
Abstract
Bioethanol is one of the main biofuels produced from the fermentation of saccharified agricultural waste; however, this technology needs to be optimized for profitability. Because the commonly used ethanologenic yeast strains are unable to assimilate cellobiose, several efforts have been made to express cellulose hydrolytic enzymes in these yeasts to produce ethanol from lignocellulose. The C. flavigenabglA gene encoding β-glucosidase catalytic subunit was optimized for preferential codon usage in S. cerevisiae. The optimized gene, cloned into the episomal vector pRGP-1, was expressed, which led to the secretion of an active β-glucosidase in transformants of the S. cerevisiae diploid strain 2-24D. The volumetric and specific extracellular enzymatic activities using pNPG as substrate were 155 IU L−1 and 222 IU g−1, respectively, as detected in the supernatant of the cultures of the S. cerevisiae RP2-BGL transformant strain growing in cellobiose (20 g L−1) as the sole carbon source for 48 h. Ethanol production was 5 g L−1 after 96 h of culture, which represented a yield of 0.41 g g−1 of substrate consumed (12 g L−1), equivalent to 76% of the theoretical yield. The S. cerevisiae RP2-BGL strain expressed the β-glucosidase extracellularly and produced ethanol from cellobiose, which makes this microorganism suitable for application in ethanol production processes with saccharified lignocellulose.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bioethanol production from lignocellulosic biomass, also known as second-generation ethanol, has gained great importance in the last two decades (Raele et al. 2014; Tye et al. 2016), as this biofuel is not produced from primary crops that are necessary as human food or animal feed (Sun and Cheng 2002).
The conversion of cellulose into ethanol includes two main steps: hydrolysis of cellulose in the lignocellulosic materials to monosaccharides and fermentation of these monosaccharides to ethanol. However, this process has large biotechnological challenges, as the transformation of biomass to ethanol is still unprofitable and the use of cellulolytic enzymes necessary to obtain fermentable sugars is one of the main contributing factors to the high production cost (Zaldivar et al. 2001; Lynd et al. 2002).
During the enzymatic saccharification process, the cellulose polymer is hydrolyzed by endoglucanases and exoglucanases to cellooligosaccharides (mainly cellobiose), which are then converted to glucose by the action of β-glucosidase. This last enzyme is a limiting step in the complete cellulose degradation process, as β-glucosidase hydrolyzes cellobiose and some cellooligosaccharides, which inhibit the activity of exoglucanases and endoglucanases, decreasing the efficiency of the saccharification (Yan et al. 1998; Shen et al. 2008).
In recent years, the heterologous expression of β-glucosidases in ethanologenic microorganisms such as Saccharomyces cerevisiae has been carried out, which demonstrated cellobiose degradation and fermentation of the glucose in just one step, which increased the hydrolysis efficiency of cellooligosaccharides and reduced the production costs of cellulosic bioethanol to make it profitable (Gurgu et al. 2011; Wilde et al. 2012); however, the ethanol yields and productivities achieved with these recombinant yeasts must be improved and, therefore, the creation of S. cerevisiae strains for cellulosic bioethanol production is still under development.
In our group, we recently achieved the active expression of the catalytic subunit of the β-glucosidase from the Cellulomonas flavigena PN-120 strain in S. cerevisiae by the cloning of the native bglA gene in the pYEX-S1 plasmid (Barrera-Islas et al. 2007; Mendoza-Aguayo et al. 2014). However, the extracellular β-glucosidase activity in the recombinant yeast was low (0.5 IU mL−1), which caused low growth and low ethanol production when cellobiose was used as the only carbon source. This low expression of β-glucosidase from C. flavigena in S. cerevisiae may be due to the difference in codon usage between prokaryote and eukaryote organisms.
One strategy that has been described to improve ethanol yields using recombinant yeast is the optimization of the expression of heterologous proteins to increase the extracellular cellulase activity, the hydrolysis of cellulose and ultimately the amount of bioethanol (Wiedemann and Boles 2008; Shim and Withers 2013; Liu et al. 2014).
In the present work, to express β-glucosidase catalytic subunit in S. cerevisiae, the bglA gene from the C. flavigena PR-22 mutant strain (Rojas-Rejón et al. 2011) has been optimized through synthetic biology tools to include codons preferably used by S. cerevisiae and cloned in a derivative of the pRGP-1 vector. Upon transformation with the plasmid, S. cerevisiae was able to grow and produce 5 g L−1 of ethanol from cellobiose, which represented a yield 0.41 g g−1 of substrate consumed. To our knowledge, this is the first report about ethanol production using a S. cerevisiae strain expressing a codon-optimized β-glucosidase from Cellulomonas flavigena.
Materials and methods
Strains, media and culture conditions
The details of microbial strains and plasmids used in this work are summarized in Table 1. Cellulomonas flavigena PR-22 was grown at 37 °C in mineral medium and 1% alkali-pretreated sugar cane bagasse as a carbon source (Ponce-Noyola and de la Torre 1993). Escherichia coli was used to replicate the vectors developed in this work, which was grown at 37 °C on Luria Bertani (LB) broth (Difco laboratories, Detroit, MI, USA) or on LB-agar plates with 2% agar (Becton, Dickinson and Company, Franklin Lakes, NJ, USA). Recombinant strains were grown on LB supplemented with ampicillin (100 µg mL−1). Saccharomyces cerevisiae was grown at 30 °C on YPD medium (1% yeast extract, 2% bacto-peptone and 2% glucose), YPC medium (1% yeast extract, 2% bacto-peptone and 2% cellobiose), SD medium (0.67% yeast nitrogen base without amino acids, 2% glucose and 1 mL L−1 Dropout mix Ura−) and SD-C medium (0.67% yeast nitrogen base without amino acids, 2% cellobiose and 1 mL L− 1 Dropout mix Ura−) liquid or solid (2% agar).
Genetic optimization of the bglA gene
Genomic DNA of C. flavigena PR-22 was extracted with some modifications to the method reported by Sambrook et al. (1989) incubating the cell suspension for 1 h with 1 mg mL− 1 of lysozyme in TE buffer to release the genomic DNA. The sequences of the oligonucleotides used in this work are detailed in Table 2. The bglA gene from C. flavigena PR-22 (GenBank, accession number: KY290247) was amplified with EcoRI-bgl and BamHI-bgl primers, previously designed based on the sequence of the bglA native gene of C. flavigena (GenBank, accession number: JX124706), and the addition of 1% DMSO to the reaction mixture to promote denaturation of the bacterial DNA due to the high GC DNA content. The PCR product was purified with the Wizard SV Gel and PCR Clean-up Kit (Promega, Madison, WI, USA) and sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), which is based on the Sanger technique. The sequence of the bglA gene was optimized for expression in S. cerevisiae (GenScript); during optimization, the codon adaptation index, GC content and location of SacI restriction sites were modified from the gene ORF sequence. The optimized gene was cloned into the pUC57 vector and used to transform the TOP-10 E. coli strain, selecting on LB-Amp.
Construction of the pRGP-bglA OPT expression vector
Plasmid DNA was purified by alkaline lysis using a standard miniprep technique (Sambrook et al. 1989). PCR amplifications were performed with Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific), and the products obtained were purified with the Wizard SV Gel and PCR Clean-up Kit (Promega, Madison, WI, USA). Restriction enzymes, T4 DNA ligase and alkaline phosphatase were purchased from Thermo Scientific and used as recommended by the manufacturer.
The optimized bglA gene (bglA OPT) was cloned into the pRGP-1 vector, obtained by point mutation of the KEX2 site of the pYEX-S1 plasmid (Mendoza-Aguayo et al. 2014). Cloning was performed by conventional restriction and ligation techniques. The bglA OPT gene was amplified by PCR using the Fw-bglAOPT-plusG and Rv-bglAOPT-plusG2 oligonucleotides, based on the sequence of the optimized gene, and the pUC57-bglA OPT vector as a template (Table 2). The pRGP-1 plasmid was obtained by modifying the multicloning site of the pYEX-S1 vector (Clontech). The vector components were digested with SacIFastDigest (Thermo Scientific), and the pRGP-1 restricted plasmid was dephosphorylated with alkaline phosphatase FastAP (Thermo Scientific). The pRGP-1 vector and the bglA OPT insert were mixed in a 1:5 ratio to perform the ligation reaction with T4 DNA ligase. The plasmid constructed (Figure S1) was used to transform E. coli TOP-10 CaCl2-competent cells.
The purified plasmid from the transformants was digested with EcoRI and SacI FD to release the gene from the multicloning site. The presence and correct orientation of bglA gene into the pRGP-bglA OPT plasmid was confirmed by PCR and sequencing reactions of the multicloning site of vector. The amplification of the multicloning region of the plasmids was performed with the D- and R-linker-pYEXS1 oligonucleotides that hybridize at the end and the beginning of the vector promoter and terminator sequences, respectively (Table 2).
Yeast transformation
Saccharomyces cerevisiae 2-24D strain was transformed with the pRPG-1 and pRPG-bglA OPT plasmids using a modified version of the procedure described by Gietz and Woods (2006). Strain 2-24D was grown overnight in 2 mL of YPD liquid medium at 30 °C; 700 µL of this culture was added to 50 mL of fresh YPD liquid medium and incubated for 4 h at 30 °C and 200 rpm. The cells were recovered by centrifugation at 8000×g for 5 min. Thereafter, the pellet was washed twice with 20 mL of sterile deionized water and resuspended in 1 mL of sterile distilled water.
An aliquot containing 1 × 108 cells was placed in a 1.5-mL sterile microtube and centrifuged. The supernatant was discarded, and the pellet was resuspended in 360 μL of T-Mix (50% PEG-4000, 240 μL; 1 M LiAc, 36 μL; 10 mg mL−1 ssDNA carrier, 10 μL; 500–1000 ng plasmid DNA; DMSO, 4 μL; sterile distilled water as required to bring the final volume of the mix up to 360 μL). The suspension was incubated at 30 °C for 20 min and then for 40 min at 42 °C in a multi-block heater (Lab-Line instruments Inc., Melrose Park, ILL, USA). The cells were centrifuged, the pellet was resuspended in 500 μL of sterile distilled water, and 500 μL of YPD medium was added to each tube and incubated for 2 h at 30 °C and 200 rpm. Aliquots of 200 μL were spread onto SC medium with the appropriate auxotrophic marker and incubated for 2–5 days at 30 °C.
β-Glucosidase expression
Yeast transformants containing the plasmid pRGP-bglA OPT were grown on SD-C and YPC plates. Zymograms for β-glucosidase activity were performed by adding 3 mM MUG (Sigma) in phosphate buffer (25 mM, pH 6.2) over the colonies in the plates. The plates were incubated at 37 °C until fluorescent halos appeared.
Selected recombinant strains were cultured in Erlenmeyer flasks with YPC medium at 30 °C for 48 h and 200 rpm. The culture supernatants were concentrated by ultrafiltration using an Amicon membrane with a 10 kDa molecular weight cutoff, and β-glucosidase activity was measured by incubating 100 μL of concentrated supernatant with 1 mM ρ-nitrophenyl β-d-glucopyranoside (pNPG) in phosphate buffer (50 mM, pH 6.2, 42 °C) as a substrate (Barrera-Islas et al. 2007). One unit of pNPGase activity was defined as the amount of p-nitrophenol (μmol) released mL−1min−1 under optimal pH and temperature conditions.
Bioethanol production
Recombinant strains were cultured in serological bottles containing YPC medium at 30 °C for 96 h in anaerobic conditions (without shaking). The culture medium was inoculated with 0.5 g L−1 yeast biomass. Cellobiose consumption was monitored by the reducing sugars technique (Miller 1959), and the ethanol was measured by gas chromatography with a Zebron column (ZB-WAXplus, Phenomenex).
Results and discussion
Cloning and expression of β-glucosidase in S. cerevisiae
Saccharomyces cerevisiae is the yeast commonly used for bioethanol production, but it is unable to utilize cellooligosaccharides as a substrate. Thus, several studies have focused on obtaining S. cerevisiae strains capable of assimilating these compounds (Fujita et al. 2002; Yanase et al. 2010), but to date, the recombinant yeast strains obtained for fermenting complex substrates such as cellobiose have achieved ethanol yields and productivities that still require improvement. Therefore, the construction of microbial platforms for the production of second-generation ethanol is still under development. Thus, we were interested in constructing a yeast strain capable of growing in enzymatically saccharified cellulose and fermenting the cellobiose present in these syrups. The S. cerevisiae 2-24D strain was transformed with the pRGP-bglA OPT vector purified from E. coli transformants 1 and 9 (Supplementary material). The transformants of S. cerevisiae 2-24D obtained with this plasmid construction were isolated in minimal medium lacking uracil and were then evaluated for their ability to grow in media with cellobiose as the sole carbon source and for their extracellular production of β-glucosidase. At the same time, S. cerevisiae transformants with the empty pRGP-1 vector were obtained as a negative control.
Among the S. cerevisiae transformants obtained with the pRGP-bglA OPT construction, transformant 2 (RP2-BGL) had a higher growth in cellobiose on the SD-C and YPC plates and thus was selected for further experiments.
Comparing growth in plates between S. cerevisiae RP2-BGL and a transformant obtained with the pRGP-1 empty vector, it was observed that both strains exhibited a comparable growth in culture media with glucose as the carbon source. However, in medium with cellobiose, only the RP2-BGL strain had the ability to grow (Fig. 1a, b).

Comparative growth in SD medium of S. cerevisiae RP2-BGL (left) and pRGP-1 (right) with a glucose and b cellobiose as the carbon source. c Zymogram overgrowth of the transformants on YPC medium using MUG as a substrate for β-glucosidase extracellular activity and revealed under UV light
To confirm the activity of β-glucosidase in S. cerevisiae RP2-BGL, zymograms were performed with MUG on the S. cerevisiae RP2-BGL strain growing in YPC plates. The zymogram revealed a fluorescent halo on the growth of strain RP2-BGL, whereas the control strain with the empty vector pRGP-1 did not show brightness under UV light (Fig. 1c).
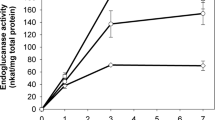
The extracellular β-glucosidase activity was quantified from the concentrated culture supernatants of the RP2-BGL and pRGP-1 transformants, with the 2-24D S. cerevisiae strain as a negative control. The activity values were compared against those in a crude cell-free extract of C. flavigena PR-22. The S. cerevisiae pRGP-1 and 2-24D strains showed no significant volumetric or specific β-glucosidase activities, as expected (approximately 2.8 IU L−1 and 2.5 IU g−1, respectively), compared with RP2-BGL, which reached volumetric and specific activities of 155 IU L−1 and 222 IU g−1, respectively. Moreover, the β-glucosidase specific activity of the crude extracts of S. cerevisiae RP2-BGL was comparable to those obtained from C. flavigena PR-22. Despite the small difference in specific activity, it is worth mentioning that the β-glucosidase in C. flavigena was intracellular, whereas the β-glucosidase in S. cerevisiae RP2-BGL was extracellular, which represents a biotechnological advantage (Fig. 2). On the other hand, the detection of extracellular activity of β-glucosidase in RP2-BGL both in zymogram and culture medium supernatant points out that K1 exportation sequence in conjunction with the KEX2 endopeptidase sequence was suitable for exporting the recombinant BGLA.

Specific β-glucosidase activity in supernatants of S. cerevisiae growing in SD medium of transformants RP2-BGL (pRGP-bglA OPT), pRGP-1 and the 2-24D strain, and a concentrated crude cell-free extract from C. flavigena PR-22 as a control
The β-glucosidase specific activity of strain RP2-BGL was more than twice of that observed by Mendoza-Aguayo et al. (2014) in S. cerevisiae PM-15, where the catalytic fraction of β-glucosidase from C. flavigena PN-120 was actively expressed but remained cell bound. This increase in specific activity could be attributed to the fact that the bglA sequence gene was optimized for expression in S. cerevisiae.
The specific activity achieved with the RP2-BGL strain (222 IU g−1) was almost twice as high as that obtained for the β-glucosidase from Saccharomycopsis fibuligera expressed in S. cerevisiae Y294[ySFI]. This strain produced a β-glucosidase with specific activities of 112 and 94 IU g−1 when grown on cellobiose and glucose, respectively (McBride et al. 2005).
In contrast, S. cerevisiae MT8-1/pBG211 and MT8-1/pBG211/pEG23u31H6 strains expressed the β-glucosidase 1 from Aspergillus aculeatus with specific β-glucosidase activities of 21 and 16 IU g−1, respectively, after aerobic cultivation in SD medium for 48 h (Fujita et al. 2002). These activities are 10 times lower than the activities achieved by S. cerevisiae RP2-BGL in our work.
In previous studies performed by our group (Rojas-Rejón et al. 2011; Mendoza-Aguayo et al. 2014), it was demonstrated that the cellulolytic enzymes of the PN-120 and PR-22 C. flavigena mutant strains were resistant to catabolic repression and product inhibition. Particularly, the catalytic subunit of β-glucosidase from C. flavigena PN-120 expressed in S. cerevisiae showed a high resistance to inhibition by glucose and ethanol (Mendoza-Aguayo et al. 2014). Thus, the S. cerevisiae transformants expressing the β-glucosidase of these C. flavigena mutants may have important biochemical advantages for bioethanol production, unlike other yeast strains expressing β-glucosidases that are more sensitive to inhibition by compounds found in these processes.
Ethanol production from cellobiose
Transformants RP2-BGL, PRGP-1 and 2-24D of S. cerevisiae were anaerobically cultured in cellobiose as the only carbon source to assess their ability to produce ethanol. In these conditions, the strain RP2-BGL that expresses the codon-optimized β-glucosidase produced 5 g L−1 of ethanol at 96 h of culture, while the pRGP-1 and 2-24D strains did not produce ethanol from cellobiose in the same culture time (Fig. 3). This ethanol production represented a yield of 0.41 g g−1 of substrate consumed, equivalent to 76% of the theoretical yield. These results were better than those obtained with S. cerevisiae PM-15 (Mendoza-Aguayo et al. 2014) which produced only 1.5 g L−1of ethanol in the same fermentation conditions (data not shown) possibly due to the low levels of expression of its β-glucosidase. Likewise the results of the present work were 2.5 times higher than those obtained for the Y294[SFI] strain which expressed the β-glucosidase from Saccharomycopsis fibuligera (van Rooyen et al. 2005), and similar to S. cerevisiae Y5/bgl1 that expresses the β-glucosidase from Aspergillus aculeatus; however, the yield was 50.2% of the theoretical yield (Yang et al. 2013).

Culture in anaerobic conditions on YPC medium of the S. cerevisiae transformants RP2-BGL (pRGP-bglA OPT), pRGP-1 and the 2-24D strain. a Production of ethanol and b cellobiose consumption. Closed circle RP2-BGL; open circle pRGP-1; closed triangle 2-24D
The S. cerevisiae RP2-BGL strain obtained in this study was competitive with other S. cerevisiae strains capable of producing ethanol from cellobiose. Additionally, the ethanol yield obtained in this study was 25% higher than those reported in other works. This makes RP2-BGL a good candidate for application in ethanol production processes using cellobiose-rich syrups as a substrate. Nevertheless, the volumetric ethanol production and ethanol productivity must be improved to increase the fermentation efficiency of the S. cerevisiae RP2-BGL strain.
Conclusion
The recombinant S. cerevisiae RP2-BGL strain obtained in this study expresses extracellularly a codon-optimized β-glucosidase which can hydrolyze cellobiose, and produce ethanol from the glucose released, making this microorganism suitable for application in ethanol production processes with saccharified lignocellulose. Cellobiose is one of the main products in the enzymatic hydrolysis of cellulose, and thus a S. cerevisiae strain with the ability to ferment this disaccharide to ethanol represents a biotechnological advantage in biofuel production processes. To our knowledge, this is the first report about ethanol production using a S. cerevisiae strain expressing a codon-optimized β-glucosidase from Cellulomonas flavigena.
Further studies will be carried out with S. cerevisiae RP2-BGLtogether with cellulase extracts that lack β-glucosidase activity, to perform simultaneous saccharification and fermentation of pretreated lignocellulosic materials, which could significantly reduce the production costs of cellulosic ethanol.
References
Barrera-Islas GA, Ramos-Valdivia AC, Salgado LM, Ponce-Noyola T (2007) Characterization of a β-glucosidase produced by a high-specific growth-rate mutant of Cellulomonas flavigena. Curr Microbiol 54:266–270
Fujita Y, Takahashi S, Ueda M, Tanaka A, Okada H, Morikawa Y, Kawaguchi T, Arai M, Fukuda H, Kondo A (2002) Direct and efficient production of ethanol from cellulosic material with yeast strain displaying cellulolytic enzymes. Appl Environ Microbiol 68:5136–5141
Gietz RD, Woods RA (2006) Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol Biol 313:107–120
Gurgu L, Lafraya A, Polaina J, Marín-Navarro J (2011) Fermentation of cellobiose to ethanol by industrial Saccharomyces strains carrying the β-glucosidase gene (BGL1) from Saccharomycopsis fibuligera. Bioresour Technol 102:5229–5236
Liu GL, Fu GY, Chi Z, Chi ZM (2014) Enhanced expression of the codon-optimized exo-inulinase gene from the yeast Meyerozyma guilliermondii in Saccharomyces sp. W0 and bioethanol production from inulin. Appl Microbiol Biotechnol 98:9129–9138
Lynd LR, Weimer PJ, vanZyl WH, Pretorius IS (2002) Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577
McBride JE, Zietsman JJ, van Zyl WH, Lynd LR (2005) Utilization of cellobiose by recombinant β-glucosidase-expressing strains of Saccharomyces cerevisiae: characterization and evaluation of the sufficiency of expression. Enzyme Microb Tech 37:93–101
Mendoza-Aguayo DJ, Poggi-Varaldo HM, García-Mena J, Ramos-Valdivia AC, Salgado LM, de la Torre-Martínez M, Ponce-Noyola T (2014) Extracellular expression of glucose inhibition-resistant Cellulomonas flavigena PN-120 β-glucosidase by a diploid strain of Saccharomyces cerevisiae. Arch Microbiol 196:25–33
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Ponce-Noyola T, de la Torre M (1993) Interactions in a mixed culture composed of Cellulomonas flavigena and Xanthomonas sp. growing in continuous culture on sugar cane bagasse. Appl Microbiol Biotechnol 40:531–534
Raele R, Boaventura JMG, Fischmann AA, Sarturi G (2014) Scenarios for the second generation ethanol in Brazil. Technol Forecast Soc Change 87:205–223
Rojas-Rejón OA, Poggi-Varaldo HM, Ramos-Valdivia AC, Martinez-Jimenez A, Cristiani-Urbina E, de la Torre-Martinez M, Ponce-Noyola T (2011) Production of cellulases and xylanases under catabolic repression conditions from mutant PR-22 of Cellulomonas flavigena. J Ind Microbiol Biotechnol 38:257–264
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual (2nd edition). Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Shen Y, Zhang Y, Ma T, Bao X, Du F, Zhuang G, Qu Y (2008) Simultaneous saccharification and fermentation of acid-pretreated corncobs with a recombinant Saccharomyces cerevisiae expressing β-glucosidase. Bioresour Technol 99:5099–5103
Shim JH, Withers SG (2013) Improvement of the expression level of β-glucosidase from Agrobacterium sp. in Escherichia coli by rare codon optimization. Food Sci Biotechnol 22:269–273
Sun Y, Cheng J (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresour Technol 83:1–11
Tye YY, Lee KT, Abdullah WNW, Leh CP (2016) The world availability of non-wood lignocellulosic biomass for the production of cellulosic ethanol and potential pretreatments for the enhancement of enzymatic saccharification. Renew Sustain Energy Rev 60:155–172
van Rooyen R, Hahn-Hagerdal B, La Grange DC, van Zyl WH (2005) Construction of cellobiose-growing and fermenting Saccharomyces cerevisiae strains. J Biotechnol 120:284–295
Wiedemann B, Boles E (2008) Codon-optimized bacterial genes improve L-arabinose fermentation in recombinant Saccharomyces cerevisiae. Appl Environ Microbiol 74:2043–2050
Wilde C, Bawa N, Gold ND, Tambor H, Mougharbel L, Storms R, Martin VJJ (2012) Expression of a library of fungal β-glucosidases in Saccharomyces cerevisiae for the development of a biomass fermenting strain. Appl Microbiol Biotechnol 95:647–659
Yan T, Lin Y, Lin C (1998) Purification and characterization of an extracellular β-glucosidase II with high hydrolysis and transglucosylation activities from Asperillus niger. J Agric Food Chem 46:431–437
Yanase S, Yamada R, Kaneko S, Noda H, Hasunuma T, Tanaka T (2010) Ethanol production from cellulosic materials using cellulase-expressing yeast. Biotechnol J 5:449–455
Yang F, Cao M, Jin Y, Yang X, Tian S (2013) Construction of a novel а-agglutinin expression system on the surface of wild-type Saccharomyces cerevisiae Y5 and genetic immobilization of β-glucosidase1. Bioenerg Res 6:1205–1211
Zaldivar J, Nielsen J, Olsson L (2001) Fuel ethanol production from lignocellulose: a challenge for metabolic engineering and process integration. Appl Microbiol Biotechnol 56:17–34
Acknowledgements
This work was supported by the Mexican Council of Science and Technology (CONACyT), Grant CB-2014/236895, and the Bioenergy Thematic Network (Red Temática de Bioenergía), Grant 260457. The authors thank Odilia Pérez-Avalos and Gustavo Medina-Mendoza for technical assistance. FJ Ríos-Fránquez received a Ph. D scholarship from CONACyT-México. The authors have no conflict of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ríos-Fránquez, F.J., González-Bautista, E., Ponce-Noyola, T. et al. Expression of a codon-optimized β-glucosidase from Cellulomonas flavigena PR-22 in Saccharomyces cerevisiae for bioethanol production from cellobiose. Arch Microbiol 199, 605–611 (2017). https://doi.org/10.1007/s00203-016-1333-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-016-1333-2