxanthin-enriched-diet reduces blood pressure and improves cardiovascular parameters in spontaneously hypertensive rats. Pharmacol Res. 2011;63:44–50.
Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension. 2001;38:606–11.
Pinto YM, Paul M, Ganten D. Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc Res. 1998;39:77–88.
Wang H, Yang Y-J, Qian H-Y, Zhang Q, Xu H, Li J-J. Resveratrol in cardiovascular disease: what is known from current research? Heart Fail Rev. 2012;17:437–48.
Chen Z-H, Hurh Y-J, Na H-K, et al. Resveratrol inhibits TCDD-induced expression of CYP1A1 and CYP1B1 and catechol estrogen-mediated oxidative DNA damage in cultured human mammary epithelial cells. Carcinogenesis. 2004;25:2005–13.
Rush JWE, Quadrilatero J, Levy AS, Ford RJ. Chronic resveratrol enhances endothelium-dependent relaxation but does not alter eNOS levels in aorta of spontaneously hypertensive rats. Exp Biol Med. 2007;232:814–22.
López-Sepúlveda R, Jiménez R, Romero M, et al. Wine polyphenols improve endothelial function in large vessels of female spontaneously hypertensive rats. Hypertension. 2008;51:1088–95.
Thandapilly SJ, Wojciechowski P, Behbahani J, et al. Resveratrol prevents the development of pathological cardiac hypertrophy and contractile dysfunction in the SHR without lowering blood pressure. Am J Hypertens. 2010;23:192–6.
Bhatt SR, Lokhandwala MF, Banday AA. Resveratrol prevents endothelial nitric oxide synthase uncoupling and attenuates development of hypertension in spontaneously hypertensive rats. Eur J Pharmacol. 2011;667:258–64.
Abboud FM. The sympathetic system in hypertension. State-of-the-art review Hypertension. 1982;4:II208–25.
Grisk O, Rettig R. Interactions between the sympathetic nervous system and the kidneys in arterial hypertension. Cardiovasc Res. 2004;61:238–46.
Pascual DW, Jin H, Bost KL, Oparil S. Interleukin-2 does not attenuate hypertension in spontaneously hypertensive rats. Hypertension. 1990;16:468–71.
Tuttle RS, Boppana DP. Antihypertensive effect of interleukin-2. Hypertension. 1990;15:89–94.
Dzielak DJ. Interleukin-2 and spontaneous hypertension. Hypertension. 1991;18:171–5.
Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–8.
Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–58.
Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1B1. Drug Metab Dispos. 2004;32:840–7.
Son Y, Cheong Y-K, Kim N-H, Chung H-T, Kang DG, Pae H-O. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct. 2011. doi:10.1155/2011/792639.
Acknowledgments
This research was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants R01-HL-19134-38 (to K.U.M) and K01-HL-96410-04 (to A.K.)]. D.E.M was supported by a Minority Access to Research Careers (MARC) summer student fellowship from the Federation for America Societies for Experimental Biology (FASEB).
We thank Dr. Edward Rosloniec and Dr. Karen Whittington (Memphis V. A. Medical Center) for assistance with the rat 10-plex cytokine panel assay, and Dr. David L. Armbruster for editorial assistance.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jennings, B.L., Montanez, D.E., May, M.E. et al. Cytochrome P450 1B1 contributes to increased blood pressure and cardiovascular and renal dysfunction in spontaneously hypertensive rats. Cardiovasc Drugs Ther 28, 145–161 (2014). https://doi.org/10.1007/s10557-014-6510-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-014-6510-4