
Abstract
Summary
We have sought the molecular diagnosis of OI in 38 Brazilian cases through targeted sequencing of 15 candidate genes. While 71% had type 1 collagen-related OI, defects in FKBP10, PLOD2 and SERPINF1, and a potential digenic P3H1/WNT1 interaction were prominent causes of OI in this underrepresented population.
Introduction
Defects in type 1 collagen reportedly account for 85–90% of osteogenesis imperfecta (OI) cases, but most available molecular data has derived from Sanger sequencing-based approaches in developed countries. Massively parallel sequencing (MPS) allows for systematic and comprehensive analysis of OI genes simultaneously. Our objective was to obtain the molecular diagnosis of OI in a single Brazilian tertiary center cohort.
Methods
Forty-nine individuals (84% adults) with a clinical diagnosis of OI, corresponding to 30 sporadic and 8 familial cases, were studied. Sixty-three percent had moderate to severe OI, and consanguinity was common (26%). Coding regions and 25-bp boundaries of 15 OI genes (COL1A1, COL1A2, IFITM5 [plus 5′UTR], SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, PLOD2, BMP1, SP7, TMEM38B, WNT1, CREB3L1) were analyzed by targeted MPS and variants of interest were confirmed by Sanger sequencing or SNP array.
Results
A molecular diagnosis was obtained in 97% of cases. COL1A1/COL1A2 variants were identified in 71%, whereas 26% had variants in other genes, predominantly FKBP10, PLOD2, and SERPINF1. A potential digenic interaction involving P3H1 and WNT1 was identified in one case. Phenotypic variability with collagen defects could not be explained by evident modifying variants. Four consanguineous cases were associated to heterozygous COL1A1/COL1A2 variants, and two nonconsanguineous cases had compound PLOD2 heterozygosity.
Conclusions
Novel disease-causing variants were identified in 29%, and a higher proportion of non-collagen defects was seen. Obtaining a precise diagnosis of OI in underrepresented populations allows expanding our understanding of its molecular landscape, potentially leading to improved personalized care in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) is a clinically and genetically heterogeneous group of hereditary bone dysplasias leading to bone fragility, deformities, and short stature [1, 2]. The severity of bone fragility ranges widely from individuals with few fractures, no deformities, and normal stature, to those with more than a hundred fractures and incapacitating bone deformities. Clinical diagnosis of OI is often made upon a history of multiple low-trauma fractures starting in childhood and low bone mass as assessed by DXA and can be aided by family history and extraskeletal features such as blue sclerae, dentinogenesis imperfecta, and hearing impairment, if present [1]. However, in face of outstanding heterogeneity, clinical diagnosis is not always straightforward, potentially resulting in missed diagnosis and inadequate counseling and follow-up.
Until the advent of massively parallel sequencing (MPS), achieving a molecular diagnosis of OI was equally challenging. Autosomal dominant OI most commonly results from quantitative or qualitative defects in type 1 collagen, but automated Sanger sequencing of the coding regions of COL1A1 and COL1A2, with 51 and 52 exons, respectively, is burdensome and has been somewhat restricted to major research centers. Moreover, the elucidation of rarer, mainly autosomal recessive cases of OI has led to the identification of several other causative factors, unraveling its complex molecular heterogeneity [3]. Currently, at least five different pathophysiological mechanisms involving 17 candidate genes are considered to lead to OI: (a) defects in the synthesis or structure of type 1 collagen due to variants in COL1A1 or COL1A2; (b) altered collagen posttranslational modification due to variants in CRTAP, P3H1 [previously known as LEPRE1] or PPIB; (c) compromised collagen processing and assembly due to variants in SERPINH1, FKBP10, PLOD2, or BMP1; (d) impaired bone mineralization due to variants in IFITM5 or SERPINF1; and (e) impaired osteoblast differentiation and function due to variants in SP7, TMEM38B, WNT1, CREB3L1, SPARC, or MBTPS2 [2]. Importantly, there is a considerable overlap in clinical presentation arising from these different molecular defects, meaning that a molecular cause cannot be assumed solely based on clinical elements, with the notable exception of autosomal dominant IFITM5-related OI, which can have distinctive clinical and radiologic features [4, 5]. Therefore, molecular analysis becomes essential for a precise diagnosis of OI.
The relative frequency of the different genetic causes of OI has been mainly ascertained based on curated molecular data compiled into the OI variant database [6, 7], and on published reports of the molecular diagnosis of OI in major research centers cohorts, mostly deriving from Sanger sequencing-based candidate gene approaches in developed countries [8,9,10]. Collectively, a notion that defects in COL1A1 and COL1A2 account for 85–90% of OI cases and that autosomal dominant IFITM5-related OI would be the main form of non-collagen OI has prevailed [2, 11]. Since 2017, studies implementing MPS gene panel approaches in underrepresented populations have revealed diverse relative frequencies of the genetic causes in these settings [12,13,14,15]. The application of MPS for the molecular diagnosis of OI is not only advantageous in terms of throughput, but also allows for systematic and simultaneous analysis of all OI candidate genes, potentially improving accuracy in the attribution of pathogenicity.
The aim of this study was to identify the molecular diagnosis in Brazilian cases of OI using an MPS gene panel, hoping to obtain a precise diagnosis of OI that could refine the clinical management of patients and their relatives and provide a better understanding of the genetic architecture of OI in the underrepresented and highly admixtured Brazilian population.
Materials and methods
Subjects
Brazilian patients with a clinical diagnosis of OI followed at the Endocrine Division of Hospital das Clinicas HCFMUSP, Faculdade de Medicina, Universidade de Sao Paulo, a major tertiary academic hospital in São Paulo, Brazil, were invited to participate and included following informed consent. The study was carried out according to the principles of the Declaration of Helsinki and was approved by the local ethics committee (Comissão de Ética para Análise de Projetos de Pesquisa, Hospital das Clinicas HCFMUSP, CAAE #43319415.2.0000.0068). Both sporadic and familial cases of OI were included. In familial cases, provided that samples could be obtained at the beginning of the study, all affected family members were individually analyzed through the MPS gene panel, aiming to identify modifying variants that could explain phenotypic variability within each family.
Clinical diagnosis of OI was based on the history of multiple fragility fractures since childhood and low bone mass (DXA Z-score < − 2.0 in the lumbar spine ot total body), associated or not to bone deformities, short stature (Z-score < − 2.0), family history, and current or previous blue sclerae or dentinogenesis imperfecta. A detailed history of nonvertebral fractures was obtained from all subjects, and vertebral fractures were ascertained through lateral thoracic and lumbar spine radiographs. OI severity was attributed according to the criteria proposed by Mrosk and colleagues in 2018, a phenotypic scoring system that integrates fracture frequency (1 to 3 points), total number of fractures (1 to 3 points) and the presence of vertebral fractures (1 point), bowing (up to 4 points) and scoliosis (1 point) [14]. Total scores between 1 and 4 render classification as mild OI, between 5 and 8 as moderate OI and from 9 to 12 as severe OI. When available, retrospective data pertaining to audiometric testing and cardiac evaluation by echocardiography were also collected.
DNA extraction
DNA was extracted from peripheral blood leukocytes using an in-house variation of the salting-out method. All DNA samples were submitted to quality control before further genetic analyses.
Massively parallel sequencing
A gene panel was designed including all coding regions and 25-bp boundaries of 15 OI candidate genes, based on human reference genome GRCh37: COL1A1 (Ensembl transcript ENST00000225964), COL1A2 (ENST00000297268), CRTAP (ENST00000320954), P3H1 (ENST00000236040), PPIB (ENST00000300026), SERPINH1 (ENST00000524558), FKBP10 (ENST00000321562), PLOD2 (ENST00000282903), BMP1 (ENST00000306385), IFITM5 (ENST00000382614), SERPINF1 (ENST00000254722), WNT1 (ENST00000293549), TMEM38B (ENST00000374692), SP7 (ENST00000536324), and CREB3L1 (ENST00000529193). The 5′untranslated region of IFITM5 was also included.
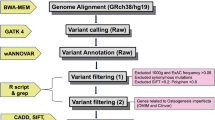
Targeted regions were captured using specifically designed probes (3× minimum tiling) and the SureSelectXT kit (Agilent Technologies, Santa Clara, CA), according to manufacturer’s instructions. Barcoded libraries were prepared from 3 μg of genomic DNA and sequenced on a NextSeq500 system (Illumina, San Diego, CA). Resulting paired-end reads were aligned to the human reference genome GRCh37/hg19 using Burrows-Wheeler alignment tool (BWA). Variant calling was performed with Platypus in all BAM files, and the resulting variants were annotated with ANNOVAR.
Variant filtering was performed to prioritize rare variants (allele frequency < 0.5% in all three databases 1000 Genomes Project [16], ExAC [17], and the Online Archive of Brazilian Mutations ABraOM [18]) expected to impact on protein function, including loss-of-function variants resulting in stop gain, frameshift, or splice site abnormalities, and nonsynonymous variants predicted to be deleterious according to SIFT [19], PolyPhen2 [20], or CADD [21]. Additional data on variants of interest were obtained from ClinVar, the Human Gene Mutation Database (HGMD), OMIM, PubMed, and the Osteogenesis Imperfecta Variation Database (OIVD) [6, 7].
CNV analysis
In cases where point variants or small indels were not identified, copy number variation (CNV) analysis of MPS data was performed using the COpy Number Targeted Resequencing Analysis (CONTRA) tool [22]. Potential CNVs were verified by BAM file visualization using the Integrative Genomics Viewer (Broad Institute). Verified CNVs were further analyzed by SNP array using CytoSNP-850 K arrays (Illumina). Briefly, DNA amplification, hybridization, staining and washing were performed according to manufacturer’s instructions, and arrays were scanned using the iScan System (Illumina). Raw data were analyzed using the BlueFuse Multi v1.1 software (Blue Gnome). CNVs confirmed by SNP array were interrogated in UCSC genome browser and the database of genomic variants (DGV) in order to retrieve previous reports.
Sanger sequencing
Automated Sanger sequencing was carried out to confirm variants of interest identified in the MPS gene panel. Starting from 50 ng of genomic DNA, PCR-amplified regions were purified enzymatically with Illustra ExoProStar (GE Healthcare Life Sciences, Little Chalfont, UK) and sequenced using the BigDye Terminator v.3.1 kit (Thermo Fisher Scientific, Waltham, MA) on an ABI 3130 × 1 automated DNA sequencer (Thermo Fisher). Oligonucleotide sequences and reaction conditions are available upon request.
Segregation analysis
First-degree relatives of study subjects were invited to participate. After informed written consent, a detailed medical history was obtained aiming to identify a history of fragility fractures, previous diagnosis of osteoporosis or low bone mass, blue sclerae, dentinogenesis imperfecta, or cardiopathies. Segregation analysis was performed using Sanger sequencing, as described above, when family samples were available. In isolated cases with a suspected autosomal dominant de novo variant, the variant was only considered to have segregated if it was not found in both unaffected parents. In cases with suspected autosomal dominant inheritance, if the variant was found in an affected parent, it was also considered to segregate, but if one of the parents was unavailable for study and the variant was not found in the other (unaffected) parent, the analysis was deemed inconclusive. In cases with suspected autosomal recessive defect and a history of consanguinity where a homozygous variant was identified, if one of the parents was unavailable but the other carried the heterozygous variant, segregation was affirmed. In familial cases, when affected individuals had the variant and unaffected ones did not, segregation was also affirmed.
Association between molecular findings and clinical features
In order to explore associations between molecular findings and clinical characteristics such as disease severity, blue sclerae, dentinogenesis imperfecta, impaired hearing, and echocardiographic abnormalities, Pearson’s Chi-squared was used in PASW Statistics v.17 (IBM). When > 20% of cells had values lower than the expected minimum, violating the test’s assumptions, Fisher’s exact test (2 × 2 tables) was used. An association was considered significant when p < 0.05.
Results
Molecular analysis with the MPS gene panel was carried out in 49 subjects, corresponding to 30 sporadic and 8 familial cases, rendering a total of 38 index cases. Clinical characteristics of the full cohort are presented on Table 1. Notably, adults composed 82% of the cohort, and the median age of subjects was 24 years (range 7–69 years). In terms of disease severity, 37% had mild OI, 30% had moderate OI, and 33% had severe OI. A higher proportion of mild OI was seen among familial cases (74%) in comparison to sporadic cases (13%), who mostly had moderate to severe disease.
Ten out of 38 cases (26%) had a history of consanguinity. Thirty-two percent of cases were originally from Sao Paulo while 58% were from other states in Brazil, and, overall, 24% were originally from small cities (population < 50,000 inhabitants).
The mean sequencing coverage ranged from 354 to 1382×, and in all samples, > 99% targeted regions were sequenced > 50×. Altogether, 42 disease-associated variants were identified (Table 2). Twenty-three of these have already been reported in association with OI, while 19 are novel variants. Point variants and small indels were confirmed by Sanger sequencing, with the exception of the PLOD2 p.(Trp561*) variant detected in 386 of 853 MPS readings in Pt 6, which, despite several attempts, could not be PCR-amplified for traditional sequencing. Seventy-four percent of identified variants were classified as pathogenic or likely pathogenic (57% and 17%, respectively) according to the ACMG-AMP guidelines [23], and 26% were classified as variants of uncertain significance (VUS). Most VUS were identified in combination, and in four cases, the molecular diagnosis was solely attributed to single or combined VUS (Pt 12, Pt 14, Pt 20, and F4).
A molecular diagnosis was obtained in 37 cases (97%). Heterozygous type 1 collagen defects were identified in 71% of cases, 47% in COL1A1 (n = 18) and 24% in COL1A2 (n = 9). All familial cases had collagen defects. In three collagen-related OI cases, a combination of variants was found: Pt 9 and F6 had combinations of two COL1A1 variants and F4 had a combination of two COL1A2 variants (Table 2). Of note, one sporadic case (Pt 14) and 3 familial cases (F3, F6, and F7) had a history of consanguinity and, still, heterozygous collagen defects were causing OI. In 10 sporadic cases, variants were found in non-collagen candidate genes: IFITM5 (n = 1, Pt 23), P3H1 (n = 1, Pt 8), SERPINF1 (n = 2, Pt 1 and Pt 5), FKBP10 (n = 2, Pt 3 and Pt 12), PLOD2 (n = 2, Pt 6 and Pt 20), TMEM38B (n = 1, Pt 16), and a combination of WNT1 + P3H1 (n = 1, Pt 17). One sporadic case remained without a molecular diagnosis (Pt 15).
The identified heterozygous variants in COL1A1 and COL1A2 were dispersed along the proteins, with variable phenotypic expression (Fig. 1). Forty-two percent of COL1A1 variants and 88% of COL1A2 variants were glycine substitutions. Two variants recurred in the cohort, in apparently unrelated cases: COL1A1 c.334-9A > G was found in Pt 30 and F1, and COL1A2 p.(Gly772Ser) was found in Pt 13, Pt 25 and F3. Individuals bearing the COL1A1 c.334-9A > G variant presented with mild OI; however, clinical variability was remarkable among bearers of COL1A2 p.(Gly772Ser): while Pt 13 had moderate OI characterized by 50 fractures and severe short stature (Z = − 5.7) at 31 years of age, 48-year-old F3d had very mild OI with only 1 fracture and normal stature (Z = − 1.6). Along similar lines, OI severity was discordant among individuals F2a and F2b bearing the COL1A2 p.(Gly319Arg) variant. In all these cases, additional sequence variants in OI candidate genes that could be modifying skeletal fragility and, thus, help explain phenotypic variability were not found.

Representation of variants identified in COL1A1 and COL1A2. Proteins are represented by gray bars, and numbers at the ends denote the initial and final residues, respectively. Variants are shown according to their relative position in the protein; the color denotes the severity of OI with which the variant was associated in the cohort, according to the legend. Above the bars are single variants identified, and below the bars are those identified in combination in the same case; the # marker preceding the variant name denotes combination pairs
The 10 sporadic cases with non-collagen defects had severe (n = 8) and moderate (n = 2) OI. In five cases, homozygous point variants were found in SERPINF1 (Pt 1 and Pt 5), FKBP10 (Pt 3 and Pt 12) and P3H1 (Pt 8); only Pt 1 did not have a history of consanguinity but his parents were both from a 11,000 inhabitants town where other OI cases resulting from the same SERPINF1 defect have been described [24]. Notably, the P3H1 c.[1080 + 1G > T];[1080 + 1G > T] variant identified in Pt 8 has been previously reported to be relatively prevalent in African Americans [25]. In two cases, without a history of consanguinity, compound heterozygous defects in PLOD2 were identified (Pt 6 and Pt 20), and in only one case (3%), the heterozygous IFITM5 c.-14C > T variant was found.
In Pt 16, born to consanguineous parents, a homozygous deletion involving whole exons 1 and 2 of TMEM38B was found. Analysis of MPS data with CONTRA indicated a CNV in the TMEM38B locus in chromosome 9, and visualization of BAM files in IGV confirmed the absence of reads for exons 1 and 2. The homozygous deletion from chr9:108,449,986 to chr9:108,474,679 was further confirmed by SNP array. Deletion of exons 1 and 2 of TMEM38B has already been described as the cause of OI in an Albanian patient [26].
One case of non-collagen OI stood out for its peculiar digenic findings. In Pt 17, who was born to nonconsanguineous parents and had severe OI, four heterozygous variants were found in two different candidate genes, two in P3H1 and two in WNT1, both genes associated to recessive OI (Fig. 2). These variants are absent or very rare in the population and predicted to impact on protein function. Segregation analysis revealed that in each gene, each variant was inherited from one of his unaffected parents, configuring a double compound heterozygote, and, potentially, a digenic cause of OI.

Potential digenic defect involving P3H1 and WNT1. Imaging of Pt 17 through total body DXA scanning in bone densitometry (upper left panel), anteroposterior spine radiography (upper central panel), and lower limb scanning (upper right panel) reveals severe bone involvement with multiple deformities and scoliosis. The table shows characteristics of the heterozygous variants identified in P3H1 and WNT1, with a peculiar inheritance pattern in double compound heterozygosity. PP2, PolyPhen2; CADDp, PHRED-like scaled C-score according to the CADD framework; D, deleterious; n/a, not available; HTZ, heterozygous; WT, wild-type
Finally, we sought to associate molecular findings to clinical characteristics, such as OI severity, blue sclerae, and dentinogenesis imperfecta (for which information was available in all subjects), hearing loss (audiometry available in 34/49 subjects) and cardiac abnormality (echocardiogram available in 39/49 subjects). Comparing collagen to non-collagen OI, a significant association with disease severity was found, with a higher prevalence of moderate to severe OI in non-collagen defects (p = 0.008, Fisher’s exact test). Within collagen defects, COL1A1 variants were more frequently associated to blue sclerae (p = 0.006, Fisher’s exact test), and glycine substitutions were more frequently associated to dentinogenesis imperfecta (p = 0.003, Fisher’s exact test). Other significant associations were not found.
Discussion
In a Brazilian tertiary service cohort, molecular analysis with an MPS gene panel including 15 candidate genes was able to identify a molecular diagnosis of OI in 37 out of 38 cases (97%). This high diagnostic yield may derive from the high-throughput approach taken and is comparable to that of recent reports implementing MPS to the molecular diagnosis of OI [12, 14, 15]. Even though most cases (71%) had COL1A1 or COL1A2 defects, a substantial 26% of cases had non-collagen OI, and defects in FKBP10, PLOD2, and SERPINF1 were prominent causes.
Subjects presented with heterogeneous clinical features, ranging from a few fractures without bone deformities to > 150 fractures and multiple deformities, but moderate to severe presentation predominated (63%). In fact, 53% had impaired mobility. The clinical setting of a referral tertiary center justifies this patient profile (referral bias), which may not necessarily reflect the severity of OI in the Brazilian population, in which large clinical and molecular studies of OI are still lacking. Another characteristic of the studied cohort is being composed predominantly by adults, with only 9 out of 49 individuals younger than 18 years of age. Most published OI studies have been performed in children and adolescents, since the disease tends to manifest more pronouncedly at these stages [8, 27]. This peculiarity of the present study may bring additional information about the clinical evolution into adulthood for the identified genetic causes of OI. While we believe that bridging the gap on adult OI in the literature is essential, we must acknowledge that historical findings in our cohort, such as number of fractures, scleral hue, and dental health during childhood and adolescence, could have been affected by recall bias, as their ascertainment depended on patient recollection.
Twenty-six percent of our cases reported a history of consanguinity. The overall prevalence of consanguineous unions in Brazil has been estimated at 1.6%, with high variability between states [28]. Even though all our cases were followed in our institution in Sao Paulo, 58% originated from other states in Brazil and from small cities in rural settings in 24%. It has been proposed that consanguinity in Brazil results mainly from geographical and social isolation, rather than cultural or religious tradition [28,29,30]. In fact, in our study, 80% of cases bearing homozygous variants were originally from small cities in remote locations.
The advent of MPS has greatly facilitated molecular diagnosis in several genetic diseases, including OI [31, 32], but also resulted in the identification of a large number of variants to which biological or clinical significance is difficult to assign [23, 33]. In this sense, the ACMG-AMP guidelines have sought to establish stringent criteria upon which the pathogenic potential of allelic variants can be derived for diagnostic reports [23]. Here, in 4 of the 37 cases for which a molecular diagnosis was attributed, single or combined VUS were found (Pt 12, Pt 14, Pt 20, and F4). In these cases, nonsynonymous variants were mainly classified as VUS because they had never been associated to OI before (novel) and segregation analysis was incomplete or unavailable. Nevertheless, several elements supported a pathogenic role: they were very rare or absent in population databases, predicted to be deleterious by in silico tools, and, in the case of two collagen variants (Pt 14 and F4), were glycine substitutions, which generally have great impact on the formation of type 1 collagen triple helix [34]. If these diagnoses were to be disregarded, the success rate of molecular diagnosis would drop from 97 to 87%. However, in view of supportive elements described above, it was considered that these VUS were potentially pathogenically implicated in these cases, and, therefore, a molecular diagnosis was attributed. It should be noted that out of the nine largest OI molecular studies published between 2015 and 2019, which will be detailed below, only one classified variants according to the ACMG-AMP guidelines, and also reported VUS in the diagnostic assignment [14].
In only one case (Pt 15) the molecular diagnosis was not identified. The clinical diagnosis of OI in this individual was reviewed and confirmed. It is unlikely that a molecular diagnosis was missed due to technical reasons since target region coverage in this sample was > 50×. It is possible, however, that the variant lies in non-coding areas (intron, promoter), which were not analyzed, or that it results from novel genetic mechanisms in OI or from variation in SPARC or MBTPS2, two novel OI candidate genes that were not represented in the MPS gene panel [35, 36].
In accordance with the literature, all mild OI cases in this cohort were due to collagen defects. Interestingly, about half of the variants leading to mild OI were quantitative type 1 collagen defects, while the other half were qualitative defects caused by non-synonymous variants, including glycine substitutions. This finding corroborates the recent molecular understanding that mild OI is not always associated to haploinsufficiency of one of the type 1 collagen chains (Forlino & Marini, 2016). Indeed, Lindahl et al. analyzed clinical and genetic characteristics of 99 patients with mild OI due to defects in COL1A1 or COL1A2, reporting 62% quantitative and 32% qualitative defects [9].
Still in regard to COL1A1 and COL1A2 variants identified here, no correlation was observed between the position of the variant and clinical presentation, since variants associated with mild, moderate and severe OI were dispersed throughout the proteins (Fig. 1). Additionally, remarkable phenotypic variability was observed among individuals bearing a similar collagen variant. In family 2, in which COL1A2 p.(Gly319Arg) was identified, subject F2a presented with mild OI while subject F2b had moderate presentation. In the 6 carriers of the recurrent COL1A2 p.(Gly772Ser) variant (Pt 13, Pt 25, and F3a to d), clinical variability was also striking. We did not find evident modifying variants in all 15 candidate genes that could explain this phenotypic variability, which also remains unexplained in the literature.
Notably, in families 3, 6 and 7 and in Pt 14 a history of consanguinity was reported, but causative heterozygous variants in COL1A1 or COL1A2 were identified, highlighting the advantage of an unbiased diagnostic approach through the MPS gene panel. Particularly in family 7, subjects F7a and F7b were the only two offspring of consanguineous parents, and no other case of OI was referred in the family, initially directing the suspicion to recessive OI. It was later discovered that their deceased father also had the COL1A1 p.(Ser271Glnfs*16) variant, and that although he had never fractured, he had short stature and cardiac valve disease, compatible with the OI spectrum. In two other cases (Pt 25 and Pt 27) the molecular diagnosis of the index individuals also allowed recognition of affected relatives, with mild manifestations and without a previous clinical diagnosis of OI. Collectively these findings demonstrate the importance of obtaining a molecular diagnosis for enabling precision medicine.
Historically, collagen-related OI is considered to account for 85–90% of cases, while 10–15% are attributed to non-collagen OI [2, 3, 32]. Since 2015, nine major studies have sought to identify the molecular diagnosis of OI in cohorts of different countries [8,9,10, 12,13,14,15, 37, 38]. As shown in Table 3, these studies were heterogeneous in terms of experimental design, methodology, candidate genes analyzed and cohort composition. Even so, the collective analysis of their results allows contextualizing our findings.
With regard to the proportion of collagen-related OI in these cohorts, the studies by Lindahl et al., Bardai et al., and Maioli et al., carried out, respectively, in Swedish, Canadian, and Italian populations, and mainly employing Sanger sequencing of COL1A1 and COL1A2 for molecular analysis, type 1 collagen defects were seen in 85 to 88% of cases [9, 10, 38]. Of note, mild OI phenotypes predominate in the studies of Lindahl et al. and Maioli et al., and approximately half of the cohort in Bardai et al. also had mild disease, potentially influencing these results.
On the other hand, studies by Liu et al., Mrosk et al., and Mohd Nawawi et al. in Chinese, Indian, and Malaysian cohorts, respectively, with predominantly moderate to severe OI and using MPS gene panels, found smaller proportions of type 1 collagen defects, from 49 to 73% [12, 14, 15], resembling our finding of 71% collagen-related cases. In this sense, it could be that ethnic and sociocultural aspects influence the relative prevalence of OI molecular causes. In addition, it is possible that the concomitant analysis of the various candidate genes by MPS allows better accuracy in attributing the molecular diagnosis. Nevertheless, it is more likely that the composition of our cohort, with 63% of moderate to severe OI, has influenced the finding of non-collagen defects in 26% of cases. In previous studies and also here, it is evident that within the cohort, moderate to severe OI was associated with non-collagen defects and mild OI to defects in COL1A1 and COL1A2 [10].
Regarding the distribution of non-collagen causes in the various cohorts, it is noteworthy the great heterogeneity among the studies (Table 3). It has been reported that IFITM5-related OI could account for 5% of cases, being the most common non-collagen cause of OI [4, 5, 11]. In our cohort, the heterozygous IFITM5 c.-14C > T variants was only found in one subject, making it a less frequent non-collagen cause than defects in SERPINF1, FKBP10, PLOD2 and P3H1. In the cohorts of Caparros-Martin et al., Mrosk et al., and Li et al., IFITM5 was also not shown to be the main non-collagen OI cause, suggesting that there is diversity in the molecular basis of OI, particularly in underrepresented populations [13, 14, 37].
Finally, OI is still regarded as a monogenic disorder [2, 32]. In our cohort, we identified one sporadic case with a potential digenic cause involving P3H1 and WNT1. On the basis of clinical presentation, we could not differentiate which candidate gene was mostly implicated in this case, because phenotypic presentation of defects in P3H1 and WNT1 overlap. Even though we do not have functional data to support that all 4 variants have a biological effect, the peculiarity of the inheritance pattern, as a double compound heterozygote, and genetic/in silico data are supportive. It may be that with the increasing availability of MPS, allowing for simultaneous analysis of all OI candidate genes, more cases of potential digenic cause will surface and change our understanding of OI.
Conclusions
A diagnostic approach with an MPS gene panel has effectively allowed establishing the molecular basis of OI in this cohort, unraveling novel disease-causing variants in 29% of cases, and potentially reflecting new aspects of OI pathogenesis in Brazil. Most cases (71%) had COL1A1 or COL1A2 defects, and phenotypic variability among individuals bearing similar collagen defects was not explained by evident modifying variants. Non-collagen defects were found in 26% of cases, with a higher prevalence of FKBP10, PLOD2, SERPINF1 defects, a potential digenic interaction involving P3H1 and WINT1, and a lower prevalence of IFITM5-related OI. Inferring the molecular diagnosis from a family history of consanguinity was misleading in this setting. Obtaining a precise diagnosis of OI in underrepresented populations allows expanding our understanding of its molecular landscape and may lead to improved personalized care in the future.
References
Rauch F, Glorieux FH (2004) Osteogenesis imperfecta. Lancet 363:1377–1385
Marini JC, Forlino A, Bächinger HP et al (2017) Osteogenesis imperfecta. Nat Rev Dis Primers 3:17052
Forlino A, Cabral WA, Barnes AM, Marini JC (2011) New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 7:540–557
Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, Park WY, Park SS, Ikegawa S, Yoo WJ, Choi IH, Kim JW (2012) A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet 91:343–348
Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, Wollnik B, Netzer C (2012) A mutation in the 5'-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91:349–357
Dalgleish R (1997) The human type I collagen mutation database. Nucleic Acids Res 25:181–187
Dalgleish R (1998) The human collagen mutation database 1998. Nucleic Acids Res 26:253–255
Patel RM, Nagamani SC, Cuthbertson D et al (2015) A cross-sectional multicenter study of osteogenesis imperfecta in North America - results from the linked clinical research centers. Clin Genet 87:133–140
Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, Kindmark A (2015) Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet 23:1042–1050
Bardai G, Moffatt P, Glorieux FH, Rauch F (2016) DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int 27:3607–3613
Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ (2000) Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 15:1650–1658
Liu Y, Asan MD et al (2017) Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int 28:2985–2995
Caparros-Martin JA, Aglan MS, Temtamy S, Otaify GA, Valencia M, Nevado J, Vallespin E, del Pozo A, Prior de Castro C, Calatrava-Ferreras L, Gutierrez P, Bueno AM, Sagastizabal B, Guillen-Navarro E, Ballesta-Martinez M, Gonzalez V, Basaran SY, Buyukoglan R, Sarikepe B, Espinoza-Valdez C, Cammarata-Scalisi F, Martinez-Glez V, Heath KE, Lapunzina P, Ruiz-Perez VL (2017) Molecular spectrum and differential diagnosis in patients referred with sporadic or autosomal recessive osteogenesis imperfecta. Mol Genet Genomic Med 5:28–39
Mrosk J, Bhavani GS, Shah H, Hecht J, Krüger U, Shukla A, Kornak U, Girisha KM (2018) Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone 110:368–377
Mohd Nawawi N, Selveindran NM, Rasat R, Chow YP, Abdul Latiff Z, Syed Zakaria SZ, Jamal R, Abdul Murad NA, Abd Aziz BB (2018) Genotype-phenotype correlation among Malaysian patients with osteogenesis imperfecta. Clin Chim Acta 484:141–147
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy M, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur D, Exome Aggregation Consortium (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291
Naslavsky MS, Yamamoto GL, de Almeida TF, Ezquina SAM, Sunaga DY, Pho N, Bozoklian D, Sandberg TOM, Brito LA, Lazar M, Bernardo DV, Amaro E Jr, Duarte YAO, Lebrão ML, Passos-Bueno MR, Zatz M (2017) Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat 38:751–763
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 76:7.20.1–7.20.41
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315
Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL (2012) CONTRA: copy number analysis for targeted resequencing. Bioinformatics 28:1307–1313
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Minillo RM, Sobreira N, de Faria Soares Mde F, Jurgens J, Ling H, Hetrick KN, Doheny KF, Valle D, Brunoni D, Perez AB (2014) Novel deletion of SERPINF1 causes autosomal recessive osteogenesis imperfecta type VI in two Brazilian families. Mol Syndromol 5:268–275
Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC (2007) Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 39:359–365
Rubinato E, Morgan A, D'Eustacchio A, Pecile V, Gortani G, Gasparini P, Faletra F (2014) A novel deletion mutation involving TMEM38B in a patient with autosomal recessive osteogenesis imperfecta. Gene 545:290–292
Rauch F, Lalic L, Roughley P, Glorieux FH (2010) Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res 25:1367–1374
Liascovich R, Rittler M, Castilla EE (2001) Consanguinity in South America: demographic aspects. Hum Hered 51:27–34
Santos S, Kok F, Weller M, de Paiva FR, Otto PA (2010) Inbreeding levels in Northeast Brazil: strategies for the prospecting of new genetic disorders. Genet Mol Biol 33:220–223
Costa-Motta FM, Bender F, Acosta A et al (2014) A community-based study of mucopolysaccharidosis type VI in Brazil: the influence of founder effect, endogamy and consanguinity. Hum Hered 77:189–196
McInerney-Leo AM, Marshall MS, Gardiner B et al (2013) Whole exome sequencing is an efficient, sensitive and specific method of mutation detection in osteogenesis imperfecta and Marfan syndrome. Bonekey Rep 2:456
Trejo P, Rauch F (2016) Osteogenesis imperfecta in children and adolescents-new developments in diagnosis and treatment. Osteoporos Int 27:3427–3437
Tucker T, Marra M, Friedman JM (2009) Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet 85:142–154
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387:1657–1671
Mendoza-Londono R, Fahiminiya S, Majewski J, Care4Rare Canada Consortium, Tétreault M, Nadaf J, Kannu P, Sochett E, Howard A, Stimec J, Dupuis L, Roschger P, Klaushofer K, Palomo T, Ouellet J, al-Jallad H, Mort JS, Moffatt P, Boudko S, Bächinger HP, Rauch F (2015) Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet 96:979–985
Lindert U, Cabral WA, Ausavarat S et al (2016) MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun 7:11920
Li L, Mao B, Li S, Xiao J, Wang H, Zhang J, Ren X, Wang Y, Wu Y, Cao Y, Lu C, Gao J, You Y, Zhao F, Geng X, Xiao Y, Jiang C, Ye Y, Yang T, Zhao X, Zhang X (2019) Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Hum Mutat 40:588–600
Maioli M, Gnoli M, Boarini M et al (2019) Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur J Hum Genet 27:1090–1100
Acknowledgments
We are grateful to Amanda Narcizo for technical assistance with massively parallel sequencing.
Funding
This work was supported by the São Paulo Research Foundation (FAPESP, Multiusuário grant 2013/02162-8). BFdS also acknowledges support from FAPESP through a Young Investigator grant (2011/12696-4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Adriana Fernandes, Manuela Rocha-Braz, Monica França, Antonio Lerario, Vivian Simões, Evelin Zanardo, Leslie Kulikowski, Regina Martin, Berenice Mendonca and Bruno Ferraz-de-Souza declare that they have no conflict of interest that could be perceived as prejudicing the impartiality of this study. Manuela Rocha-Braz has received lecture fees from EMS and Amgen Brazil. Bruno Ferraz-de-Souza has received consulting fees from Sandoz Brazil, UCB Brazil and Hypera Brazil, and lecture fees from Amgen Brazil, Sandoz Brazil, Sanofi Brazil and Hypera Brazil.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fernandes, A., Rocha-Braz, M., França, M. et al. The molecular landscape of osteogenesis imperfecta in a Brazilian tertiary service cohort. Osteoporos Int 31, 1341–1352 (2020). https://doi.org/10.1007/s00198-020-05366-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-020-05366-4