
Abstract
A green, sensitive and accurate dispersive liquid–liquid microextraction (DLLME) method was used to preconcentrate four selected pesticides in dam lake water samples for determination by gas chromatography–mass spectrometry (GC–MS). Conditions of the DLLME method were comprehensively investigated and optimized according to type/volume of extraction solvent, type/volume of dispersive solvent, and type/period of mixing. The developed method was validated according to the limits of detection and quantitation, accuracy, precision and linearity. Under the optimum conditions, limit of detection values calculated for alachlor, acetochlor, metolachlor and fenthion were 1.7, 1.7, 0.2 and 7.8 µg/kg (mass based), respectively. The method recorded 202, 104, 275 and 165 folds improvement in detection power values for acetochlor, alachlor, metolachlor and fenthion, respectively, when compared with direct GC–MS measurements. In order to evaluate the accuracy of the developed method, real sample application with spiking experiments was performed on dam lake water samples, and satisfactory percent recovery results in the range of 81%–120% were obtained.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
In the past few decades, investigation of micropollutants in environmental samples has been widely conducted in attempt to enhance the detection power of analytical instruments to allow trace level determinations. Micropollutants consist mainly of anthropogenic chemicals such as personal care products, illicit drugs, pharmaceuticals and pesticides (Noguera-Oviedo and Aga 2016). Pesticides are considered according to their properties such as persistence in the environment, toxicity, chemical structure and usage. Generally, they are categorized as three main groups according to target organism, namely, herbicides, insecticides and fungicides (Nasiri et al. 2020). Application of pesticides in agricultural fields has been a superior control system to decrease the loss of crops, improve crop quality, increase yields and extend postharvest life by controlling fungi, weeds, insects and other unwanted pests (Al-Nasir et al. 2020). After application, pesticides may remain on the surface of crops, leach down the soil into groundwater, or be transported with runoff water into other water bodies. Contaminated water bodies have the tendency to cause negative effects on aquatic life and human health (Yuan et al. 2020).
Alachlor, acetochlor and metolachlor are obsolete herbicides widely used to improve the crop yields by controlling broadleaf weeds and annual grasses on farmlands (Wang et al. 2019; Lou et al. 2020; Sun et al. 2020). Fenthion is an obsolete organophosphate insecticide employed against biting and sucking pests such as Eurygaster cereal bugs, stem borers, mosquitoes and fruit flies (Kyriakidis et al. 2005). These pesticides are obsolete because of their harmful effects. Thus, sensitive methods are required to monitor environmental and food samples to ensure that they are not being applied illegally. Their persistence in the environment also requires monitoring even after their usage is stopped (Shah and Devkota 2009).
Surplus usage of these pesticides poses a threat to human health as chronic exposure could lead to neurological effects, cancer tumors, birth defects and allergic reactions (Kanashina et al. 2020). In recent years, attention has been drawn to water contamination, especially in surface and groundwater sources (Hoff et al. 2019). Thus, it is of great importance to develop highly sensitive and accurate analytical methods to determine these contaminants in water samples.
There are many reported techniques for the determination of pesticides in water samples such as liquid chromatography–mass spectrometry (LC–MS) (Picó et al. 2004), gas chromatography–mass spectrometry (GC–MS) (Alder et al. 2006) and high-performance liquid chromatography (HPLC) (Aulakh et al. 2005). For example, a study reported by Chen and Da (2006) aimed to develop a dynamic hollow fiber-protected liquid-phase microextraction method coupled with gas chromatography–mass spectrometry (GC–MS) to detect ethoprop, diazinon, disulfoton and fenthion in lake water samples. An automated solid phase extraction (SPE) GC–MS method for the determination of acetochlor in reagent water, surface water, and groundwater samples was reported by Lindley et al. (1996). In another study, Brooks et al (1989) reported a SPE method for the simultaneous determination of alachlor, atrazine and metolachlor in groundwater samples.
Among these, GC–MS has been widely used for volatile organic species, and it distinguishes between compounds based on their different retention times and ion fragmentations (mass to charge ratio, m/z). In addition, SIM (specific m/z) and Scan (defined range of m/z) modes of data acquisition can be performed for the determination of specific or selected compounds, respectively (Bulgurcuoğlu et al. 2018). However, this technique lacks the proper sensitivity to determine most organic species at relatively low concentration levels. Therefore, GC–MS is combined with sample pretreatment methods to further improve its sensitivity.
The conventional liquid–liquid extraction (LLE) (Holadová and Hajšlová 1995) and solid phase extraction (SPE) (Rodríguez et al. 2000) methods have several drawbacks such as being time consuming, expensive, labor-intensive and they involve the consumption of large organic solvent volumes. Researches in recent years have focused on the development of economical, efficient and miniaturized sample pretreatment techniques in order to overcome the setbacks of the conventional methods (Xue et al. 2020). In this regard, liquid phase microextraction (LPME) (Vidal et al. 2010), solid-phase microextraction (SPME) (Negreira et al. 2009) and hollow fiber liquid phase microextraction (HF-LPME) (Ge and Lee 2012) methods have been developed. Dispersive liquid–liquid microextraction (DLLME) method is a very trending eco-friendly sample pretreatment method for various metal ions and organic compounds due to its advantages such as simplicity, cheapness, quick operation, high output and being environmentally friendly (Mashayekhi et al. 2010). This method is based on the dispersion phenomenon, where the extraction solvent is dispersed in the aqueous solution with the help of a disperser solvent (Zari et al. 2020). Fine microdroplets of extraction solvent is formed throughout the aqueous solution by rapid injection with the disperser solvent, leading to an enhanced contact surface area between the aqueous solution and extraction solvent. This allows the rapid mass transfer of the target analytes to the extractor phase (Xue et al. 2020).
Herein, a DLLME method was developed to preconcentrate alachlor, acetochlor, metolachlor and fenthion from dam lake water samples for determination by GC–MS at trace levels with high accuracy. In order to determine the optimum conditions for selected pesticides, effecting parameters on GC–MS instrument and DLLME method were tested; and the optimal combination of parameters was determined.
Materials and Methods
Acetochlor (≥ 95.0%, purity), fenthion (≥ 95.0%, purity) and metolachlor (≥ 95.0%, purity) standards were attained from Sigma-Aldrich (Darmstadt – Germany). Alachlor (99.99%) and bisphenol A-d16 (98.0%) standards were purchased from Dr. Ehrenstorfer GmbH (Augsburg – Germany). Chloroform, dichloromethane, 1–2 dichloroethane, methanol, ethanol, isopropyl alcohol, acetone and acetonitrile were all purchased from Merck (Germany, Darmstadt) and used for developing the DLLME method. Main stock solutions of acetochlor, fenthion, metolachlor and alachlor were gravimetrically prepared in acetonitrile. Mixed standard solution (100.0 mg/kg) of the four analytes was gravimetrically prepared in acetonitrile and diluted to prepare working and calibration standards gravimetric preparation has higher accuracy than volumetric preparation and also possess traceability. Preparation of aqueous standards and washing purposes were done with deionized water obtained from an Elga Flex 3 Water Purification System (Resistivity 18.2 Ω.cm).
A gas chromatograph (Agilent, HP 6890) system with a capillary column (HP-5MS: 30 m length × 0.25 mm internal diameter × 250 nm film thickness) was used for chromatographic separation of the analytes. The gas chromatography system was equipped with a mass spectrometry (Agilent, HP 5973) detector for qualitative and quantitative determination of the analytes. Helium was employed as mobile phase with a constant flow rate of 2.60 mL/min. Temperature of the inlet port was set at 280°C, and 1.0 µL volume of sample/standard was injected in the spitless mode. The oven temperature was programmed to increase from 70 to 240°C (30°C/min), then to 260°C (120°C/min) and a final ramp to 300°C (30°C/min). Temperature/energy of ionization source, temperature of quadrupole and transfer line were 230°C/70 eV, 150°C and 280°C, respectively. The retention time, mass to charge ratio (m/z) of qualifier fragmentations and quantifier ions (underlined values) for acetochlor, alachlor, metolachlor and fenthion are summarized in Table 1.
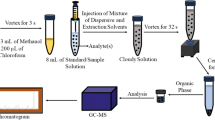
A mixture of dichloromethane (300 µL) and methanol (1.5 mL) was rapidly injected into 8.0 mL of standard/sample solution in a 15 mL centrifuge tube. The cloudy solution obtained after the injection was placed on a mechanical shaker and mixed for 45 s to augment interactions between the analytes and extraction solvent. After the mixing process, the solution was centrifuged for 2.0 min at 3461 g to separate the aqueous and organic phases from each other. The upper phase (aqueous solutions) was advertently transferred into a waste container and lower organic phase (approximately 100 µL) was taken into an insert vial sent to the instrument for the determination of the analytes.
Water samples were collected from Altinapa Dam, which is supplying drinking water for the City of Konya, and the effluent from the Konya Drinking Water Treatment Facility. The water sampled from the lake (Altınapa dam) and treated water were labelled as influent sample (InSa) and effluent sample (EfSa). All samples were kept into polyethylene terephthalate containers, which were thoroughly rinsed with the respective samples. Blank analysis was performed for the samples and spike recovery experiments were used to validate the accuracy of the method.
Results and Discussions
Two main optimization experiments were performed in this study: instrumental optimization and DLLME method optimization. Temperature program and carrier gas flow rate were optimized to obtain symmetrical peak signals. The univariate optimization approach was utilized for the DLLME method to augment the signal to noise ratio and boost extraction yield for the analytes. Tukey’s method based post hoc comparisons were applied to evaluate the difference in pairwise comparisons in all optimization studies. Lower-case letters such as a, b, c, d were used to represent the difference between the variables.
Different temperature programs and carrier gas flow rates were tested to obtain good chromatographic separation and symmetrical signals for the analytes. In the first step, a single ramp temperature program (70–300°C, 5.0°C/min) and 1.0 mL/min of carrier gas flow rate were tested. While acetochlor and alachlor were well-separated from each other, metolachlor and fenthion produced overlapping peaks. Moreover, a broad peak signal was obtained for fenthion with this temperature program. Peak signals of metolachlor and fenthion did not interfere with each other when selected ion monitoring (SIM) mode was used to acquire data. When the temperature program and helium flow rate explained in Instrumental Section was used, better symmetrical signals and higher signal to noise ratios were obtained for all analytes. Total ion and extracted ion chromatograms taken from GC–MS ChemStation Software are presented in Fig. 1.

Total ion chromatogram of 100 mg/kg of mixed standard solution (a) and extracted ion chromatograms of 146 (acetochlor), 160 (alachlor), 162 (metolachlor) and 278 (fenthion) ion fragment (b)
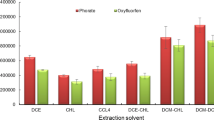
The basic requirements of an ideal extraction solvent in DLLME are immiscibility with aqueous solution, miscibility with disperser solvent, to have different density from aqueous solution, and to have good solubility for the analyte (Chormey and Bakirdere 2018). For this reason, chloroform, dichloromethane and 1–2 dichloroethane were tested to find the most efficient extraction solvent. Dichloromethane gave the highest peak area values for all four analytes as presented in Fig. 2, therefore; dichloromethane was chosen as optimum extraction solvent. In addition, there was also a significant difference between dichloromethane and other extraction solvents with respect to statistical test.

Optimization of extraction solvent type (lower-case letters present significant difference at p < 0.05 for n = 3)
Extraction solvent volume is an important parameter in this extraction protocol since it directly affects the preconcentration factor of the analytes. Hence, dichloromethane volumes were tested in the range of 250–350 µL to investigate the effect of these volumes on preconcentration factor. Phase separation was not observed for the 250 µL volume because of complete solubility of dichloromethane in water (13,200 mg/L at 25°C) by reason of dispersion with methanol (Yalkowsky et al. 2016). The highest peak area values were recorded for 300 µL of dichloromethane (Figure S1), and it was selected as optimum one.
A dispersive solvent should be miscible with both aqueous solution and extraction solvent. Generally, mid polarity solvents are selected as disperser solvent because of their high solubility in both aqueous and organic solvents. This makes them efficient in distributing extraction solvents as nano/micro particles throughout the whole aqueous solution for high surface area contact (Chormey and Bakirdere 2018). In this optimization step, five solvents including methanol, ethanol, isopropyl alcohol, acetone and acetonitrile were employed as disperser solvent to evaluate their effects on extraction efficiency for the analytes. Dichloromethane was dispersed more efficiently in aqueous solution by methanol for all analytes as shown in Fig. 3, and it was therefore selected as optimum disperser solvent.

Optimization of disperser solvent type (lower-case letters present significant difference at p < 0.05 for n = 3)
Three different volumes (1.0, 1.5 and 2.0 mL) of dispersive solvent were tested to determine optimum methanol volume. Although the 2.0 mL methanol volume provided higher peak area results than the other volumes (for all analytes except fenthion), the organic phase formed after the centrifugation process was very low. Collection of the organic phase for instrumental reading was very challenging even for ultrapure deionized water, and this was expected to be more difficult for real samples. Although there was no significant difference between results of 1.0 and 1.5 mL methanol, 1.5 mL of methanol provided better results than 1.0 mL of methanol for all analytes except metolachlor (Figure S2). Thus, it was selected as the optimum value.
In the DLLME protocol, sample agitation could have a positive effect on mass transfer of an analyte(s) from aqueous solution into an organic phase. Agitation further enhances the extraction of the analyte(s) by increasing the movement of the extraction solvent through the aqueous solution, thus, enhancing the pickup of the desired analytes. Therefore, hand shaking (up-down), mechanical shaking (left–right), ultrasonication and vortex were tested to examine effects on mass transfer of the analytes. There was no significant difference between the results of hand shaking and mechanical shaking for acetochlor and alachlor, but mechanical shaking made an important impact on the results of metolachlor and fenthion as shown in Fig. 4. For this reason, mechanical shaking was selected as optimum mixing type.

Optimization of mixing type (lower-case letters present significant difference at p < 0.05 for n = 3)
The final optimization of this study was period of mechanical shaking, and periods between 15 and 60 s were tested and compared to an experiment performed without mixing (0.0 s). The highest result was obtained for 45 s of mechanical shaking period (Figure S3), and it was chosen as optimum value.
A series of mixed standard solutions (in acetonitrile) prepared in the range of 0.25–100.00 mg/kg (ten different concentrations) were directly sent to the GC–MS to evaluate the analytical performance of the system. Six replicate measurements were carried out for each standard solution under the optimum chromatographic conditions. The limit of detection (LOD) and limit of quantification (LOQ) for the analytes were calculated in the range of 0.07–1.28 and 0.22–4.27 mg/kg, respectively. In addition to the LOD and LOQ values, linear range, percent relative standard deviation (%RSD) and coefficient of determination (R2) were used to validate the GC–MS system as summarized in Table 2.
Mixed standard solutions were prepared in the range of 0.25–500 µg/kg (eleven different concentrations) and analyzed under optimum conditions to determine analytical performance values of the DLLME-GC–MS system. In addition, 250 µg/kg bisphenol A-d16 as internal standard was added to all calibration standard solutions to eliminate errors arising from the extraction procedure and GC–MS measurement. Calibration plots for each analyte were constructed utilizing concentration versus the ratio of analyte peak area to internal standard peak area. LOD and LOQ values for the developed system were determined in the ranges of 0.24–7.8 and 0.82–26 µg/kg, respectively. The detection power of the GC–MS system was enhanced by 104–275 folds (Table 2) for the analytes. Enhancement was calculated by comparing LOD values of the GC–MS system to the DLLME-GC–MS method. LOD, LOQ, dynamic range, %RSD and R2 for DLLME-GC–MS system are presented in Table 2. LOD and LOQ values were calculated using the following equations;
where SDLC is standard deviation (six measurements) of the lowest concentration in linear range, m is calibration plot slope.
The developed method is comparable with the literature findings in terms of limit of detection and sample pre-treatment procedures. For instance, in a study, a method for the determination of 251 pesticide and degradation product residues in vegetables and fruit samples by gas chromatography–mass spectrometry (GC–MS) and by liquid chromatography fluorescence detection (solid phase extraction) was reported. The LOD values obtained for iron fenthion, alachlor and metolachlor were 30, 20 and 20 μg/L, respectively (Fillion et al. 2000). A DLLME method combined with gas chromatography-flame ionization detection (GC-FID) was developed for the determination of multiclass pesticides in tomato and cucumber samples and the detection limits were found to be 3.0 μg/L for both alachlor and fenthion (Khor and Chai 2014). In another study, LOD values of 0.01, 0.02 and 0.03 μg/L were obtained for acetochlor, alachlor and metolachlor, respectively by gas chromatography-electron capture detector (GC-ECD) after solid phase extraction (SPE) using multiwalled carbon nanotubes as adsorbents (Dong et al. 2009). The detection limit values reported for acetochlor, alachlor, and metolachlor using solid-phase microextraction coupled with gas chromatography–mass spectrometry (SPME–GC–MS) were found to be 0.25 μg/L (Rocha et al. 2008). In a study, metolachlor, acetochlor, alachlor and dimethenamid were extracted using C-18 solid-phase extraction (SPE) column before their determinations by liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI–MS/MS) (Yokley et al. 2002). Determination of corn herbicides (atrazine, alachlor, metolachlor, and pendimethalin) by GC–MS and GC-NPD in soil and water samples were performed in another study (Sánchez-Brunete et al. 1994). If the developed method in this study is compared with literature studies, it offers a simple and time saving extraction procedure and mitigates the long sample pre-treatment processes with high sample volume and toxic organic solvent usage. A green extraction method can be applied to complex matrices without the need for highly toxic organic solvents with the presented method. It also provides low detection limits with low %RSD values which indicates high precision. A solid phase microextraction (SPME) method was applied for the determination of priority pesticides in river samples by gas chromatography with flame thermionic detection (GC-FTD) and the mean detected concentrations for alachlor and fenthion were found to be 0.08 and 0.02 µg/L, respectively (Albanis et al. 2003). Detected concentration values are lower than the LOD values obtained with the presented method in this study. However, the detection limits can be further improved by using gas chromatography-tandem mass spectrometry (GC–MS/MS) and gas chromatography-high-resolution mass spectrometry (GC-HRMS).
Spike recovery experiments were performed with InSa and EfSa samples to check the accuracy and applicability of the DLLME method. Blank analysis using the optimized method did not produce signals at the retention times of the analytes, indicting the analytes were absent or below the specified LOD values. The samples were then spiked at different concentrations, taking into consideration acetonitrile content of the aqueous standard solutions. All samples and standards were prepared to contain 1.0% acetonitrile, to obtain an equal effect from acetonitrile as a disperser solvent. The standard solutions were prepared between 50.0 and 500 µg/kg and spiked with 250 µg/kg of bisphenol A-16. The external calibration method was used to calculate the concentration of the analytes in the spiked samples. Relative recovery results for the spiked influent and effluent samples were found to be in the range of 81%–120% (Table 3) and 80%–115% (Table 4), respectively. Relative recovery results were calculated by employing the following equation;
CE: experimental concentration of the analyte.
CT: theoretical concentration of the analyte.
In this study, a DLLME method was proposed for simultaneous preconcentration of acetochlor, alachlor, metolachlor and fenthion for the determination by GC–MS system. Variable parameters of the DLLME method such as type/volume of extraction solvent, type/volume of disperser solvent, and mixing type/period were successfully optimized to enhance the signal to noise ratio of the analytes. After the optimization steps, system analytical performance was evaluated, and a good linearity was attained for all analytes. The LOD values for acetochlor, alachlor, metolachlor and fenthion were found to be 1.7, 1.7, 0.2 and 7.8 µg/kg, respectively. The analytes were determined at trace levels because high enhancement in detection power values (104–275 folds) were obtain employing the DLLME method for the analytes prior to GC–MS system. Spiking experiments were used to check applicability/accuracy of the proposed DLLME method. The results obtained certified that the developed method could be conveniently used to determine the analytes at trace level in lake water samples with high accuracy.
References
Albanis TA, Lambropoulou DA, Sakkas VA, Hela D (2003) Monitoring of priority pesticides using SPME (solid phase microextraction) in river water from Greece. Water science and technology: water supply. IWA Publishing, London, pp 335–342
Alder L, Greulich K, Kempe G, Vieth B (2006) Residue analysis of 500 high priority pesticides: better by GC-MS or LC-MS/MS? Mass Spectrom Rev 25:838–865. https://doi.org/10.1002/mas.20091
Al-Nasir FM, Jiries AG, Al-Rabadi GJ et al (2020) Determination of pesticide residues in selected citrus fruits and vegetables cultivated in the Jordan Valley. LWT. https://doi.org/10.1016/j.lwt.2019.109005
Aulakh JS, Malik AK, Kaur V, Schmitt-Kopplin P (2005) A review on solid phase micro extraction—high performance liquid chromatography (SPME-HPLC) analysis of pesticides. Crit Rev Anal Chem 35:71–85. https://doi.org/10.1080/10408340590947952
Brooks MW, Jenkins J, Jimenez M et al (1989) Rapid method for the determination of alachlor, atrazine and metolachlor in groundwater by solid-phase extraction. Analyst 114:405–406. https://doi.org/10.1039/AN9891400405
Bulgurcuoğlu AE, Yılmaz B, Chormey DS, Bakırdere S (2018) Simultaneous determination of estrone and selected pesticides in water medium by GC-MS after multivariate optimization of microextraction strategy. Environ Monit Assess. https://doi.org/10.1007/s10661-018-6625-3
Chen PS, Da HS (2006) Determination of ethoprop, diazinon, disulfoton and fenthion using dynamic hollow fiber-protected liquid-phase microextraction coupled with gas chromatography-mass spectrometry. In: Talanta. https://www.sciencedirect.com/science/article/pii/S0039914005007289. Accessed 15 Jul 2020
Chormey DS, Bakirdere S (2018) Principles and recent advancements in microextraction techniques. Compr Anal Chem 81:257–294. https://doi.org/10.1016/bs.coac.2018.03.011
Dong M, Ma Y, Zhao E et al (2009) Using multiwalled carbon nanotubes as solid phase extraction adsorbents for determination of chloroacetanilide herbicides in water. Microchim Acta 165:123–128. https://doi.org/10.1007/s00604-008-0109-z
Fillion J, Sauvé F, Selwyn J (2000) Multiresidue method for the determination of residues of 251 pesticides in fruits and vegetables by gas chromatography/mass spectrometry and liquid chromatography with fluorescence detection. Oxford University Press, Oxford
Ge D, Lee HK (2012) Ionic liquid based hollow fiber supported liquid phase microextraction of ultraviolet filters. J Chromatogr A 1229:1–5. https://doi.org/10.1016/j.chroma.2011.12.110
Hoff R, Echeverria AD, Hoff GD et al (2019) Efficiency of a low-cost pyramid-shaped solar still for pesticide removal from highly contaminated water. Chemosphere 234:427–437. https://doi.org/10.1016/j.chemosphere.2019.06.062
Holadová K, Hajšlová J (1995) A comparison of different ways of sample preparation for the determination of phthalic acid esters in water and plant matrices. Int J Environ Anal Chem 59:43–57. https://doi.org/10.1080/03067319508027635
Kanashina D, Pochivalov A, Timofeeva I, Bulatov A (2020) Mixed surfactant systems based on primary amine and medium-chain fatty acid: Micelle-mediated microextraction of pesticides followed by the GC–MS determination. J Mol Liq. https://doi.org/10.1016/j.molliq.2020.112906
Khor L, Chai MK (2014) Development and validation of dispersive liquid-liquid microextraction for the determination of multiclass pesticide residues in complex food matrix. Malaysian J Sci 33:89–98
Kyriakidis NV, Pappas C, Athanasopoulos P (2005) Degradation of fenthion and fenthion sulfoxide on grapes on the vines and during refrigerated storage. Food Chem 91:241–245. https://doi.org/10.1016/j.foodchem.2003.10.022
Lindley CE, Stewart JT, Sandstrom MW (1996) Determination of low concentrations of acetochlor in water by automated solid-phase extraction and gas chromatography with mass-selective detection. Oxford University Press, Oxford
Lou YY, Geneste F, Soutrel I et al (2020) Alachlor dechlorination prior to an electro-Fenton process: influence on the biodegradability of the treated solution. Sep Purif Technol. https://doi.org/10.1016/j.seppur.2019.115936
Mashayekhi HA, Abroomand-Azar P, Saber-Tehrani M, Husain SW (2010) Rapid determination of carbamazepine in human urine, plasma samples and water using DLLME followed by RP-LC. Chromatographia 71:517–521. https://doi.org/10.1365/s10337-009-1456-6
Nasiri M, Ahmadzadeh H, Amiri A (2020) Sample preparation and extraction methods for pesticides in aquatic environments: a review. Trends Anal Chem 123:115772
Negreira N, Rodríguez I, Ramil M et al (2009) Sensitive determination of salicylate and benzophenone type UV filters in water samples using solid-phase microextraction, derivatization and gas chromatography tandem mass spectrometry. Anal Chim Acta 638:36–44. https://doi.org/10.1016/j.aca.2009.02.015
Noguera-Oviedo K, Aga DS (2016) Lessons learned from more than two decades of research on emerging contaminants in the environment. J Hazard Mater 316:242–251
Picó Y, Blasco C, Font G (2004) Environmental and food applications of LC-tandem mass spectrometry in pesticide-residue analysis: an overview. Mass Spectrom Rev 23:45–85
Rocha C, Pappas EA, Huang CH (2008) Determination of trace triazine and chloroacetamide herbicides in tile-fed drainage ditch water using solid-phase microextraction coupled with GC-MS. Environ Pollut 152:239–244. https://doi.org/10.1016/j.envpol.2007.04.029
Rodríguez I, Llompart MP, Cela R (2000) Solid-phase extraction of phenols. J Chromatogr A 885:291–304
Sánchez-Brunete C, Martínez L, Tadeo JL (1994) Determination of corn herbicides by GC-MS and GC-NPD in environmental samples. J Agric Food Chem. https://doi.org/10.1021/jf00046a025
Shah BP, Devkota B (2009) Obsolete pesticides: their environmental and human health hazards. J Agric Environ 10:60–66. https://doi.org/10.3126/aej.v10i0.2130
Sun Y, Zhao L, Li X et al (2020) Response of soil bacterial and fungal community structure succession to earthworm addition for bioremediation of metolachlor. Ecotoxicol Environ Saf. https://doi.org/10.1016/j.ecoenv.2019.109926
Vidal L, Chisvert A, Canals A, Salvador A (2010) Ionic liquid-based single-drop microextraction followed by liquid chromatography-ultraviolet spectrophotometry detection to determine typical UV filters in surface water samples. Talanta 81:549–555. https://doi.org/10.1016/j.talanta.2009.12.042
Wang H, Meng Z, Zhou L et al (2019) Effects of acetochlor on neurogenesis and behaviour in zebrafish at early developmental stages. Chemosphere 220:954–964. https://doi.org/10.1016/j.chemosphere.2018.12.199
Xue L, Chen L, Dong J et al (2020) Dispersive liquid-liquid microextraction coupled with surface enhanced Raman scattering for the rapid detection of sodium benzoate. Talanta. https://doi.org/10.1016/j.talanta.2019.120360
Yalkowsky S, He Y, Jain P (2016) Handbook of aqueous solubility data. CRC Press, Boca Raton
Yokley RA, Mayer LC, Ben HS, Vargo JD (2002) Analytical method for the determination of metolachlor, acetochlor, alachlor, dimethenamid, and their corresponding ethanesulfonic and oxanillic acid degradates in water using SPE and LC/ESI-MS/MS. Anal Chem 74:3754–3759. https://doi.org/10.1021/ac020134q
Yuan YY, Wang ST, Liu SY et al (2020) Green approach for simultaneous determination of multi-pesticide residue in environmental water samples using excitation-emission matrix fluorescence and multivariate calibration. Spectrochim Acta A. https://doi.org/10.1016/j.saa.2019.117801
Zari N, Hassan J, Tabar-Heydar K, Ahmadi SH (2020) Ion-association dispersive liquid–liquid microextraction of trace amount of gold in water samples and ore using Aliquat 336 prior to inductivity coupled plasma atomic emission spectrometry determination. J Ind Eng Chem 86:47–52. https://doi.org/10.1016/j.jiec.2017.01.038
Acknowledgements
This work was supported by The Scientific and Technological Research Council of Turkey (TÜBİTAK) with a Grant Number of 118Y402.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interest among the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bodur, S., Borahan, T., Ates, N. et al. Sensitive Determination of Acetochlor, Alachlor, Metolachlor and Fenthion Utilizing Mechanical Shaking Assisted Dispersive Liquid–Liquid Microextraction Prior to Gas Chromatography–Mass Spectrometry. Bull Environ Contam Toxicol 105, 460–467 (2020). https://doi.org/10.1007/s00128-020-02965-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-020-02965-z