
Abstract
Summary
The achievement of more accurate diagnosis would greatly benefit the management of patients with osteogenesis imperfecta (OI). In this study, we present the largest OI sample in China as screened by next generation sequencing. In particular, we successfully identified 81 variants, which included 45 novel variants. We further did a genotype-phenotype analysis, which helps make a better understanding of OI.
Introduction
This study aims to reveal the gene mutation spectrum and the genotype-phenotype relationship among Chinese OI patients by next generation sequencing (NGS).
Methods
We developed a NGS-based panel for targeted sequencing of all exons of 14 genes related to OI, and performed diagnostic gene sequencing for a cohort of 103 Chinese OI patients from 101 unrelated families. Mutations identified by NGS were further confirmed by Sanger sequencing and co-segregation analysis.
Results
Of the 103 patients from 101 unrelated OI families, we identified 79 mutations, including 43 novel mutations (11 frameshift, 17 missense, 5 nonsense, 9 splice site, and 1 chromosome translocation) in 90 patients (87.4%). Mutations in genes encoding type I collagen, COL1A1 (n = 37), and COL1A2 (n = 29) accounts for 73.3% of all molecularly diagnosed patients, followed by IFITM5 (n = 9, 10%), SERPINF1 (n = 4, 4.4%), WNT1 (n = 4, 4.4%), FKBP10 (n = 3, 3.3%), TMEM38B (n = 3, 3.3%), and PLOD2 (n = 1, 1.1%). This corresponds to 75 autosomal dominant inherited (AD) OI patients and 15 autosomal recessive (AR) inherited patients. Compared with AD inherited OI patients, AR inherited patients had lower bone mineral density (BMD) at spine (P = 0.05) and less frequent blue sclera (P = 0.001). Patients with type I collagen qualitative defects had lower femoral neck BMD Z-score (P = 0.034) and were shorter compared with patients with type I collagen quantitative defects (P = 0.022).
Conclusion
We revealed the gene mutation spectrum in Chinese OI patients, and novel mutations identified here expanded the mutation catalog and genotype and phenotype relationships among OI patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) is a rare congenital skeletal dysplasia that is characterized by increasing bone fragility, recurrent fracture, and subsequent growth retardation. OI patients also share some extraskeletal symptoms, such as blue sclera, hearing deficits, dentinogenesis imperfecta (DI), and valvular heart disease [1].
The widely used Sillence classification divides OI into four types on the basis of clinical and radiological features [2]. In 2014, van Dijk then made some modification to the phenotype assessment [3]. Type I OI is the mildest type, and patients usually have variable fractures and a normal stature. Type II OI is fatal, wherein patients with this type of OI have difficulty surviving perinatal period due to skeletal deformities and respiratory compromise. Type III is the most severe type in patients, in terms of those that are able to survive, as patients often sustain from multiple fractures and have a severely short stature. Patients with moderate skeletal deformities are classified into type IV OI. Finally, type V is distinguished by apparent hypercallus formation and intraosseous membrane calcification [3].
Although the diagnosis of OI largely relies on clinical evaluation and pedigree analysis, molecular analysis can help achieve higher levels of diagnostic accuracy. Currently, it is known that about 85 to 90% OI patients are caused by autosomal dominant (AD) mutations of genes that encode type I collagen, COL1A1, and COL1A2 [1]. Now, with the development of molecular diagnosis, a total of 20 genes have been identified as the causative candidate gene of OI, including IFITM5, SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, PLOD2, WNT1, CREB3L1, WNT1, SP7, CREB3L1, and MBTPS2 [4,5,6,7,8,9,10,11,12,13,14]. However, the absence of gene-specific symptoms and the large number of candidate genes hamper the molecular diagnosis. In addition, the current gold standard method, Sanger sequencing, is time-consuming and costly, especially when applied to large genes such as COL1A1 and COL1A2 [15]. As a result, compared to traditional methods, next generation sequencing (NGS) is more cost-effective and more efficient in its ability to identify the causal mutation of diseases with genetic heterogeneity [16].
Additionally, although over 1600 pathogenic variants had been identified, the genotype-phenotype correlations in OI patients have only partially been elucidated. Previous studies revealed that phenotypic severity was associated with affected collagen helical location and types of amino acid substitution. However, these genotype-phenotype studies focused more on AD inherited OI and less is known about autosomal recessive (AR) inherited OI. Moreover, genotype-phenotype studies are elusive and are often limited to a relatively small size in China.
Herein, we developed a novel panel of genes related to OI and other skeletal dysplasia disorders. Given that most reports on the mutation spectrum of OI have been restricted to Western populations and only a few OI-related genetic studies have been conducted in China, we applied the panel in a cohort of Chinese OI patients not only to prove the efficiency of NGS in the genetic analysis of OI but also to gain more population-specific insight into OI. Furthermore, by analyzing the mutation spectrum and bone morphologic findings of OI patients, we also aim to investigate the genotype-phenotype relationship.
Materials and methods
Patients
The study includes 103 patients with OI who were evaluated at Peking Union Medical College Hospital (PUMCH) from 2010 to 2014. Clinical diagnoses of OI were based on the following: a history of more than one fracture under minor trauma and an age-adjusted and sex-adjusted BMD Z-score of less than −1.0 or more for either lumbar spine or femoral neck or with a family history of OI or an adjusted areal BMD Z-score of −2·0 or less irrespective of a history of fractures [2]. Patients were further classified into OI subtypes based on Sillence classification[2]. Patients were regarded as type V OI if they had history of hyperplastic callus formation or had radiological apparent calcification of forearm intraosseous membrane [17]. Physical examination revealed hypermobile joints and mild skin hyperextensibility. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethic Committee of PUMCH. Written informed consent was obtained from parents or legal guardians of children younger than 18 years old.
Design of targeted capture panel
A capture panel of targeted DNA was designed by selecting 14 well-known OI-related genes from previous studies and OMIM, which are COL1A1, COL1A2, IFITM5, SERPINF1, CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1, SP7, PLOD2, TMEM38B, BMP1, and WNT1. The panel also included a cluster of 708 possible candidate genes with expression patterns that have been associated with congenital rickets, such as FGF23, PHEX, DMP1, ENP11, CLCN5, and SLC34A3 [18]. Genes related to cartilage problems were also included. The targeted panel included a total of 722 genes, which compromised 9712 exons.
Paneled exome sequencing
A minimum of 3 μg of genomic DNA was extracted from each blood sample and was sheared into 200-bp fragments to construct the DNA library. The DNA fragment was ligated to illumine adaptors with barcoded oligonucleotides using Klenow exonuclease. Pooled, barcoded libraries were clonally amplified using the Illumina systems. The targeted DNA was captured, washed, and recovered using streptavidin-coated magnetic beads. Captured libraries were then subjected to NGS using an Illumina Solexa HiSeq 2000 sequencer as 100-bp paired-end reads, following the manufacturer’s protocol.
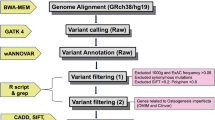
In-depth bioinformatics analysis
Sequence reads acquired from raw data were converted to qseq, which were aligned to the reference human genome using the SOAPaligner program. Single-nucleotide polymorphisms were initially identified by the SOAPsnp program (http://soap.genomics.org.cn/) after filtering polymerase chain reaction duplicates using SAMtools (http://samtools.sourceforge.net/). Sequence variants were reported following the Human Genome Variation nomenclature guidelines (http://www.hgvs.org/mut-nomen/). Insertions or deletions were analyzed using BWA (http://bio-bwa.sourceforge.net/) and GATK programs (https://www.broadinstitute.org/gatk/). Variants were filtered with dbSNP135, 1000 Genome, and HapMap databases as well as an internal control database from the Beijing Genomics Institute. PoplyPhen-2, SIFT (score <0.05), and PhyloP (score >2.5) were used to assess missense variants. Mutations known to cause osteogenesis imperfecta were identified by searching against collagen-related disease database (http://www.le.ac.uk/ge/collagen).
Sanger sequencing validation
The genomic DNA was extracted from peripheral blood using Qiagen Midi Kit (Qiagen, Hilden, Germany). All of the candidate mutations were confirmed by Sanger sequencing following standard protocol. Primers were designed using Primer3 (http;//www.primer3.ut.ee, version 4.0). To ensure the quality of Sanger sequencing, the amplicons were designed to have a boundary around 100 bp away from the mutation. Then, the amplicons (~400 bp) were Sanger sequenced on Applied Biosystems 3730xl capillary sequencer (Applied Biosystems Inc., Foster City, CA, USA) using standard method.
Evaluation of phenotype
The medical history was collected, and physical examination was completed in detail. The previous fractures were documented including site, degree of trauma, frequency, and time of the initiation. Body height was measured with a Harpenden stadiometer and was adjusted to age- and sex-specific Z-scores on the basis of reference data of the Chinese National Centers for Disease Control and Prevention [19]. BMD at lumbar spine and proximal femur was measured by dual-energy x-ray absorptiometry (DXA) (Lunar, GE, USA). BMD phantom scan was measured daily by a DXA instrument and detected no significant machine drifts during the 5-year study. Areal BMD values were converted to age and sex-specific Z-scores using data of previous studies [20, 21].
Statistical analysis
Data that was normally distributed such as age, BMD, and height were presented as mean ± standard deviation, while those of abnormal distribution were expressed as median and quartiles. Difference between two groups (for example, age, gender, inheritance pattern, quality versus quantity defect of type I collagen) was analyzed by Student’s t test. Differences between the three groups (for example, Sillence classification, and genotype of OI) were evaluated using analysis of variance. Qualitative data was compared by the chi-square test. Analysis was also performed by Mann-Whitney U test as appropriate. The statistical analyses were performed using SPSS 21.0 (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
Phenotypes of OI patients
We included 103 patients from 101 unrelated non-consanguineous families. The patients ranged from neonates to 40 years old at assessment, and their average age was 9.9 years old at diagnosis of OI. According to Sillence classification, our cohort includes 29 type I, 39 type III, and 35 type IV OI patients. Sixty-six patients were male, 37 patients were female (Table 1). A positive family history was reported in 23 patients. Type I OI patients had the highest BMD while type III OI patients had the lowest BMD at both lumbar spine and femoral neck (Table 1). Compared with children OI, height Z-scores of adult patients were significantly lower (Z-score −13.6 ± 11.4 in adult versus −2.9 ± 4.1, P = 0.012, respectively). In type III OI, male patients were found to have lower lumbar spine BMD (Z-score −4.4 ± 1.2) than female patients (Z-score − 3.3 ± 1.7, P = 0.046, respectively). Femoral BMD Z-scores were higher in adult than in children (Z-score −6.6 ± 3.5 in children versus −3.5 ± 2.1 in adults, P = 0.041, respectively).
Next generation sequencing summary
The sequencing yield was on average 288× per sample. On average, 547,696 of the reads mapped to the intended targets and 98.79% of the targets had at least 20 reads per base, with an average of 200 reads per base. In order to test the validity, we also tested ten patients with previous sequencing by Sanger analysis. The variation in these patients was identical between NGS and Sanger sequencing.
Identification of mutation
Definite genetic diagnosis was achieved in 90 out of 103 patients. Among the 90 patients, a total of 79 mutations were identified (15 frameshift, 6 nonsense, 40 missense, 15 splice site, 1 new start codon, 1 chromosome translocation, 1 whole gene deletion), 43 of which were novel variants that had not previously been reported (11 frameshift, 5 nonsense, 17 missense, 9 splice site, 1 chromosome translocation; shown in Table 2). The other 52 mutations have been reported in previous studies (4 frameshift, 1 nonsense, 23 missense, 6 splice site, 1 new start codon, and 1 whole gene deletion; shown in Supplemental materials). Nine mutations were found in more than two unrelated families.
Molecular diagnosis
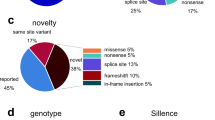
The genetic etiologies found in this cohort were as follows: COL1A1 (n = 37), COL1A2 (n = 29), TMEM38B (n = 3), FKBP10 (n = 3), PLOD2 (n = 1), IFITM5 (n = 9), SERPINF1 (n = 4), and WNT1 (n = 4). The details of the different mutation types are shown in Fig. 1.

Mutation spectrum in Chinese OI patients. a Mutation spectrum in Chinese OI patients by causative genes. b Mutation spectrum of type I OI patients. c Mutation spectrum of type III OI patients. d Mutation spectrum of type IV OI patients. e Mutation spectrum of Chinese osteogenesis imperfecta patients by mutation effects
The majority of the variants were due to COL1A1 and COL1A2 mutations, of which 37 patients had COL1A1 variants, followed by 29 patients with COL1A2 mutations. Fifteen patients had COL1A1 missense mutations, 9 patients had COL1A1 frameshift mutations, 9 patients had splice site mutations, 3 patients had COL1A1 nonsense mutations, and 1 patient harbored whole gene deletions (family 39). A total of 25 patients had COL1A2 missense mutations, with the glycine to serine being the most frequent amino acid substitutions. Three patients had COL1A2 splice site mutations, and one patient had a deletion in chr5:146294373 and chr7:94038710 that resulted in chromosome translocation t(5;8)(q32;q21).
As the third most common cause of OI, IFITM5 mutations have been recognized in nine patients. The recurrent IFITM5 c.-14C>T mutation was found in eight patients, and IFITM5 c.119C>T mutation was found in one patient.
For AR inherited OI, four individuals carried SERPINF1 mutations, three of which were compound heterozygous and one was homozygous mutation. The majority of SERPINF1 variants lead to the production of truncated protein, and the missense mutations were predicted to affect the secondary structure of the protein encoded by SERPINF1.
Mutations in TMEM38B were identified in three patients from two unrelated families. In two affected siblings from a non-consanguineous family, we identified a nonsense mutation that leads to premature termination codon. The c.455-7T>G in TMEM38B was found in another family which led to an insertion of two amino acids p. G152_A153insVL and affected a key domain of the TRIC-B protein.
Genes that are involved in collagen folding and cross-liking (FKBP10, PLOD2) had been identified to cause OI in four patients. Most of the mutations in FKBP10 are predicted to result in loss of function. One patient was identified to be compound heterozygous for splice mutation and missense mutation in PLOD2.
Patients from four families were either reported homozygous (n = 1, c.506dupG) or compound heterozygous (n = 3, [c.110 T>C] + [c.505G>T] and [c.385G>A] + [c.506 G>A], [c.506 G>A] + [c.506dupG]) for alterations of the WNT1 gene. All of the novel missense mutations were predicted to affect the N-terminal domain of the Wnt1 protein.
Genotype-phenotype correlation
Mutations leading to an early stop codon or frameshift in COL1A1 were regarded as the quantitative group (haploinsufficiency). Mutations causing amino acid substitutions in the triple helical domain of COL1A1 or COL1A2 were classified into the collagen qualitative defect group. As the effect of splice site mutation was difficult to predict, we did not include splice site mutations in the genotype-phenotype correlation of type I collagen.
BMD
AR inherited patients were prone to have lower lumbar spine BMD (Z-score − 3.6 ± 2.1) compared with AD inherited patients (Z-score −2.6 ± 1.9, P = 0.05). Compared with the quantitative change in collagen, patients with triple helical mutations had significantly lower femoral BMD (−3.1 ± 2.1 for quantitative and −5.0 ± 3.2 for qualitative, P = 0.034, Table 3).
Height
Patients with qualitative mutations were significantly shorter in height than patients with quantitative defects (−1.2 ± 4.8 for quantitative and −6.5 ± 8.2 for qualitative, P = 0.022). There was no difference between quantitative and any qualitative mutation in the α1- versus α2-chain. Nor was there any difference in the inheritance pattern (Table 3).
Blue sclera
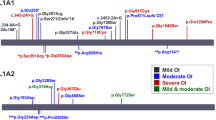
Patients with AD OI had blue sclera significantly more frequently than patients with AR OI (71.1% in AD versus 26.7% in AR, P = 0.001). All patients with haploinsufficiency of type I collagen had blue sclera, compared with 75% of qualitative collagen defect patients. The position of glycine within a1(1) triple helix domain was associated with the presence of blue sclera (Table 3). There is a relationship between the triple helical position of glycine mutations in COL1A1 and COL1A2 and the manifestation of blue sclera and dentinogenesis imperfecta, from the N- to C-terminal. All patients with mutations affecting the N-terminal of Gly154 (p.G332) had blue sclera (Fig. 2).

Relationship between the triple helical position of glycine mutations in COL1A1 and COL1A2 and the manifestation of blue sclera and dentinogenesis imperfecta, from the N- to C-terminal. The colored boxes suggest that all patients with mutations affecting N-terminal of Gly154(p.G332) had blue sclera with an absence of dentinogenesis imperfecta
Dentinogenesis imperfecta
DI was more prevalent in patients with collagen qualitative defects than quantitative defects (0 versus 27.5%, P = 0.023). Concerning N- to C-terminal location, none of the individuals with helical glycine substitutions in the N-terminal of p.Gly332 in the α1-chain had DI. The presence of DI was not associated with OI-type inheritance pattern or types of defects in type I collagen. No significant difference was found in patients who had glycine mutations in the triple helical domain between COL1A1 and COL1A2 (Table 3).
Discussion
To the best of our knowledge, our study was the largest OI cohort in Asian patients. The results of this study outlined a significant mutation spectrum in Chinese patients. By using a self-designed panel with 722 candidate genes, we identified 81 variants in 90 probands which included 45 novel variants.
The accurate and reliable molecular diagnosis of OI is important, because it enables appropriate therapeutic interventions and facilitates a more precise description of prognosis. Our panel sequenced 722 genes spanning 9712 exons simultaneously. The depth of sequencing coverage was high for over 200× of the amplicons, indicating that the majority of amplicons were covered sufficiently. In total, the panel was able to identify 90 probands with gene mutations in the patients analyzed (detection rate 87.4%). While further validation in a larger population is needed, our panel in itself proves the versatility of NGS for variant detection in OI patients.
Our medical center is a tertiary referral center in China, and therefore, our study includes more patients with moderate and severe types of OI, especially in adult patients. Compared with children and adolescent OI patients, adult patients were found to be shorter than people of the same age. This was similar to what was reported by Lindahl et al., and may be related to the existence of multiple fractures, leading to severe skeletal deformities and a shorter statue [22]. All of the clinical information was collected before the initiation of treatment with bisphosphonates. Compared with children and adolescents, the average interval of diagnosis for adults was significantly longer, and therefore likely to result in more severe phenotypes to be present in adults and suggesting that early diagnosis is of great importance in these patients [22]. Moreover, type III and type IV OI are more prevalent in adult patients. To limit this bias, we further conducted analyses on different types of OI.
Similar to previous studies, COL1A1 and COL1A2 mutations were dominant in our cohort [23]. However, the percent of COL1A1 and COL1A2 mutations was lower than expected, accounting for only 73.3% of all variations. We further analyzed the gene mutations that were observed in different types of OI and found that COL1A1 and COL1A2 explained 81% of type I OI and 71% of moderate to severe OI. The detection rate in moderate and severe OI is similar to previous studies, whereas the detection rate in type I OI is lower. The reason for this may stem from the definition of type I OI, as Bardai et al. used the definition of typical type OI, which was defined as having both extra-skeletal characteristics and bone fragility [24]. In contrast, we used only bone fragility to differentiate OI types and included patients with TMEM38B, PLOD2, and IFITM5 in our type I OI analysis. This is similar to a recent study, which found that 12 out of 28 individuals with mild bone fragility and an absence of extraskeletal manifestation, who were identified to have gene mutations, carried mutations in COL1A1 and COL1A2 [25].
As for genotype-phenotype analysis, we confirmed that patients with collagen qualitative defects had lower BMDs than patients with collagen quantitative defects, especially at the femoral neck. As previous studies mainly focused on the axial skeleton and BMD is known to vary by location in OI patients, our results provide valuable information about the appendicular skeleton of OI patients [26,27,28]. Consistent with previous studies, we found that patients with triple helical mutations were taller than patients with haploinsufficiency of type I collagen. Similarly, previous studies have reported that the triple helical position of glycine in COL1A1 was associated with the presence of blue sclera [22, 28], which we also observed, as all patients with mutations affecting the first 152 amino acids in COL1A1 presented blue sclera with the absence of DI.
As the third most common mutation in our cohort, mutations in IFITM5 were detected in nine patients, eight with c.-14C>T and one with c.119C>T. The patient with c.-14C>T was quite similar to what was reported previously in large phenotype variability, typical calcification, and hypercallus formation. Similar to previous studies, patients with IFITM5 c.119C>T had severe skeletal deformities [29]. However, we observed no significant difference in BMD, height, sclera hue, and dentinogenesis imperfecta between type V and other types of OI. The heterogeneity of IFITM5, which has been shown to be related to OI, may explain the similarity in these clinical phenotypes.
To date, there have been limited studies focusing on AR OI in Chinese patients. We successfully detect 15 patients with AR OI. As such, here, we further extend the known mutation spectrum to include WNT1, SERPINF1, PLOD2, FKBP10, and TMEM38B, thus contributing to a better understanding of the genotype that is present in Chinese patients. In our cohort, SERPIFN1 and WNT1 mutations were the most common causes of AR OI. This is different from what was reported by Bardai et al., who found SERPINF1 and CRTAP to be more prevalent in Canadian OI patients [24]. As our study focused on a Chinese population, we attributed this discrepancy to a difference in ethnicity. In addition, we further detected three patients with FKBP10 and one patient with PLOD2 mutation. In contrast to previous studies, which reported that FKBP10 and PLOD2 were responsible for Bruck syndrome, we found that joint contracture was absent in patients with a FKBP10 mutation but was present in a patients with a PLOD2 mutation [30].
Compared with AD OI patients, AR OI tends to have more severe skeletal deformities and were less likely to have blue sclera. This finding is similar to what was reported previously. The reason for this phenomenon has yet to be defined; however, we hypothesize that this may be due to the fact that AD OI is usually associated with collagen defects, whereas AR OI tends to affect the post-translational process of collagen or bone mineralization.
Despite successful detection of most mutations in our patients, we failed to identify any related gene mutation in 12 patients. This may have occurred because probands carry mutations in genes not targeted by this study given that we chose to examine the most highly implicated genes in skeletal dysplasia causation. For instance, PLS3, a gene that encodes a protein in the formation F-actin and associated with juvenile osteoporosis, was not included at the time we designed our panel [31]. One of the test-negative patients was later found to have a large deletion involving intron 9 to the 3′-UTR (E10-E16 del) in PLS3. For those patients without a detected mutation, further whole exome sequencing may be able to resolve some of these possibilities [32]. Other possible reasons for the detection of false negatives may be poor coverage due to high GC content and the difficulties associated with the detection of large deletions [33]. Alternative methods like comparative genome hybridization or multiplex ligation-dependent probe amplification would be needed to investigate further [34].
Conclusions
The achievement of more accurate diagnosis would greatly benefit the management of patients with congenital skeletal dysplasia. In this study, we present the largest OI sample in China as screened by NGS. In particular, we successfully identified 81 variants, which included 45 novel variants. The novel variants that were identified in these Chinese OI patients enrich the known pathogenic spectrum of OI. Finally, our analysis of genotype-phenotype correlation helps make a differential diagnosis and helps predict the prognosis of OI.
Change history
02 November 2017
In Table 2:
References
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387:1657–1671
Sillence DO, Senn A, Danks DM (1979) Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16:101–116
Van Dijk FS, Sillence DO (2014) Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164A:1470–1481
Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E (2012) First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact 12:183–188
Semler O, Garbes L, Keupp K et al (2012) A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91:349–357
Homan EP, Rauch F, Grafe I et al (2011) Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Miner Res Off J Am Soc Bone Miner Res 26:2798–2803
Baldridge D, Schwarze U, Morello R et al (2008) CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat 29:1435–1442
Barnes AM, Carter EM, Cabral WA et al (2010) Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N Engl J Med 362:521–528
Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH (2010) Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet 86:389–398
Kelley BP, Malfait F, Bonafe L et al (2011) Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res Off J Am Soc Bone Miner Res 26:666–672
Laine CM, Joeng KS, Campeau PM et al (2013) WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med 368:1809–1816
Lapunzina P, Aglan M, Temtamy S et al (2010) Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet 87:110–114
Symoens S, Malfait F, D'Hondt S, Callewaert B, Dheedene A, Steyaert W, Bachinger HP, De Paepe A, Kayserili H, Coucke PJ (2013) Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis 8:154
Lindert U, Cabral WA, Ausavarat S et al (2016) MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun 7:11920
Donahue LR, Chang B, Mohan S et al (2003) A missense mutation in the mouse Col2a1 gene causes spondyloepiphyseal dysplasia congenita, hearing loss, and retinoschisis. J Bone Miner Res Off J Am Soc Bone Miner Res 18:1612–1621
Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, Soong R (2012) Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol 71:5–14
Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ (2000) Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res Off J Am Soc Bone Miner Res 15:1650–1658
Ruppe MD, Brosnan PG, Au KS, Tran PX, Dominguez BW, Northrup H (2011) Mutational analysis of PHEX, FGF23 and DMP1 in a cohort of patients with hypophosphatemic rickets. Clin Endocrinol 74:312–318
Li H, Ji CY, Zong XN, Zhang YQ (2009) Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Chin J Pediatr 47:487–492
Xu H, Zhao Z, Wang H, Ding M, Zhou A, Wang X, Zhang P, Duggan C, Hu FB (2013) Bone mineral density of the spine in 11,898 chinese infants and young children: a cross-sectional study. PLoS One 8:e82098
Khadilkar AV, Sanwalka NJ, Chiplonkar SA, Khadilkar VV, Mughal MZ (2011) Normative data and percentile curves for dual energy X-ray absorptiometry in healthy Indian girls and boys aged 5-17 years. Bone 48:810–819
Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, Kindmark A (2015) Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Human Genet: EJHG 23:1112
Marini JC, Forlino A, Cabral WA et al (2007) Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 28:209–221
Bardai G, Moffatt P, Glorieux FH, Rauch F (2016) DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int 27:3607–3613
Bardai G, Ward LM, Trejo P, Moffatt P, Glorieux FH, Rauch F (2017) Molecular diagnosis in children with fractures but no extraskeletal signs of osteogenesis imperfecta. Osteoporos Int
Rauch F, Lalic L, Roughley P, Glorieux FH (2010) Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur J Human Genet: EJHG 18:642–647
Rauch F, Land C, Cornibert S, Schoenau E, Glorieux FH (2005) High and low density in the same bone: a study on children and adolescents with mild osteogenesis imperfecta. Bone 37:634–641
Rauch F, Lalic L, Roughley P, Glorieux FH (2010) Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res Off J Am Soc Bone Miner Res 25:1367–1374
Hoyer-Kuhn H, Semler O, Garbes L, Zimmermann K, Becker J, Wollnik B, Schoenau E, Netzer C (2014) A nonclassical IFITM5 mutation located in the coding region causes severe osteogenesis imperfecta with prenatal onset. J Bone Miner Res Off J Am Soc Bone Miner Res 29:1387–1391
Zhou P, Liu Y, Lv F, Nie M, Jiang Y, Wang O, Xia W, Xing X, Li M (2014) Novel mutations in FKBP10 and PLOD2 cause rare Bruck syndrome in Chinese patients. PLoS One 9:e107594
van Dijk FS, Zillikens MC, Micha D et al (2013) PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med 369:1529–1536
Rauch F, Lalic L, Glorieux FH, Moffatt P, Roughley P (2014) Targeted sequencing of a pediatric metabolic bone gene panel using a desktop semiconductor next-generation sequencer. Calcif Tissue Int 95:323–331
Benjamini Y, Speed TP (2012) Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40:e72
van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, Rohrbach M, Symoens S, Sistermans EA, Pals G (2012) EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta. Eur J Human Genet: EJHG 20:11–19
Acknowledgements
We wish to thank the patients and their family for participating in this study. We thank the National Natural Science Foundation of China and the Tianjin Municipal Science and Technology Special Funds for Enterprise Development for the support of funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the Ethic Committee of PUMCH.
Conflicts of interest
None.
Additional information
This study was supported by the National Natural Science Foundation of China (81570802) and Tianjin Municipal Science and Technology Special Funds for Enterprise Development (14ZXLJSY00320).
A correction to this article is available online at https://doi.org/10.1007/s00198-017-4250-6.
Electronic supplementary material
ESM 1
(DOCX 47 kb)
Rights and permissions
About this article

Cite this article
Liu, Y., Asan, Ma, D. et al. Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int 28, 2985–2995 (2017). https://doi.org/10.1007/s00198-017-4143-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-017-4143-8