
Abstract
Aims/hypothesis
Diabetic retinopathy is a common microvascular complication of diabetes mellitus and is initiated by inflammation and apoptosis-associated retinal endothelial cell damage. Prostaglandin E2 (PGE2) has emerged as a critical regulator of these biological processes. We hypothesised that modulating PGE2 and its E-prostanoid receptor (EP2R) would prevent diabetes mellitus-induced inflammation and microvascular dysfunction.
Methods
In a streptozotocin (STZ)-induced rat model of diabetes, rats received intravitreal injection of PGE2, butaprost (a PGE2/EP2R agonist) or AH6809 (an EP2R antagonist). Retinal histology, optical coherence tomography, ultrastructure of the retinal vascular and biochemical markers were assessed.
Results
Intravitreal injection of PGE2 and butaprost significantly accelerated retinal vascular leakage, leucostasis and endothelial cell apoptosis in STZ-induced diabetic rats. This response was ameliorated in diabetic rats pre-treated with AH6809. In addition, pre-treatment of human retinal microvascular endothelial cells with AH6809 attenuated PGE2- and butaprost-induced activation of caspase 1, activation of the complex containing nucleotide-binding domain and leucine rich repeat containing family, pyrin domain containing 3 (NLRP3) and apoptosis-associated speck-like protein containing a C-terminal caspase-activation and recruitment domain (ASC), and activation of the EP2R-coupled cAMP/protein kinase A/cAMP response element-binding protein signalling pathway.
Conclusions/interpretation
The PGE2/EP2R signalling pathway is involved in STZ-induced diabetic retinopathy and could be considered as a potential target for diabetic retinopathy prevention and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
Diabetic retinopathy, a prevalent complication of diabetes, is a leading cause of visual impairment and blindness in the adult population. However, the biochemical and molecular mechanisms are not well understood [1, 2]. Early clinical symptoms include retinal microvascular endothelial cell (RMEC) dysfunction and vascular dysfunction [3, 4]. Emerging evidence indicates that high-glucose-induced para-inflammation, characterised by a chronic low level of inflammation and a disordered immune response, is involved in the onset and progression of RMEC damage [5, 6]. The retinas from animal models of diabetes show inappropriate activation of the nucleotide-binding domain and leucine rich repeat containing family, pyrin domain containing 3 (NLRP3) inflammasome, which is a molecular complex of NLRP3, apoptosis-associated speck-like protein containing a C-terminal caspase-activation and recruitment domain (ASC) and pro-caspase 1 [7]. These diabetic retinas also express high levels of proinflammatory cytokines such as IL-1β [8].
Two signalling pathways are believed to be involved in the generation and release of IL-1β induced by high glucose levels and the accumulation of advanced glycation end-products [9]. The first pathway is triggered by glucose abnormalities, which induce IL-1β transcription and stimulate the production of IL-1β precursor pro-IL-1β. The second signalling pathway induces conformational changes in the NLRP3 inflammasome platform and activates caspase 1 to convert pro-IL-1β into the mature secreted form of IL-1β. The NLRP3 inhibitor MCC950 has been shown to inhibit high-glucose-induced RMEC dysfunction, consistent with the promising clinical effects of an IL-1 receptor antagonist for the treatment of diabetic retinopathy [5].
Prostaglandin E2 (PGE2) is a potent inflammatory mediator that is a crucial IL-1β inducer and causes fever [10]. PGE2 is biosynthesised from arachidonic acid by cyclooxygenase enzyme and stimulates its G-protein-coupled plasma membrane receptors (E-prostanoid1–4 receptors [EP1–4Rs]), activating multiple signal transduction pathways leading to downstream responses [11]. The EP1 receptor mainly couples to the Gq protein and upregulates the level of intracellular calcium; the EP2 and EP4 receptors couple to the Gs protein, activate adenylate cyclase and increase the production of intracellular cAMP. In contrast, the EP3 receptor couples to the Gi protein, inactivates adenylate cyclase and decreases the formation of intracellular cAMP [12].
Recent research findings have verified that cyclooxygenase-2 (COX-2) and PGE2 are involved in the pathogenesis of diabetic retinopathy. Significantly higher than normal PGE2 levels have been detected in the vitreous fluid of individuals with complications from proliferative diabetic retinopathy and also in animal models of diabetic retinopathy [13]. In addition, progression of retinopathy can be prevented or delayed by prostaglandin inhibitors [14, 15]. In receptor combination patterns, PGE2 shows various biological effects and the specific E-prostanoid receptors of PGE2 that regulate endothelium impairment and vascular dysfunction have not been well illustrated. These findings prompted us to determine whether the PGE2/EP2R cascade mediates RMEC damage in diabetic retinopathy and to investigate the underlying molecular mechanisms.
Methods
Human vitreous fluid
Participants with type 1 diabetes who had undergone vitrectomy owing to proliferative diabetic retinopathy were recruited from Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, China. The research followed the tenets of the Declaration of Helsinki. The protocol for sample collection was approved by the hospital ethics committee and the study participants gave informed consent. For further details, see electronic supplementary materials (ESM) Methods.
Cell culture
Human RMECs (hRMECs) were obtained from BeNa Culture Collection (Beina Chuanglian Biotechnology Institute, Beijing, China) and cultured in DMEM supplemented with 10% FBS (vol./vol.) and 1% antimycotics and antibiotics (vol./vol.). Mycoplasma contamination was not tested. For further details, see ESM Methods.
Animals and treatments
Eight-week-old homozygous male Sprague Dawley rats (220–250 g) were randomly divided into six groups. Diabetes was induced with an i.p. injection of streptozotocin (STZ; 60 mg/kg in 10 mmol/l citrate buffer at pH 4.6), as previously reported [16]. The rats had blood glucose levels >16.7 mmol/l, indicating that diabetes had been successfully established. The STZ-treated rats were given an intravitreal injection of 5 mmol/l PGE2, butaprost (a PGE2/EP2R agonist) or AH6809 (an EP2R antagonist), all mixed with saline solution (154 mmol/l NaCl) 1:1, at a total volume of 6 μl for each eye. A vehicle control was prepared by mixing one volume of DMSO with one volume of saline solution. The six experimental groups were as follows: control; untreated STZ; STZ + PGE2; STZ + butaprost; STZ + AH6809 and STZ + DMSO. All studies adhered to the institutional guidelines for humane treatment of animals, Principles of Laboratory Animal Care (National Institutes of Health [NIH], Bethesda, MD, USA) and to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. For further details, see ESM Methods.
Intravitreal injection
Rats were ventilated after being anaesthetised with a mixture of ketamine (80 mg/kg, i.p.) and xylazine (4 mg/kg, i.p.). A volume of about 6 μl of the designated mixture was delivered into the vitreous cavity using a 33-gauge needle. Rats received an intravitreal injection every 3 weeks.
Retinal imaging
Rats were anaesthetised (ketamine/xylazine) and their pupils were dilated with Cyclomydril (Alcon, Fort Worth, TX, USA). Spectral domain optical coherence tomography (OCT) was performed using the image-guided OCT system (Micron IV; Phoenix Research Labs, Pleasanton, CA, USA) with the guidance of a bright-field live fundus image.
Permeability measurement
The permeability of the blood–retina barrier in rats was quantified with Evans Blue, which binds to the plasma albumin, using the method described by Shan and colleagues [16] (see ESM Methods). Digital images of rat retinal flat mounts were examined under an Olympus BX-51 light microscope (Olympus, Tokyo, Japan) to check for Evans Blue extravasation from the retinal vessels.
Retinal trypsin digestion assay
Eyes of rats were enucleated, fixed in 4% paraformaldehyde (wt/vol.) for 24 h, equatorially bisected and the retinas were removed. The retinas were incubated with 3% trypsin (wt/vol.) at 37°C for 3 h; they were then gently shaken to free the vessel network, washed and mounted onto glass slides to dry. Retinal vasculature was stained with Periodic acid–Schiff and Haematoxylin. Digital images were examined under an Olympus BX-51 light microscope (Olympus, Tokyo, Japan).
H&E staining
Eyes of rats were enucleated and fixed in 4% paraformaldehyde (wt/vol.) for 24 h. The retina and sclera were dehydrated in a graded ethanol series and embedded in paraffin. For H&E staining, 5 μm thick sections were taken along the vertical meridian and observed under an Olympus BX-51 light microscope (Olympus, Tokyo, Japan).
Immunofluorescence analysis
Standard immunofluorescence analysis was performed to localise NLRP3 and ASC expression in retina of rats as previously described [17]. For TUNEL analysis, the retina sections of rats were stained using a fluorescein-conjugated TUNEL in situ cell death detection kit (Roche Diagnostics, Mannheim, Germany). Images were acquired using a confocal microscope (Leica, Heidelberg, Germany).
Lectin labelling of the adherent retinal leucocytes
Leucostasis assay was performed using a perfusion labelling technique, as previously reported [18] (see ESM Methods). FITC-coupled concanavalin A lectin (Vector Labs, Burlingame, CA, USA) was used to label the adherent leucocytes and the vascular endothelial cells in rat retinas. Images were examined under a confocal microscope (Leica, Heidelberg, Germany).
Transmission electron microscopy analysis
Retinal sections of rats, approximately 2 mm × 3 mm, were isolated from each eyecup following the protocol described previously [19] (see ESM Methods). The ultrastructure of the retina tissues was observed using a transmission electron microscope (Tecnai G2 Spirit Bio TWIN; FEI, Hillsboro, OR, USA).
Immunoprecipitation and western blot analysis
Immunoprecipitation and western blotting were performed as described previously [17] using NLRP3, ASC, caspase 1, caspase 3, CREB, p-CREB, EP1R, EP2R, EP3R, EP4R, COX-2, Epac1, β-actin and Laminb antibodies (see ESM Methods).
RNA quantification
The relative expression levels of mRNA of rat Icam1, rat Il-1β, human IL-1β, human NLRP3, human EP1R, human EP2R, human EP3R or human EP4R were quantified by quantitative RT-PCR [17] (see ESM Methods). The data were analysed by using the \( {2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \) method and normalised to endogenous control GAPDH or Gapdh mRNA. The primers used are detailed in ESM Table 1.
Tissue and serum biochemical measurements
Serum lactate dehydrogenase (LDH) levels in the cell culture supernatant fractions of the hRMECs were measured using commercially available assays (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Flow cytometry
The hRMECs were suspended in 400 μl of binding buffer (422201, Biolegend, San Diego, CA, USA) and stained with 5 μl of annexin V–FITC at 4°C in the dark. After 15 min, the cells were incubated with 10 μl of propidium iodide buffer for 5 min at 4°C in the dark. The cell apoptosis rates were evaluated by a Cytomics FC500 instrument (Beckman Coulter, Miami, FL, USA).
IL-1β and PGE2 assay
IL-1β and PGE2 in the cell culture supernatant fractions of the hRMECs was measured using commercial ELISA kits (Elabscience Biotechnology Co., Wuhan, China; Cayman Chemical Company, Ann Arbor, MI, USA, respectively). The cell culture supernatant fraction was concentrated tenfold by ultrafiltration centrifugation (Amicon Ultra-0.5; Millipore, Billerica, MA, USA).
Statistical analysis
The experiments were not performed blind. However, an effort was made to simulate the conditions of blinded assays. All the samples were obtained via the same procedures and treated in the same way. All the data was obtained via direct recording of the physiological variables so that the analysis did not include any subjective evaluations. The results are expressed as the mean ± SEM. Significance was established between two groups using Student’s t test (paired t test), while ANOVA was used for multiple group comparisons followed by Tukey’s post hoc test. Tukey’s post hoc test was run only if the F value achieved p < 0.05 and there was no significant variance in homogeneity. The data was analysed with the GraphPad Prism-5 statistical software (Prism v5.0; GraphPad Software, La Jolla, CA, USA). Differences were considered statistically significant at p < 0.05.
Results
PGE2/EP2R signalling mediates the impairment of retinal vessels in rat model of type 1 diabetes
The potent proinflammatory and angiogenic cytokine PGE2 was upregulated in the vitreous fluid of individuals with diabetic retinopathy vs healthy individuals (ESM Fig. 1a). We cultured hRMECs in high-glucose medium to mimic diabetic conditions in vitro. High-glucose stress resulted in a significant increase in COX-2 and EP2R expression compared with the control medium (ESM Fig. 1b–d). Moreover, EP2R (also known as PTGER2) mRNA levels were significantly increased in hRMECs treated with ATP and lipopolysaccharide (important risk factors for endothelial cell injury in diabetic retinopathy) (ESM Fig. 1e). This data suggests a potential role for EP2R in RMECs experiencing high-glucose stress.
Diabetic retinopathy is typically characterised by an abnormal change in the retinal microvasculature, resulting in retinal non-perfusion, increased vasopermeability and pathological intraocular proliferation of the retinal vessels [20]. We used intravitreal injection of an EP2R agonist (butaprost) and antagonist (AH6809) in STZ-induced diabetic rats for 3 months to investigate whether EP2R is a potential regulator of diabetes-induced microvascular complications. Evans Blue leakage assay indicated that PGE2 and butaprost increased diabetes-induced retinal vascular leakage; these symptoms were alleviated in rats that were pre-treated with AH6809 (Fig. 1a–c). Retinal trypsin digestion assay indicated that diabetes-associated pericyte loss and capillary degeneration were more severe in the retinas of PGE2- and butaprost-treated rats (Fig. 1d,e). Inhibition of EP2R partially reduced this detrimental effect (Fig. 1d,e).

PGE2/EP2R signalling mediates retinal vascular leakage and capillary degeneration in diabetic rats. (a–c) Rats were infused with Evans Blue dye for 2 h. Red fluorescence dots in the flat-mounted retina indicated retinal vascular leakage. The fluorescence signal was detected using an Olympus BX-51 light microscope at ×4 objective (a); scale bar, 100 μm. The area (b) and the quantity (c) of the Evans Blue leakage were determined (n=6). (d, e) Retinal trypsin digestion was used to detect changes in the pericytes and the acellular capillaries; scale bar, 25 μm. Representative images are shown (d). Original magnification ×200 using an Olympus BX-51 light microscope; scale bar, 25 μm. Red arrows indicate acellular capillaries. Acellular capillaries were quantified in 30 random fields per retina and averaged (n=5) (e). Results are presented as means ± SEM; *p<0.05 and **p<0.01 for each pair of groups indicated. Con, vehicle control-treated non-diabetic rats
PGE2/EP2R signalling influences morphological changes in diabetic rat retina
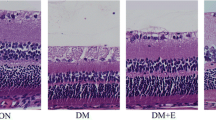
The fundus images taken in PGE2- and butaprost-treated diabetic rats showed that the intraretinal microvascular abnormalities occurred as early as 6 weeks after diabetes was successfully established (Fig. 2a). At that time, the deposition of extravasated lipoproteins in each group of rats was not yet clearly visible by fundus photography. OCT showed an increased retinal thickness in the PGE2- and butaprost-treated diabetic rats. The number of hyperreflective dots (arrows) in the superficial portion of the inner retina dramatically increased in the PGE2- and butaprost-treated diabetic rats (Fig. 2a,b). In contrast, the AH6809 pre-treated group showed significantly improved morphology of the retinal layers under diabetic conditions (Fig. 2a,b). We continued to monitor the retinal oedema and detachments later in the course of the disease model. Three months after diabetes was successfully established, histological examination showed that the retinal tissue in the control rats housed under normal conditions was intact and that the layers of the retina were clear and regularly arranged (Fig. 2c). By comparison, the retinal oedema in the STZ-treated rats was remarkable, and in the PGE2- and butaprost-treated rats, the disordered retinal structure, retinal oedema and neovascularisation were potently exacerbated (Fig. 2c–e). Meanwhile, rats pre-treated with AH6809 demonstrated improved histological retinal changes under diabetic conditions (Fig. 2c–e).

PGE2/EP2R signalling influences morphological changes in the eyes of diabetic rats. (a, b) Retinal images at 6 weeks and 9 weeks after diabetes was successfully established in the rat model (a). Note the morphological changes in the colour fundus images and the OCT images. The number of hyperreflective dots in the OCT images (arrows) was determined (b). (c–e) H&E staining in paraffin sections of rat retinas 3 months after establishment of the diabetes model (c); scale bar, 25 μm. GCL–IPL and retinal thickness were evaluated in the H&E-stained sections (d, e). The results are presented as means ± SEM; n=6, *p<0.05 and **p<0.01 for each pair of groups indicated. Con, vehicle control-treated non-diabetic rats; GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer
PGE2/EP2R signalling regulates endothelial cell apoptosis in the retina of STZ-induced diabetic rats
In diabetic retinopathy, endothelial cell injury and apoptosis are thought to be one of the initial pathological changes responsible for breakdown of the blood–retina barrier and the subsequent vascular hyperpermeability [20, 21]. In advanced diabetic retinopathy, there is a more severe loss of endothelial cells due to retinal hypoperfusion and hypoxia, as well as the aberrant formation of new blood vessels [22]. Three months after diabetes was successfully established in our rat model, the PGE2- and butaprost-treated groups displayed significantly increased amounts of diabetes-induced cell apoptosis as assessed by TUNEL assay; compared with these groups, the rats treated with the EP2R antagonist AH6809 showed decreased levels of apoptosis (Fig. 3a,b). Electron-microscopic examination revealed that PGE2 induced apoptotic nuclear condensations in the endothelial cells when compared with cells from the untreated diabetic rats (Fig. 3c). In addition, the endothelium was partially detached from the basal membrane in the PGE2-treated group. At the same time point, the diabetic rats treated with AH6809 showed attenuated cellular injury (Fig. 3c), which correlated with the TUNEL assay data.

PGE2/EP2R signalling regulates endothelial cell apoptosis in the retina of STZ-induced diabetic rats. (a) Apoptotic cells in the retinal sections were detected by TUNEL assay. Retinal vessels were counterstained with isolectin B4 (IB4, green); scale bar, 25 μm. (b) Quantification of TUNEL-positive cells in the outer nuclear layer, inner nuclear layer and ganglion cell layer. The results are presented as means ± SEM; n=6, *p<0.05 for each pair of groups indicated. (c) Representative electron micrographs of retinal vascular endothelial cells in diabetic rats treated with intravitreal injection of PGE2 or AH6809, showing samples from two control rats; scale bar, 2 μm. Arrows indicate retinal vascular endothelial cells. Con, vehicle control-treated non-diabetic rats; EC, endothelial cell; GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; nu, nucleus of retinal vascular endothelial cell; ONL, outer nuclear layer; OPL, outer plexiform layer
PGE2/EP2R signalling is engaged in leucocyte adhesion in the diabetic rat retina
Chronic subclinical inflammatory response is thought to play a critical role in the pathogenesis of diabetic retinopathy [18, 23]. Leucostasis, a main characteristic of inflammation in diabetic retinopathy, is reported to contribute to retinal capillary closure, non-perfusion, capillary dropout and local ischaemia [24, 25]. In our rat model, adherent retinal leucocytes were labelled in situ with FITC-linked concanavalin A. Six weeks after diabetes was successfully established, retinal flat mounts were prepared and the adherent leucocytes were counted in the blood vessels. Compared with the diabetic retina, a 3.6-fold (n = 7, p < 0.01) and 3.3-fold (n = 7, p < 0.01) increase in the number of adherent leucocytes was seen in the PGE2- and butaprost-treated diabetic retinal capillaries, respectively (Fig. 4a,b). A 0.7-fold (n = 7, p < 0.01) decrease in the number of adherent leucocytes was seen in the retinas of the AH6809-treated rats vs untreated diabetic rats (Fig. 4a,b).This was consistent with the mRNA level of Icam1 in the retinas (Fig. 4c).

PGE2/EP2R signalling is engaged in leucocyte adhesion and inflammation in the retina of diabetic rats. (a) Representative images of retina sections showing adherent leucocytes labelled with FITC–concanavalin A within the diabetic rat retinal vessels; scale bar, 50 μm. (b) Quantification of adherent leucocytes in the retina sections. (c) Icam1 mRNA expression in retina (fold normalised to Gapdh) was determined by real-time PCR. The results are presented as means ± SEM; n=6, *p<0.05 and **p<0.01 for each pair of groups indicated. Con, vehicle control-treated non-diabetic rats
PGE2/EP2R signalling is involved in the activation of the NLRP3 inflammasome in vivo
NLRP3 inflammasome activation has been reported in diabetic retinopathy [26]. Its effector molecule, IL-1β, mediates leucostasis and apoptosis in retinal capillary endothelial cells [27]. The NLRP3 inflammasome comprises the cytoplasmic receptor NLRP3, the adaptor molecule ASC and pro-caspase 1. Assembly of the NLRP3 inflammasome requires the association of NLRP3 with ASC oligomers via homotypic pyrin domain interactions. In our study, the PGE2-treated diabetic rats displayed augmented formation of the diabetes-induced NLRP3–ASC complex, while AH6809 blocked the STZ-related association of NLRP3 with the ASC oligomers (Fig. 5a). Moreover, this was consistent with Il-1β (also known as Il1b) and Nlrp3 mRNA levels in the retina (Fig. 5b,c).

PGE2/EP2R signalling is involved in the activation of the NLRP3 inflammasome in vivo. (a) Representative images of the immunofluorescence staining of NLRP3 and ASC in the retinal sections of each group of rats; scale bar, 25 μm. (b, c) Il-1β and Nlrp3 mRNA expression in retina (fold normalised to Gapdh) was measured by real-time PCR. The results are presented as means ± SEM; n=6, *p<0.05 and **p<0.01 for each pair of groups indicated. Con, vehicle control-treated non-diabetic rats; GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer
PGE2/EP2R signalling mediates activation of the NLRP3–ASC complex inflammasome in hRMECs
Endothelial cells are recognised as the primary cellular targets for diabetes-induced vascular damage [20, 28, 29]. We selected an RMEC line to study the mechanistic aspects and the functional significance of PGE2/EP2R signalling alteration in vitro. The association of NLRP3 with the ASC oligomers significantly increased in the PGE2- and butaprost-treated cells; the association was inhibited by AH6809 pre-treatment (Fig. 6a). To further assess the impact of the PGE2/EP2R signalling alteration on inflammasome activation, we examined the maturation of pro-caspase 1, which is cleaved into active 10 kDa or 20 kDa fragments that then enzymatically cleave pro-IL-1β to produce mature IL-1β. Treatment with high glucose or LPS + ATP increased both the caspase 1 cleavage and the levels of IL-1β and NLRP3 mRNA in hRMECs (ESM Fig. 2a,b). A similar effect was seen in hRMECs treated with increasing concentrations of PGE2 (1–20 μmol/l) (ESM Fig. 2c). Caspase 1 cleavage in the hRMECs was increased by both PGE2 and butaprost treatment; the effect was diminished in the cells pre-treated with AH6809 (Fig. 6b). AH6809 administration also significantly attenuated PGE2-induced increases in the mRNA levels of IL-1β and NLRP3 (Fig. 6c). We next addressed whether IL-1β administration could mediate apoptosis of hRMECs. IL-1β (10 ng/ml) stimulation significantly increased the activation of caspase 3, consistent with the number of apoptotic cells measured by flow cytometry analysis (Fig. 6d,e). As previously mentioned, pyroptosis is initiated by caspase 1 activation and leads to membrane pore formation and the leakage of cellular contents. To confirm that the cell death induced by PGE2 and butaprost was pyroptosis, the caspase 1 inhibitor, z-YVAD-fmk (z-VAD), was used. z-VAD pre-treatment effectively abolished the LDH release and the caspase 3 activation induced by PGE2 and butaprost exposure (Fig. 6f,g).

PGE2/EP2R signalling mediates the activation of the NLRP3–ASC complex in hRMECs. The hRMECs were treated with 1 μmol/l PGE2 for 24 h, with or without 30 min AH6809 pre-treatment. (a) Immunoprecipitation was performed with anti-NLRP3 antibody followed by immunoblotting with ASC antibody; n=5. (b) The levels of pro-caspase 1 and cleaved caspase 1 (P20) were determined by western blot; n=5. (c) NLRP3 and IL-1β mRNA expression in hRMECs (fold normalised to GAPDH) was detected by real-time PCR; n=4. (d, g) The levels of caspase 3 and cleaved caspase 3 in response to different treatments as indicated were determined by western blot; n=5. (e) Apoptosis of the hRMECs was determined by annexin V–FITC/propidium iodide flow cytometry; n=6. (f) LDH activity was measured in each group; n=4. Representative blots are shown, with quantification. Data are presented as means ± SEM. *p<0.05 and **p<0.01 for each pair of groups indicated. AV, annexin V; Buta, butaprost; Con, vehicle control; gprot, gram protein; PI, propidium iodide
EP2R-coupled cAMP/protein kinase A/cAMP response element-binding protein signalling mediated NLRP3 activation and pyroptosis in RMECs
We next examined the signalling pathways involved in regulating the PGE2/EP2R-mediated NLRP3 activation and pyroptosis in hRMECs. Recent studies have shown that PGE2 activates EP2R, stimulating cAMP/protein kinase A (PKA) signalling [30, 31]. Indeed, in our study, pre-treatment of the hRMECs with either SQ22536 (an adenylate cyclase inhibitor) or H89 (a PKA inhibitor) dramatically inhibited the PGE2-induced NLRP3 activation (Fig. 7a,b). This was consistent with the decreased IL-1β and NLRP3 mRNA levels, as well as the attenuated IL-1β production (Fig. 7c,d). Epac1 and PKA are both intracellular receptors of cAMP. However, Epac1 expression was significantly inhibited by high glucose (30 mmol/l) compared with normal glucose (5 mmol/l) treatment (ESM Fig. 3a,b). Epac1 agonist had no effect on PGE2-induced NLRP3 inflammasome activation in hRMECs (ESM Fig. 3c,d). Under the circumstances, diabetic retinopathy and high-glucose stress-associated PGE2/EP2R-mediated NLRP3 activation may mainly result from the cAMP/PKA/cAMP response element-binding protein (CREB) signalling pathway. It has been reported that activated PKA transfers into the cell nucleus and phosphorylates the transcription factor CREB protein, regulating gene expression. Therefore, we examined the phosphorylation of CREB at Ser133, which is critical for CREB transcriptional activation. Stimulation of hRMECs with PGE2 and butaprost led to a significant increase in CREB phosphorylation, which was blocked by AH6809 pre-treatment (Fig. 7e). PGE2 induced the phosphorylation and activation of CREB in rats and this was also blocked by AH6809 pre-treatment (ESM Fig. 4a,b). We also investigated the effects of AH6809 on the activation of NF-κB, another key transcription factor implicated in PGE2/EP2R signalling pathways in hRMECs. There was no significant difference noted between each group of hRMECs (ESM Fig. 5a,b). Moreover, both the adenylate cyclase inhibitor (SQ22536) and the PKA inhibitor (H89) inhibited CREB phosphorylation (Fig. 7f). Taken together, this data suggests that the cAMP/PKA/CREB signalling pathway is involved in EP2R-mediated NLRP3 activation and pyroptosis in hRMECs.

EP2R-coupled cAMP/PKA/CREB signalling mediates NLRP3 activation and pyroptosis in hRMECs. (a) Immunoprecipitation was performed with anti-NLRP3 antibody followed by immunoblotting with ASC antibody; n=4. (b) The levels of pro-caspase 1 and cleaved caspase 1 (P20) were determined by western blot; n=5. (c) NLRP3 and IL-1β mRNA expression in hRMECs (fold normalised to GAPDH) was determined by real-time PCR; n=4. (d) IL-1β was detected in the cell culture supernatant fractions of hRMECs by ELISA; n=3. (e, f) The levels of p-CREB and CREB were determined by western blot. Laminb antibody was used to confirm equal nuclear protein loading among samples; n=5. Representative blots are shown, with quantification. The results are presented as means ± SEM. *p<0.05 and **p<0.01 for each pair of groups indicated. Con, vehicle control
Discussion
In the present study, we demonstrated the important role of PGE2/EP2R signalling in diabetes-associated hRMEC dysfunction and vascular dysfunction. We found that the diabetes-associated inflammation and hRMEC damage were ameliorated by inhibition of the PGE2/EP2R and coupled cAMP–PKA–CREB signalling pathways. This was associated with an amelioration of NLRP3 inflammasome activation.
Animals in the STZ-induced diabetes model displayed symptoms of diabetic retinopathy such as endothelium impairment, vascular leakage and capillary degeneration, leading to increased acellular vessels [32,33,34]. We showed that the retinas of STZ-induced diabetic rats developed phenotypical and histopathological features consistent with diabetic retinopathy. PGE2, butaprost, AH6809 and DMSO were administered to the rats by intravitreal injection, suggesting that their retinal effects are independent of any systemic activity. We observed that both PGE2 and butaprost aggravated the deleterious effects of STZ-induced diabetes, while the effects were ameliorated in the animal group that received AH6809.
OCT is a non-invasive imaging modality that enables quantitative measurement of retinal thickness and evaluation of morphological changes in eyes with diabetic retinopathy and diabetic macular oedema [35]. Consistent with the histological examination, the OCT images revealed retinal oedema and an increased number of hyperreflective dots in the inner retina of PGE2- and butaprost-treated diabetic rats. The hyperreflective dots in diabetic retina delineated on spectral domain OCT represent activated microglia cells and it has been reported that the number increases with progressing retinopathy [36, 37]. These hyperreflective dots have been described in inflammatory retinal conditions and may present in diabetic eyes even when clinical retinopathy is undetectable [38, 39]. Therefore, hyperreflective dots in OCT imaging of diabetic retinas are a prominent feature of the disease process and may be used to closely monitor diabetic retinopathy in clinical practice.
In the early stages of diabetic retinopathy, hyperglycaemia and chronic inflammation damage the retinal endothelium and play a key role in further vascular leakage, pericyte loss, increased acellular vessels and the eventual manifestation of clinical diabetic retinopathy symptoms [40, 41]. Leucostasis is a characteristic of diabetic retinopathy inflammation [25, 42]. Our results demonstrate that PGE2 and butaprost treatment of hRMECs increases diabetes-induced leucocyte infiltration, Icam1 expression and apoptosis of RMECs. These effects were suppressed by AH6809 pre-treatment. However, the sample size of the AH6809 + STZ group in our study was not large enough to draw an irrefutable conclusion regarding Icam1 expression.
Recent studies have implicated the inappropriate activation of the NLRP3 inflammasome in diabetic retinopathy [26]. Glucose abnormalities have also been reported to be an important trigger of the sterile inflammatory response mediated by the NLRP3 inflammasome [43]. We observed that PGE2 and butaprost promoted diabetes-induced activation of NLRP3 inflammasome in vivo and in vitro and that this could be prevented by AH6809 administration. Moreover, exogenous administration of IL-1β led to an elevation in hRMEC apoptosis. Alternatively, however, it was also reported that PGE2 inhibits NLRP3 inflammasome activation through EP4R and intracellular cAMP in human macrophages [44]. This difference might be due to the dose and the way in which PGE2 was used in those studies (PGE2 was used after LPS priming). The differences in our findings might also arise from cell type and epoprostanoid expression. This data suggests that the PGE2/EP2R signalling pathway mediates NLRP3 inflammasome activation in diabetic retinopathy.
The G protein consists of α, β and γ subunits, and the α subunit is divided into Gαs, Gαi and Gαq, among others. According to previous reports, EP2R couples to the Gαs subunit, activates adenylate cyclase, increases cytoplasmic cAMP and induces PKA activation [12]. Our data showed that the PKA inhibitor H89 and the adenylate cyclase inhibitor SQ22536 suppressed the increase in IL-1β and NLRP3 mRNA levels, as well as the increase in IL-1β levels, induced by PGE2. This demonstrates that cAMP and PKA are involved in the signalling pathway mediated by PGE2/EP2R.
The transcription factors CREB and NF-κB play pivotal roles in the PGE2 signalling pathway and the development of diabetic retinopathy [45,46,47,48]. Toll-like receptor-mediated NF-κB activation is associated with an acute activation of the NLRP3 inflammasome and production of IL-1β in macrophages. After these acute and initial signals are received, adenosine further regulates IL-1β production by activating the cAMP–PKA–CREB signalling cascade, resulting in the upregulation of pro-IL-1β and NLRP3, further activating caspase 1, without the need for any other initiating signals [49, 50]. This signalling pathway has an established important role in several chronic inflammatory diseases. Likewise, we observed in hRMECs that the ability of the PGE2/EP2R–cAMP–PKA signalling pathway to upregulate pro-IL-1β was dependent on CREB activation, while there was no significant difference in the activation of NF-κB.
In summary, we demonstrated that disruption of the PGE2/EP2R signalling pathway contributes to the attenuation of diabetic retinopathy. The underlying mechanism is multifold. First, long-term exposure to high glucose concentrations and other diabetes risk factors increases the expression and activation of COX-2, subsequently promoting the PGE2/EP2R–cAMP–PKA signalling pathway. PKA transfers into the cell nucleus and phosphorylates the transcription factor CREB, upregulating the transcription of NLRP3 and pro-IL-1β. Although the molecular basis for the activation of the PGE2/EP2R cascade in diabetic retinopathy remains to be delineated, the present study implies that the PGE2/EP2R signalling pathway is a target for new therapeutic strategies to prevent and treat diabetic retinopathy.
Data availability
All relevant data are included in the article and/or the ESM files.
Abbreviations
- ASC:
-
Apoptosis-associated speck-like protein containing a C-terminal caspase-activation and recruitment domain
- COX-2:
-
Cyclooxygenase-2
- CREB:
-
cAMP response element-binding protein
- EP1–4R:
-
E-prostanoid1–4 receptor
- hRMEC:
-
Human RMEC
- LDH:
-
Lactate dehydrogenase
- NLRP3:
-
Nucleotide-binding domain and leucine rich repeat containing family, pyrin domain containing 3
- OCT:
-
Optical coherence tomography
- PGE2 :
-
Prostaglandin E2
- PKA:
-
Protein kinase A
- RMEC:
-
Retinal microvascular endothelial cell
- STZ:
-
Streptozotocin
- z-VAD:
-
Z-YVAD-fmk
References
Zheng Y, He M, Congdon N (2012) The worldwide epidemic of diabetic retinopathy. Indian J Ophthalmol 60(5):428–431. https://doi.org/10.4103/0301-4738.100542
Duh EJ, Sun JK, Stitt AW (2017) Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2(14). https://doi.org/10.1172/jci.insight.93751
Li AF, Roy S (2009) High glucose-induced downregulation of connexin 43 expression promotes apoptosis in microvascular endothelial cells. Invest Ophthalmol Vis Sci 50(3):1400–1407. https://doi.org/10.1167/iovs.07-1519
Monaghan K, McNaughten J, McGahon MK et al (2015) Hyperglycemia and diabetes downregulate the functional expression of TRPV4 channels in retinal microvascular endothelium. PLoS One 10:e128359
Zhang Y, Lv X, Hu Z et al (2017) Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis 8(7):e2941. https://doi.org/10.1038/cddis.2017.308
Liu K, Liu H, Zhang Z, Ye W, Xu X (2016) The role of N-glycosylation in high glucose-induced upregulation of intercellular adhesion molecule-1 on bovine retinal endothelial cells. Acta Ophthalmol 94(4):353–357. https://doi.org/10.1111/aos.13028
Sutterwala FS, Haasken S, Cassel SL (2014) Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319(1):82–95. https://doi.org/10.1111/nyas.12458
Chen W, Zhao M, Zhao S et al (2017) Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res 66(2):157–166. https://doi.org/10.1007/s00011-016-1002-6
Zhang Y, Yang X, Qiu C, Liu F, Liu P, Liu Z (2018) Matrine suppresses AGE-induced HAEC injury by inhibiting ROS-mediated NRLP3 inflammasome activation. Eur J Pharmacol 822:207–211. https://doi.org/10.1016/j.ejphar.2018.01.029
Zaslona Z, Palsson-McDermott EM, Menon D et al (2017) The induction of pro-IL-1beta by lipopolysaccharide requires endogenous prostaglandin E2 production. J Immunol 198(9):3558–3564. https://doi.org/10.4049/jimmunol.1602072
Hata AN, Breyer RM (2004) Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther 103(2):147–166. https://doi.org/10.1016/j.pharmthera.2004.06.003
Wu T (2005) Cyclooxygenase-2 and prostaglandin signaling in cholangiocarcinoma. Biochim Biophys Acta 1755(2):135–150. https://doi.org/10.1016/j.bbcan.2005.04.002
Schoenberger SD, Kim SJ, Sheng J, Rezaei KA, Lalezary M, Cherney E (2012) Increased prostaglandin E2 (PGE2) levels in proliferative diabetic retinopathy, and correlation with VEGF and inflammatory cytokines. Invest Ophthalmol Vis Sci 53(9):5906–5911. https://doi.org/10.1167/iovs.12-10410
Kern TS, Miller CM, Du Y et al (2007) Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes 56(2):373–379. https://doi.org/10.2337/db05-1621
Ayalasomayajula SP, Kompella UB (2003) Celecoxib, a selective cyclooxygenase-2 inhibitor, inhibits retinal vascular endothelial growth factor expression and vascular leakage in a streptozotocin-induced diabetic rat model. Eur J Pharmacol 458(3):283–289. https://doi.org/10.1016/S0014-2999(02)02793-0
Shan K, Liu C, Liu BH et al (2017) Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation 136(17):1629–1642. https://doi.org/10.1161/CIRCULATIONAHA.117.029004
Wang X, Gao Y, Song J et al (2017) The TIR/BB-loop mimetic AS-1 prevents non-alcoholic steatohepatitis and hepatic insulin resistance by inhibiting NLRP3-ASC inflammasome activation. Br J Pharmacol 174(12):1841–1856. https://doi.org/10.1111/bph.13786
Noda K, Nakao S, Zandi S, Sun D, Hayes KC, Hafezi-Moghadam A (2014) Retinopathy in a novel model of metabolic syndrome and type 2 diabetes: new insight on the inflammatory paradigm. FASEB J 28(5):2038–2046. https://doi.org/10.1096/fj.12-215715
Zhang J, Wu Y, Jin Y et al (2008) Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci 49(2):732–742. https://doi.org/10.1167/iovs.07-0721
Watson EC, Grant ZL, Coultas L (2017) Endothelial cell apoptosis in angiogenesis and vessel regression. Cell Mol Life Sci 74(24):4387–4403. https://doi.org/10.1007/s00018-017-2577-y
Feenstra DJ, Yego EC, Mohr S (2013) Modes of retinal cell death in diabetic retinopathy. J Clin Exp Ophthalmol 4:298
Antonetti DA, Klein R, Gardner TW (2012) Diabetic retinopathy. N Engl J Med 366(13):1227–1239. https://doi.org/10.1056/NEJMra1005073
Wu H, Hwang DK, Song X, Tao Y (2017) Association between aqueous cytokines and diabetic retinopathy stage. J Ophthalmol 2017:9402198
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158(1):147–152. https://doi.org/10.1016/S0002-9440(10)63952-1
Rangasamy S, McGuire PG, Franco NC, Monickaraj F, Oruganti SR, Das A (2014) Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One 9(10):e108508. https://doi.org/10.1371/journal.pone.0108508
Chen H, Zhang X, Liao N et al (2018) Enhanced expression of NLRP3 inflammasome-related inflammation in diabetic retinopathy. Invest Ophthalmol Vis Sci 59(2):978–985. https://doi.org/10.1167/iovs.17-22816
Zhu X, Xie M, Wang K et al (2014) The effect of puerarin against IL-1beta-mediated leukostasis and apoptosis in retinal capillary endothelial cells (TR-iBRB2). Mol Vis 20:1815–1823
Zhang W, Chen S, Liu ML (2018) Pathogenic roles of microvesicles in diabetic retinopathy. Acta Pharmacol Sin 39(1):1–11. https://doi.org/10.1038/aps.2017.77
Eshaq RS, Aldalati A, Alexander JS, Harris NR (2017) Diabetic retinopathy: breaking the barrier. Pathophysiology 24(4):229–241. https://doi.org/10.1016/j.pathophys.2017.07.001
Brudvik KW, Tasken K (2012) Modulation of T cell immune functions by the prostaglandin E(2) - cAMP pathway in chronic inflammatory states. Br J Pharmacol 166(2):411–419. https://doi.org/10.1111/j.1476-5381.2011.01800.x
Luo X, Zhu Q, Zhang J, Huang Q, Xie Z, Cheng Y (2017) The double roles of the prostaglandin E2 EP2 receptor in intracerebral hemorrhage. Curr Drug Targets 18(12):1377–1385. https://doi.org/10.2174/1389450117666151209122826
Olivares AM, Althoff K, Chen GF et al (2017) Animal Models of Diabetic Retinopathy. Curr Diab Rep 17(10):93. https://doi.org/10.1007/s11892-017-0913-0
Lai AK, Lo AC (2013) Animal models of diabetic retinopathy: summary and comparison. J Diabetes Res 2013:106594
Fong DS, Aiello L, Gardner TW et al (2003) Diabetic retinopathy. Diabetes Care 26(1):226–229. https://doi.org/10.2337/diacare.26.1.226
Sun JK, Lin MM, Lammer J et al (2014) Disorganization of the retinal inner layers as a predictor of visual acuity in eyes with center-involved diabetic macular edema. JAMA Ophthalmol 132(11):1309–1316. https://doi.org/10.1001/jamaophthalmol.2014.2350
Vujosevic S, Bini S, Midena G, Berton M, Pilotto E, Midena E (2013) Hyperreflective intraretinal spots in diabetics without and with nonproliferative diabetic retinopathy: an in vivo study using spectral domain OCT. J Diabetes Res 2013:491835
Garner A (1981) Developments in the pathology of diabetic retinopathy: a review. J R Soc Med 74(6):427–431. https://doi.org/10.1177/014107688107400607
Zeng HY, Green WR, Tso MO (2008) Microglial activation in human diabetic retinopathy. Arch Ophthalmol 126(2):227–232. https://doi.org/10.1001/archophthalmol.2007.65
Ogino K, Murakami T, Tsujikawa A et al (2012) Characteristics of optical coherence tomographic hyperreflective foci in retinal vein occlusion. Retina 32(1):77–85. https://doi.org/10.1097/IAE.0b013e318217ffc7
Dejana E, Tournier-Lasserve E, Weinstein BM (2009) The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 16(2):209–221. https://doi.org/10.1016/j.devcel.2009.01.004
Ogura S, Kurata K, Hattori Y et al (2017) Sustained inflammation after pericyte depletion induces irreversible blood-retina barrier breakdown. JCI Insight 2:e90905
Vestweber D (2015) How leukocytes cross the vascular endothelium. Nat Rev Immunol 15(11):692–704. https://doi.org/10.1038/nri3908
Zhang Y, Lv X, Hu Z et al (2017) Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis 8(7):e2941. https://doi.org/10.1038/cddis.2017.308
Sokolowska M, Chen LY, Liu Y et al (2015) Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J Immunol 194(11):5472–5487. https://doi.org/10.4049/jimmunol.1401343
Devi TS, Singh LP, Hosoya K, Terasaki T (2011) GSK-3β/CREB axis mediates IGF-1-induced ECM/adhesion molecule expression, cell cycle progression and monolayer permeability in retinal capillary endothelial cells: Implications for diabetic retinopathy. Biochim Biophys Acta 1812(9):1080–1088. https://doi.org/10.1016/j.bbadis.2011.04.007
Graves DT, Kayal RA (2008) Diabetic complications and dysregulated innate immunity. Front Biosci 13(13):1227–1239. https://doi.org/10.2741/2757
Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M (2015) CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A 112(51):15642–15647. https://doi.org/10.1073/pnas.1519644112
Steer SA, Corbett JA (2003) The role and regulation of COX-2 during viral infection. Viral Immunol 16(4):447–460. https://doi.org/10.1089/088282403771926283
Baron L, Gombault A, Fanny M et al (2015) The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis 6(2):e1629. https://doi.org/10.1038/cddis.2014.576
Ouyang X, Ghani A, Malik A et al (2013) Adenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1α pathway. Nat Commun 4(1):2909. https://doi.org/10.1038/ncomms3909
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 81770941), Jiangsu Key Medical Disciplines (ZDXKC2016008), Technology Development Fund (CSE12N1701) and Wuxi Eminent Medical Talents (JCRC005).
Author information
Authors and Affiliations
Contributions
XW and YY made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. MW, YW, TX and PZ made substantial contributions to acquisition of data, analysis and interpretation of data and revising the article critically for important intellectual content. JZ, XN, JLi, YL, QS and JLeng made substantial contributions to conception and design, analysis and interpretation of data and revising the article critically for important intellectual content. JShao, MZ and CT made substantial contributions to conception and design, acquisition of data and revising the article critically for important intellectual content. JT, YD and JSun made substantial contributions to conception and design, analysis and interpretation of data and revising the manuscript critically for important intellectual content. All authors gave final approval of the version to be published. XW had full access to all the data in this study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material
ESM
(PDF 894 kb)
Rights and permissions
About this article

Cite this article
Wang, M., Wang, Y., Xie, T. et al. Prostaglandin E2/EP2 receptor signalling pathway promotes diabetic retinopathy in a rat model of diabetes. Diabetologia 62, 335–348 (2019). https://doi.org/10.1007/s00125-018-4755-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4755-3