
Abstract
The transcription factor GATA2 is involved in human diseases ranging from hematopoietic disorders, to cancer, to infectious diseases. GATA2 is one of six GATA-family transcription factors that act as pioneering transcription factors which facilitate the opening of heterochromatin and the subsequent binding of other transcription factors to induce gene expression from previously inaccessible regions of the genome. Although GATA2 is essential for hematopoiesis and lymphangiogenesis, it is also expressed in other tissues such as the lung, prostate gland, gastrointestinal tract, central nervous system, placenta, fetal liver, and fetal heart. Gene or transcriptional abnormalities of GATA2 causes or predisposes patients to several diseases including the hematological cancers acute myeloid leukemia and acute lymphoblastic leukemia, the primary immunodeficiency MonoMAC syndrome, and to cancers of the lung, prostate, uterus, kidney, breast, gastric tract, and ovaries. Recent data has also linked GATA2 expression and mutations to responses to infectious diseases including SARS-CoV-2 and Pneumocystis carinii pneumonia, and to inflammatory disorders such as atherosclerosis. In this article we review the role of GATA2 in the etiology and progression of these various diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
GATA2 is one of six transcription factors (GATA1–GATA6) that bind to the DNA motif “GATA” through two zinc finger domains [1, 2]. Historically, mammalian GATA family members were grouped into two subfamilies: GATA1, 2 and 3 which were thought to be restricted to the hematopoietic compartment, and GATA4–GATA6 which were thought to be expressed in endodermal and cardiovascular tissues [3,4,5]. This view of GATA subfamilies is no longer considered correct, as many are expressed outside of their “canonical” locations. Indeed, GATA2 is expressed constitutively in several non-hematopoietic sites including the lymphatic endothelium, and is upregulated in myeloid cells responding to certain inflammatory and infectious stimuli. Within the hematopoietic system, GATA2 is crucial for the proliferation and maintenance of hematopoietic stem cells (HSC) and hematopoietic progenitor cells (HPC) [6]. While GATA2 is downregulated in HPCs that commit to the lymphoid lineage, expression is highest in early myeloid progenitors, in megakaryocyte and eosinophilic precursors, and is expressed by mast cells [7, 8]. Although GATA2 is most notably expressed in the hematopoietic compartment, it is also expressed in lymphatic endothelial cells, V2 interneurons within the central nervous system, and can be found expressed in multiple tissues during development including in the placenta, fetal liver, and fetal heart [9,10,11,12,13,14,15].
GATA transcription factors tend to work cooperatively with other transcription factors, allowing for the gene expression profile driven by a particular GATA protein to vary between cell types, with these differences dictated by the expression patterns of cooperating and competing transcription factors. These interactions can allow GATA2 to induce or suppress expression of the same gene depending on which GATA2-interacting partners are present [16]. GATA2 regulates genes both directly—by binding to promoters of target genes—and indirectly, by regulating the expression of other transcription factors. In this review, we described GATA2’s regulation, post-translational modifications, and its role in the development and progression of several diseases.
GATA2 gene and structure
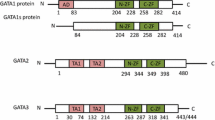
The GATA2 gene is situated at 21.3 cM on the long arm of human chromosome 3, and was first cloned by Lee et al. as a 13,760 kb loci [17]. Three GATA2 transcripts have been identified, expressed from two different promoters. The first GATA2 promoter (specific/IS promoter) is found 5′ to the first exon, with expression from the IS promoter occurring in hematopoietic and neuronal cells [9, 18]. A second internal (general/IG) promoter is found between the first and second exons and is used in all tissues that express GATA2. The IS and IG promoters contain vastly different sets of predicted transcription factor binding sites [19, 20]. Combined with alternative splicing, these promoters allow for three different mRNAs to be produced, each differing in the 5′ UTR but sharing the same translation start site, protein coding region, and 3′ UTR (Fig. 1A–B). Alternative splicing of these mRNAs can produce the short GATA2 isoform, which lacks the first 14 residues of the second zinc finger, thus reducing its size from 480 to 466 amino acids (50.5 kDa to 49.0 kDa, Fig. 1B–C) [20, 21]. Unlike many putative protein isoforms, which may represent erroneous mRNA splicing events that do not result in an expressed protein, immunoblotting of HSCs has identified a doublet GATA2 band consistent in size with the long and short isoforms [22]. Interestingly, the lower-weight band in this doublet disappears once these cells have differentiated into mature monocytes and macrophages, indicating that the short isoform may be selectively expressed in certain cell types or at specific stages of hematopoiesis [23]. Whether this short isoform has a function different from that of the long isoform, and what regulates the formation of the short isoform, remains unknown. Note that in the hematopoiesis and hematological cancer fields, it is convention to number the GATA2 exons based on their inclusion and order in the IS promoter-derived transcript (Transcript 2, NM_032638). While this is the predominant mRNA isoform expressed in the hematopoietic compartment, the transcript produced from the IG promoter (Transcript 1, NM_001145661) is the predominant transcript outside of the hematopoietic compartment and includes two exons that form the 5′ UTR that are unnumbered in this convention. As such, care must be taken when interpreting the GATA2 literature as exon numbering can be inconsistent between studies, especially in studies outside of the hematopoietic compartment. For clarity, in this review, we have used the Transcript 2 nomenclature, numbering the as exons that are alternatively spliced to create different 5′ UTRs as exons 1A, 1B, and 1C, numbered based on their order in the genome (Fig. 1A). All references to SNPs and other mutations refer either to exons numbered in this manner, or where possible, refer to the specific amino acids or protein domains that are affected.

Transcriptional regulation and structure of GATA2. A GATA2 is regulated from two separate promoters—IS (specific promoter) and IG (general promoter). The IS promoter is active in hematopoietic and neuronal tissue, while IG is active in most GATA2-expresing tissues. Exons are color-coded, numbered, and drawn to scale. Enhancers are shown as unfilled boxes. Introns are not illustrated, but their size in base pairs (bp) is indicated at the top of the panel. B Three mRNA transcripts are produced from the GATA2 gene via the IS and IG promoter and alternative splicing. All three mRNA transcripts can undergo additional alternative splicing at the junction of the first and second zinc finger, producing a 42 bp deletion. C Human GATA2 contains multiple functional domains, including a nuclear localization signal (NLS), two zinc-finger domains (N/C-ZnF), N- and C-terminal transactivation domains (TAD), and a negative regulatory domain (NRD). The short isoform of GATA2 lacks amino acids 340–353 of the C-ZnF. Domains are numbered by amino acid, starting at the N-terminus, and the coding of these domains within the GATA2 exon structure is indicated
Transcriptional regulation of GATA2
As mentioned in the previous section, the GATA2 gene is regulated by two promoters: IS and IG. The IS promoter, which produces transcript variant 2, is selectively utilized in hematopoietic and neuronal tissues. The IG promoter, which produces transcript variant 1, is active in all other tissues where GATA2 is expressed [20, 21, 24]. While the transcription factors which bind to the IS and IG promoters are not fully elucidated, the ENCODE database identified EGR1, GATA1, GATA2, GATA3, Myc, MAX, E2F1/F4/F6, ELF1, RAD21, and HDAC2/5 as factors which bind the IS promoter, while AP-1, STAT1, IRF-1, NF-kB, Myc, HDAC1, MAX, TCF12, and E2F4/F6 were found bound to the IG promoter (Fig. 1A) [25]. Experimental data have linked additional transcription factors to the regulation of GATA2, including ETS1 [26], BMP4 [27], NOTCH1 [28, 29], PU.1 [30], and EVI1 [31, 32]. Upstream of the IG and IS promoters is a − 110 kb intronic enhancer, and a second intronic enhancer can be found at the + 9.5 kb location [33, 34]. These enhancers have a significant role in the regulation of GATA2 expression, with GATA2 binding the − 110 kb enhancer in an autoregulatory fashion, along with SP1, and CTCF, while AP-1, STAT5a, GATA1, and GATA2 bind to the + 9.5 enhancer. There is a consensus sequence in + 9.5 enhancer where the hematopoietic heptad regulatory unit, comprised of GATA2, SCL, LYL1, LMO2, RUNX1, ERG, and FLI-1, which bind in this region to regulate GATA2 expression [35].
The − 110 kb and + 9.5 kDa enhancers are required for human GATA2 expression during hematopoiesis [36,37,38]. An E box GATA element present in the + 9.5 kDa enhancer scaffolds a complex including GATA2 that maintains the + 9.5 site in an open chromatin state that is required for GATA2 expression [39]. A comparison of the human and xenopus GATA2 promoters identified conserved ets binding sites in the ~ 800 bp region upstream of the GATA2 transcription start site [19]. In zebrafish, the GATA2 promoter spans over 7 kb, with tissue-specific regulation driven by discrete regions of the promoter, although these sites are not conserved in mammals [40]. The mouse and human GATA2 promoters contain GATA-response elements (GATA-RE) at − 1.8 kb, − 2.8 kb, and − 3.9 kb, and both the IS and IG promoters are autoregulated by GATA2 via these GATA2-REs, [41, 42]. In addition to transcription factor binding sites, methylation within exons 3 and 4 may negatively regulate GATA2 expression, although this regulation has only been observed in patients which hypermethylated this region due to SNPs in the methylase DNMT3A [43].
GATA2 protein structure and its post-translational modification
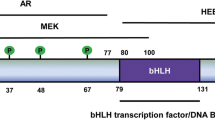
GATA2 has a conserved DNA-binding domain made of two multifunctional zinc-finger domains (ZnF): N-ZnF/ZfN1 and C-ZnF/ZfN2 (Fig. 1C) [12]. The N-ZnF domain is encoded by exon 4, and C-ZnF by exon 5, with the short form of GATA2 lacking 14 amino acids from C-ZnF. Although no functional differences between the long and short GATA2 isoforms has been identified, the short isoform lacks part of C-ZnF domain and therefore may have modified DNA or protein binding characteristics [44]. Both the N-ZnF and C-ZnF domains regulate interactions with other transcription factors. For example, C-ZnF is required for GATA2 interactions with PU.1 [45] and FOG1 [46], while both the C-ZnF and N-ZnF domains are required for interactions with PIASy [47], HDAC3 [48], RARα [5], c-MYB [4], PLZF [49], and PPARγ2 [50]. Not all inter-protein interactions occur via these zinc fingers—for example, a region proximal to the C-ZnF domain is required to interact with C/EBPA [51]. The exact size of the C-ZnF has not been resolved at high resolution, with deletion studies suggesting that it may extend from amino acid 349 through to the putative nuclear localization signal (NLS), while other studies and protein structure prediction identify a smaller zinc-finger domain that extends between amino acid 349 and 373 [52, 53]. Similarly, the localization of the NLS has not been mapped at high resolution, with Hsu et al. mapping it to the c-terminal half of GATA2 based on mutations identified in patients bearing GATA2 mutations [1, 54]. As NLS sequences are conserved across all six members of the GATA gene family, and are always found c-terminal to the second zinc finger, Hsu et al. were able to further refine the position of the GATA2 NLS to between the second zinc finger and the c-terminus, with deletion mapping by Visvader et al. refining this further to a region between amino acids 335 and 413 [2, 3, 55]. Consistent with these studies, using NLStradamus, we have identified a probable NLS between amino acids 396 and 409 (RNRKMSNKSKKSKKP), with this sequence being the only region in human GATA2 exhibiting the classical polybasic and lysine-rich structure of an NLS [4, 5, 16, 56, 57]. In addition to the zinc-finger and NLS domains, GATA2 has two transactivation domains (TAD), a negative regulatory domain (NRD), and regions that are important for GATA2 protein stability and degradation (Fig. 1C) [58, 59]. The function of these regions is not as well understood as the zinc finger domains; however, the NRD is homologous to the NRD of GATA3 which has transactivating activity [58, 60]. The N-terminal TAD is required for erythroid progenitor development, while the C-terminal TAD is required for megakaryocyte development [61]. GATA2 engages in concentration-dependent dimerization, with monomeric versus dimeric GATA2 interacting with unique DNA sites and transcription co-factors [62].
While GATA2 expression levels represents the major mechanism of GATA2 regulation, GATA2’s activity is also regulated by post-translational modifications. After translation, GATA2 is modified by acetylation, phosphorylation, and sumoylation, while uniquitination induces its rapid degradation [63, 64]. GATA2 has several evolutionarily conserved lysine-rich motifs where it is acetylated, with acetylation increasing both DNA-binding and transactivation activity. Mutation of any of these lysine residues reducing its transactivating activity [65]. GATA2 activity is also regulated by phosphorylation, with HPC proliferation requiring the constitutive phosphorylation of GATA2 [64]. This phosphorylation is regulated by a MAPK-dependent pathway that responds to developmental cytokines such as IL-3 [66, 67]. GATA2 is phosphorylated by the insulin dependent-PI3K/AKT pathway in preadipocytes and prevents GATA2 from translocating into the nucleus. The resulting loss of GATA2 nuclear activity allows preadipocyte to exit cell cycle and differentiate into adipocytes, and reduces the inflammatory activity of preadipocytes [68].
GATA2 expression varies during cell-cycle, peaking in S phase and declining through G1/S and M phase, with GATA2 protein levels largely regulated by phosphorylation-dependent changes in protein stability [69]. During cell cycle, GATA2 is phosphorylated and thus stabilized by Cdk2/cyclin A and Cdk4/cyclin D. Cdk2/cyclin A phosphorylates GATA2 during S-phase, which is required for protease-dependent degradation of GATA2 later in M phase. Control of GATA2 expression and degradation by its phosphorylation by the Cdk/cyclin systems regulates HSCs self-renewal and differentiation [69]. SUMO conjugation is another post-translational modification frequently found on GATA2. In endothelial cells, GATA2 is modified by SUMO2 addition mediated by the SUMO E3 ligase PIASy [47]. SUMOylated GATA2 acts as a transcriptional repressor by reducing Endothelin-1 expression in endothelial cells [47]. The half life of the GATA2 protein is short, typically around 30 min, although this can vary across cell types. GATA2 is turned over rapidly by the ubiquitin–proteasome pathway, in which poly-ubiquitination of GATA2 in the C- and N-terminal TADs triggers its degradation [63].
GATA2 as a pioneer transcription factor
Genome-wide location analysis demonstrated that the bulk of nuclear DNA is inaccessible due to being compacted into heterochromatin, meaning that most putative DNA-binding sites are inaccessible to transcription factors and other regulators [70]. Pioneer transcription factors have a special ability to bind these inaccessible DNA regions, in order to open the chromatin at these sites and make them accessible to other transcription factors [71, 72]. Pioneer transcription factors first gain access to compacted DNA by opening small regions of closed chromatin, allowing them to bind to their cognate DNA binding site. Once bound, pioneers then facilitate the binding of other transcription factors, co-factors, chromatin remodeling modifiers, and other modifying proteins (e.g. acetyltransferases) which facilitate the opening of the chromatin to an accessible state amenable to gene expression [72]. GATA2 is one of the first discovered pioneer transcriptions factors (Fig. 2). Once bound to heterochromatin, GATA2 recruits chromatin remodeling complexes such as the SWI/SNF complex and BRG1 [39, 73]. Once bound, these remodeling complexes modify histones and alter nucleosome positioning to open the chromatin structure and allow other transcription factors access to the DNA.

Model of GATA2’s pioneer function. Heterochromatin (closed chromatin, top) is usually inaccessible to transcription factors and is therefore not transcribed. GATA2 can bind to GATA2 binding sites within heterochromatin (middle), after which GATA2 recruits histone- and chromatin-modifiers which decompact the chromatin, forming accessible euchromatin. GATA2 can then facilitate the binding of additional transcription factors and regulators, leading to gene expression (bottom)
GATA2 can both activate or suppresses gene expression by facilitating the reorganization of the chromatin proximal to the GATA2 binding site [72]. For example, GATA2 opens the androgen receptor (AR) promoter by establishing an accessible local chromatin environment within the androgen receptor enhancer. Here, GATA2 recruits the histone acetyl transferase p300 that acetylates histone H3, producing H3K27 acetylation marks which open the chromatin structure to allow binding of other transcription factors which then enables AR expression [74]. While the mechanisms by which GATA2 opens chromatin are beginning to be uncovered, it is unclear how GATA2 inactivates genes through inducing heterochromatin formation, despite clear examples of GATA2-dependent heterochromatin formation. For example, using CUT-and-tag and ATAC-Seq, Jung et al. investigated the occupancy of GATA2 on GATA2-activated versus GATA2-suppressed genes [75]. GATA2 was found to occupy euchromatin at the GATA2-activated Far2, Gata1, Hdc, and Kit loci. In marked contrast, GATA2 was absent from the Ifi209, Tifab, Nlrp1a, and Trem1 loci, despite ATAC-seq demonstrating that GATA2 was required for the formation of heterochromatin at these loci. These data suggest that GATA2-mediated heterochromatin formation occurs via an indirect mechanism, but at this time, this mechanism has yet to be elucidated.
In addition to modifying chromatin, GATA2 also interacts with other transcription factors to regulate and target their activity, and indeed, most of GATA2’s transcriptional activity is though to occur via interactions with other transcription factors. For example, HSC survival and self-renewal is mediated by GATA2 interactions with GATA1, CEBPA, SPI1 (PU.1), and ZFPM1 (FOG1) [24], and indirect interactions with SPI1 (PU.1), FLI1, TAL1 (SCL), LMO2, LYL1, MEIS1, ERG, FLI-1, GFI1B, and RUNX1 [76,77,78]. Genome-wide analysis of HSC/HPCs demonstrated that GATA2 also forms heterodimeric transcriptional complexes with SCL/LMO2, LMO2, ERG, and FLI-1. Moreover, GATA2 binds to more than 1000 loci through cooperating with other transcription factors to form a core heptad regulatory unit in hematopoietic cells [79]. This heptad regulatory unit consist of GATA2, SCL, LYL1, LMO2, RUNX1, ERG, and FLI-1 and mainly targets microRNAs, lncRNAs, and lineage- and maturation-stage specific genes [79,80,81,82]. Interestingly, the GATA2 loci contains a lncRNA (GATA2-AS1) that is divergently transcribed with GATA2. GATA2-AS1 modifies the expression of over 1300 RNAs and contributes to the hypoxia response in endothelial cells [81].
This heptad controls cell fate during hematopoiesis through cooperative binding of heptad members within their promoters and regulatory regions. The exact mechanisms through which this complex web of interactions regulates hematopoietic cell fate are incompletely understood, but two mechanisms through which this heptad regulates hematopoietic fate determination have been identified. The balance of GATA2, RUNX1, and SCL expression controls several genes that are critical for determining the balance between HSC quiescence and proliferation, with increased GATA2 expression promoting HSC quiescence—likely in anticipation of the HSC committing to terminal differentiation [83]. This GATA2 upregulation was recently shown to promote commitment to the erythroid lineage via a subcircuit within the heptad. Recently discovered by Thoms et al., this subcircuit allows HSCs to enter the erythroid developmental lineage through the GATA2-dependent upregulation of TAL1 and the concordant GATA2-independent downregulation of ERG. Amplifying this circuit through ectopic overexpression of GATA2 or knockdown of ERG promoted commitment to the erythrocyte lineage, while ectopic overexpression of ERG maintained progenitors in a self-renewing HSC-like state [84]. Given that this same lineage commitment also leads to the development of megakaryocytes, myeloid cells, and mast cells—and that these lineages share a similar dependency on GATA2 expression—it is likely that similar GATA2-directed heptad subcircuits are involved in the development of these cell types.
The GATA switch
A GATA switch, in which one GATA-family transcription factor displaces another GATA-family factor in a promoter, is an important element of cell fate determination during hematopoiesis. A GATA2/GATA1 switch controls the relative expression of these two transcription factors, with the altering balance between GATA1 and GATA2 defining lineage choice by regulating HSC renewal, differentiation of progenitors cells, and inducing terminal differentiation of erythrocytes, megakaryocytes, and mast cells [42, 85]. GATA2 and GATA1 engage in a dynamic reciprocal regulation of each others expression, which for example, can initiate HSC to erythrocyte development by reducing GATA2 levels and elevating GATA1 expression. The change of expression of GATA transcription factors is regulated by cis-acting GATA motifs, which in HSCs and HPCs are bound by GATA2, which is highly expressed in these cells. Later in hematopoiesis, GATA1 expression increases and displaces GATA2 from these cis-acting motifs, thus inducing terminal differentiation into erythrocytes and megakaryocytes [7, 8, 86]. The high levels of GATA2 in HPCs are mediated by a positive feedback loop in which GATA2 promotes its own expression through binding GATA motifs in its promoter and enhancer regions [42]. Once a progenitor cell commits to the erythrocytes or megakaryocytic lineage, GATA1 expression increases and forms a heterodimer with FOG-1. This GATA1/FOG1 heterodimer displaces GATA2 from the GATA2 gene promoter and enhancers, thus ceasing GATA2 expression and allowing for GATA1-mediated terminal differentiation [42, 85]. Conversely, during mast cell development GATA2 downregulates FOG-1 expression, allowing for mast cell development via a GATA2-mediated genetic program [87]. The GATA2/GATA1 switch also regulates expression of embryonic and fetal haemoglobins, with GATA2 expression driving expression of γ- and ε-globin, with increases in GATA1 expression suppressing expression of both GATA2 and ε-globin [88]. In addition, GATA2 expression prevents embryonic lethality due to GATA1 deficiency by allowing for embryonic and perinatal hematopoiesis. While this allows embryonic lethality to be bypassed, the requirement for GATA1 activity in adult hematopoiesis results in death shortly after birth [89].
Although the above studies indicate that GATA2 is expressed earlier than GATA1 during hematopoiesis, and GATA1 “switches” with GATA2 on target genes through a competition mechanism [90], a recent publication has challenged this GATA switch model. Using absolute quantification of mRNA and protein levels in HSCs and erythroid progenitors, Gillepsie et al. determined that while the pattern of GATA1 and GATA2 mRNA levels were consistent with the switch model, the protein levels of GATA1 and GATA2 were not. Unexpectedly, GATA1 protein was found at much higher abundance at early timepoints than its mRNA level would suggest, exceeding GATA2 protein levels at very early stages in differentiation. At later timepoints, GATA1 protein levels were up to 50 times higher than GATA2, even though the mRNAs for each protein was present at similar copy numbers. Moreover, GATA1 protein levels were consistent during erythropoiesis despite three to fourfold changes in its mRNA abundance, while GATA2 protein levels dropped below detection shortly after its mRNA levels began to decrease. As a consequence, at the time point where HSCs had committed to the erythrocyte lineage, little-to-no GATA2 protein was present in the cells while GATA1 protein levels remained high. This indicates that in some circumstances, the GATA2/GATA1 switch may be mediated simply by a loss of GATA2 protein rather than competition for promoter binding with GATA1 [91].
GATA2 in hematopoietic disease
GATA2 regulates the transition of embryonic endothelial cells into the first HSCs, and consequently, GATA2 knockout causes embryonic lethality due to severe anemia [7]. In the bone marrow, GATA2 is expressed in HSCs, HPCs, erythroid precursors, eosinophilic and megakaryocytic progenitors, and in mast cells. In HSCs/HPCs, GATA2 is required for commitment to the myeloid lineage [92], with GATA2 expression decreasing once these precursor cells enter terminal differentiation, although some GATA2 expression is maintained in eosinophilic progenitors, and throughout the megakaryocytic and mast cell lineages [93]. GATA2 is present in the hematopoietic tissues of adult organisms [20, 94] where it continues to have an essential role in the maintenance of HSCs [6]. Knockdown of GATA2 reduces the self-renewing capacity of HSCs [92, 95], with inactivation of even one GATA2 allele causes insufficient HSC self-renewal without affecting the entry of HSCs into the myeloid differentiation pathway is unaffected [96, 97]. Not all GATA2 mutations affect HSC self-renewal, with some mutations resulting in monocytopenia and neutropenia due to failed differentiation following HSC commitment to the myeloid lineage [98]. Conversely, overexpression of GATA2 inhibits hematopoiesis by trapping HSCs in a self-renewal loop and by blocking myeloid and erythroid differentiation [83, 99]. GATA2 can also decrease the sensitivity of HSCs to apoptosis, through promoting the expression of the anti-apoptotic protein BCL2L1 [100].
Several GATA2 mutations have been identified that cause hematopoietic disorders. Mutations which decrease GATA2 expression, or which result in non-functional protein cause cytopenias including monocytopenia and mycobacterial infection (MonoMAC) syndrome and dendritic cell, monocyte, B and NK lymphoid deficiency syndrome (DCML). Similar mutations have been associated with inherited myelodysplastic syndrome [101]. Because of GATA2’s central role in hematopoiesis, in addition to the primary hematopoietic deficiencies observed in patients with GATA2 mutations, these patients are also prone to develop cytogenetic abnormalities and acute or chronic leukemias including acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and rarely, acute lymphoblastic leukemia (Fig. 3)[102]. GATA2 mutations can occur as both germline and stomatic mutations, with the latter typically arising as secondary mutations in response to an initiating oncogenic event. In addition, patients with mutations in GATA2 are also prone to lymphedema, due to the requirement for GATA2 in the development of the lymphatic system [103]. While many GATA2 mutations affecting the coding region are correlated with the severity of pediatric and adult AML, mutations can also be found in the non-coding enhancer regions that impart their oncogenic effect by altering GATA2 expression levels [52, 104].

Patterns of GATA2 expression in the onset of various GATA2-associated diseases. Green arrows indicate diseases promoted by elevated GATA2 expression, and red arrow indicates diseases promoted by lower levels of GATA2 expression or by inactivating mutations in GATA2
Monocytopenia and mycobacterial infection (MonoMAC) and related syndrome
Germline mutations in GATA2 cause a range of syndromes including MonoMAC, DCML, familial MDS/AML, and primary Emberger syndrome, while both germline and spontaneous mutations can lead to classic NK cell deficiency [54, 103, 105,106,107,108,109]. While these syndromes are all unified by sharing GATA2 mutations as their root cause, the symptoms exhibited by these patients and the specific cell populations that are affected vary. How mutations in GATA2 cause the different symptoms that define these syndromes is unknown, but the mutations driving MonoMAC syndrome are the best characterized. Patients with MonoMAC syndrome experience monocytopenia, NK- and B-lymphocytopenia, and severe infections with M. avium complex, and have an increased risk of developing myelodysplasia and AML [110]. Patients with MonoMAC syndrome have a 28% mortality rate and typically suffer from severe and recurrent nontuberculous mycobacterial infections, infections by opportunistic fungi, reoccurring human papilloma virus infections, and opportunistic bacterial infections [105, 111]. This syndrome is caused by GATA2 germline mutations [54, 112], often occurring as a result of rare autosomal dominant mutations such as (E.G. c.1186C > T;p.R396W), resulting in loss of function in one allele of GATA2 [54, 113]. These mutations can occur across the GATA2 gene, with causal mutations reported in both TAD domains, the NRD domain, the NLS, and in both zinc finger domains (Fig. 1C) [114,115,116,117,118,119,120,121,122,123,124,125]. MonoMAC syndrome can also be caused by frameshift, nonsense, splice-site mutations, and deletion mutations, with these mutations typically inactivating one GATA2 allele [124, 125]. Historically, MonoMAC patients were often misdiagnosed as having some form of myelodysplastic syndrome, especially those with rarer GATA2 mutations where the underlying germline mutation had not yet been identified. Recent work by Shen et al. has shown that next-generation sequencing can be used to effectively identify MonoMAC patients, even those with rare or unique mutations. For example, using this approach, a 65-year-old myelodysplastic syndrome patient who had a unique, heterozygous mutation in GATA2 at exon 6:c.1126_1128del:p.K376del was identified [126]. MonoMAC syndrome develops with low GATA2 mutational burden, emphasizing the sensitivity of hematopoiesis to GATA2 mutations [54, 112]. Untreated, around 50% of the patients with MonoMAC syndrome will be diagnosed with myelodysplastic syndrome or AML [105, 111]. Allogenic HSC transplantation can be an effective solution to MonoMAC syndrome, by reconstituting the defective hematopoietic compartments with HSCs from an unaffected donor [111, 127]. HSC transplantation of MonoMAC syndrome has survival rate of 57% at 36 months, making transplantation a risky therapeutic approach [128].
Acute myeloid leukemia
AML is the most common form of acute leukemia in adults, accounting for around 80% of leukemia cases [129, 130]. AML is a heterogeneous disease caused by mutations in the genes that regulate hematopoietic cell proliferation and differentiation [131,132,133]. Abnormal proliferation and differentiation cause the accumulation of malignant and poorly differentiated myeloid cells within the bone marrow and peripheral blood, ultimately leading to AML. The main pathogenesis of AML is the excessive proliferation, lack of differentiation, and reduced apoptosis of myeloid stem cells, eventually leading to the replacement of normal myeloid precursors with malignant leukemia cells, thus preventing production of other hematopoietic cell types [134,135,136]. AML patients suffer from bone marrow failure, which in some patients is complicated by leukocytosis. If AML remains untreated, patients die due to infection or uncontrolled bleeding [130, 137]. GATA2 haploinsufficiency can be a cause of AML. GATA2 haploinsufficiency is most often caused by frameshift, nonsense and missense mutations which results in a non-functional GATA2 allele, and by mutations in GATA2’s regulatory regions that result in uniallelic expression [102].
Both somatic and germline mutations in GATA2 can lead to AML, with the former contributing to familial AML. Shiba et al. found that 5% of pediatric AML patients had somatic mutations in GATA2, 0.6% carried a germline GATA2 mutation, and these were often biallelic mutations with concordant mutations in CEBPA [138]. In some patients, chromosomal rearrangement in inv(3)/t(3;3) causes repositioning of the − 110 kb GATA2 enhancer, leading to GATA2 gene silencing [139, 140]. Hahn et al. showed that a heterozygous c.1061C > T(p.Thr354Met) missense mutation predisposed members of three families to AML and identified a similar predisposing mutation [c.1063_1065delACA(p.Thr355del)] in a fourth family. These mutations all occurred in the C-ZnF of GATA2 [107]. Another study found heterozygous missense mutations in both the N-ZnF and C-ZnF domains within an AML patient cohort [141]. These mutations are thought to abrogate GATA2 activity through impairing DNA and transcriptional partner binding. Chemotherapy and allogeneic hematopoietic stem cell transplantation are most often the treatment for AML [141].
Myelodysplastic syndrome (MDS)
Germline mutations of GATA2 are one cause of myelodysplastic syndrome (MDS), an age-related hematopoietic malignancy characterized by abnormal blood cell maturation and a high frequency of leukemic transformation [142]. Patients with MDS suffer from chronic fatigue due to a lack of functional red blood cells, bleeding and bruising due to thrombocytopenia, and recurrent infections due to a lack of mature leukocytes. Although MDS and AML have similar clinical symptoms, they differ in the peripheral blood cell count and in the pathological presentation of patients’ bone marrow. Patients with MDS are diagnosed with myelodysplasia (< 20% myeloblasts in the bone marrow) and cytopenia of one or more myeloid lineage in the peripheral blood. In comparison, AML patients have > 20% myeloblasts in their bone marrow [143, 144].
There is a significant difference in the presentation of primary MDS between children and adults. Pancytopenia and hypocellular bone marrow are present in children and adolescents [145], and monosomy 7 is the most frequent karyotype aberration in children whereas other aberrations are more common in adult MDS [146]. Wlodarski et al., in a cohort study, found that GATA2 mutations are the most common germline defect that predispose to pediatric MDS, and that GATA2 mutations occur at high prevalence in adolescents with monosomy 7. GATA2 deficiency is responsible for 7% of all primary MDS cases and is present in two third of adolescents with monosomy 7 [147]. In addition, Donadieu et al. determined that patients with GATA2 deficiencies have an elevated risk of developing MDS, with this risk increasing from 6% at 10 years of age to 80% at 40 years of age [124].
Acute lymphoblastic leukemia
Acute lymphoblastic leukemia (ALL) is the malignant transformation and uncontrolled proliferation of lymphoid progenitor cells. As with AML, this results in the replacement of normal hematopoietic progenitors by malignant cells, which in ALL results in an increase in the number of circulating leukemic cells and infiltration of these cells to the central nervous system and testes. Both children and adults can develop ALL, with the highest incidence observed in children under 4 years of age [148]. Although the majority of ALL develops in healthy persons, genetic susceptibility and common allelic variants can predispose patients to ALL [149]. Abnormal, constitutive activation of transcription factors is one of the main drivers of ALL [150, 151]. There are two primary types of ALL: B-cell acute lymphoblastic leukemia (B-ALL) and T-cell acute lymphoblastic leukemia (T-ALL). GATA2 is a very rare driver of ALL, likely because GATA2 is not normally expressed by lymphoid progenitor cells [1, 152]. Even so, Wang et al. identified GATA2 expression in 20.5% of pediatric B-ALL patients. This expression of GATA2 was found across a range of B-ALL subtypes, and there was no single mechanism that accounted for GATA2 expression in these patients. Interestingly, however, GATA2 expression was associated with KMT2A-USP2 fusion in a subset of patients, while a single patient was identified with a somatic focal deletion of a regulatory region downstream of GATA2, indicating that both cis- and trans-activation of GATA2 can contribute to B-ALL [153]. This aberrant expression of GATA2 in these patients produced B-ALL cells that exhibited myeloid-like gene expression patterns, including activation of genes that induce myeloid proliferation, thus contributing to leukemogenesis [153].
GATA2 in non-hematological cancers
Normally, GATA2’s expression is restricted to the hematopoietic compartment, lymphatic endothelial cells, the central nervous system, and fetal development of the placenta, liver, and heart [9,10,11,12,13]. In adults, aberrant expression outside of these compartments can led to various cancers including prostate, breast, lung, endometrial, urothelial, and renal cancer (Fig. 3).
Prostate cancer
Prostate cancer is one of the most common cancers of men. Many prostate cancers are confined to the prostate gland and grow slowly, but aggressive forms can be highly metastatic. The transition from a slowly growing non-invasive cancer to rapidly-growing invasive metastatic disease is often driven by a transition from AR-dependent to AR-independent tumor growth, with GATA2 contributing to both forms of disease [154]. Overexpression of GATA2 increases cellular proliferation, tumorigenicity, cellular motility, and invasiveness, and can contribute to resistance to standard prostate cancer therapies [154]. Inhibition of GATA2 reduced prostate cancer proliferation [155], with GATA2 driving prostate cancer progression in both AR-dependent and AR-independent disease [156]. The transition of prostate cancer from AR-dependent to AR-independent growth is accompanied by an increase in invasiveness and metastatic potential, with a GATA2-driven regulatory network driving proliferation in both the AR-dependent and -independent stages of disease [74, 157]. Moreover, GATA2 expression may contribute to progression to the more lethal AR-independent state [158]. GATA2 drives prostate cancer metastasis, with higher GATA2 expression correlating to greater metastatic potential, higher invasiveness, and increased cell motility in primary tumor samples [158,159,160]. Conversely, silencing GATA2 in LNCap cells (an AR-dependent human prostate adenocarcinoma) decreased expression of multiple genes required for migration, invasion, and proliferation, and was accompanied by decreases in cell migration and tissue invasion, and increased adhesion [159].
GATA2 drives AR-dependent prostate cancer progression via three mechanisms. Firstly, GATA2 binds to the regulatory region upstream of the AR promoter, thus increasing AR expression. If required, GATA2 also acts as a pioneer transcription factor at this site to open the chromatin to other transcription factors [161]. Secondly, GATA2 acts as a pioneer transcription factor that generates an accessible local chromatin environment in the AR enhancers to further promote AR expression. Thirdly, GATA2 increases the expression of multiple AR-regulated genes by recruiting the co-regulatory complex subunit MED1 to form and maintain a regulatory chromatin loop between AR-bound distal enhancers and the promoters of AR target genes [74]. In addition to upregulating AR-dependent genes, GATA2 also promotes prostate cancer through mediating AR-independent expression of IGF2, PAK4, FOXM1, GLNT7, and ARRDC3 [158]. GATA2 can be upregulated by the EGFR/ERK/Elk-1 signaling axis, with GATA2 expression decreased by inhibition of EGFR signaling using EGFR or ERK inhibitors [159]. Upregulation of GATA2 contributes to chemotherapy resistance by regulating genes that are associated with resistance to chemotherapy, and GATA2 knockdown-sensitized prostate cancer cells to chemotherapy by increasing sensitivity to apoptosis [158]. Inhibition of Elk-1 expression or inhibition of the EGFR/ERK pathway may be a target for limiting GATA2 tumor-inducing activity, and inhibition of this pathway may be a potential therapeutic target for prostate cancer [162].
Breast cancer
Breast cancer is one of the most common cancers of women and affects some men. Expression of GATA2 by breast carcinoma cells promotes metastasis by stimulating their proliferation and migration [163]. This increase in proliferation occurs via a GATA2-driven enhancement of PI3K/AKT signaling, through regulation of the inhibitory protein phosphatase and tensin homolog (PTEN). There is a negative correlation between GATA2 expression and expression PTEN in breast cancer cells, which has several effects on breast cancer growth and metastasis. Firstly, GATA2 directly inhibits PTEN expression by acting as a transcriptional repressor at the PTEN promoter [163]. This reduction in PTEN expression alleviates PTEN’s suppression of the PI3K/AKT signaling pathway, with the resulting elevated AKT activity inhibiting cancer cell apoptosis and enhancing cell survival [163]. Activated AKT also increases GATA2 expression, producing a positive feedback loop which further inhibits PTEN expression and enhances AKT activity [163]. Secondly, GATA2 inhibits AR-dependent expression of PTEN by preventing the nuclear translocation of dihydrotestosterone-ligated AR [164, 165]. GATA2 likely has additional effects on breast cancer development, with the results of Ercelylan et al. demonstrating that GATA2 is one of 59 core genes that forms a regulatory network that may be a target for breast cancer therapies [166]. Germline mutations in GATA2 are not commonly considered to be risk factors for breast cancer; however, in a screen of GATA2 mutations of patients with familial aggregations of hematological malignancies, a single patient with AML and breast cancer was found with a p.Arg396Gln mutation in GATA2, suggesting that some germline GATA2 mutations may increase a patients risk of developing breast cancer [167]. As with prostate cancer, GATA2 may be a viable target for future breast cancer therapies.
Gastric cancer
Gastric cancer is one of the most prevalent cancers worldwide, with most gastric cancers starting within the stomach’s inner lining and developing into epithelial dysplasia and gastric adenomas. Unlike breast and prostate cancer, it is the loss of GATA2 expression that creates a favorable environment for the growth and survival of gastric tumors. This is largely due to the upregulation of GATA6 expression that follows the loss of GATA2 [168]. Hypermethylation of the GATA2 promoter silences GATA2 expression in gastric cancer cells, with the loss of GATA2 allowing for GATA2/GATA6 switch that constitutes one of the first steps that leads to gastric cancer. Normally, GATA2 suppresses transcription of GATA6 by stabilizing polycomb repressive complex 2 on the GATA6 promoter [168]. Depletion of GATA2 removes this suppression, allowing for GATA6 expression. While the signals that initiate the hypermethylation of the GATA2 promoter in gastric cells are unknown, it has been shown that this process is independent of Helicobacter pylori infection [168]. Once gastric cancer has formed, re-expression of GATA2 is associated with chemoresistance. Sublethal chemotherapy induces GATA2 expression, thereby increasing the GATA2-dependent upregulation of the α-subunit of glycoprotein hormone (CGA). Upregulated CGA binds to and activates EGFR, thereby promoting survival of gastric cancer cells [169]. Through this mechanism, CGA/EGFR/GATA2 act as a positive feedback loop that induce resistance to chemotherapy in established gastric cancers [169]. While GATA2 is not likely a viable therapeutic target for gastric cancer, detection of epigenetic silencing of GATA2 may serve as a biomarker for precancerous or early-stage disease.
Colorectal cancer
Colorectal cancer is one of the most lethal cancers globally. Elevated GATA2 expression is associated with poor survival of colorectal patients, especially in those with concomitant mutations in KRAS [170]. Moreover, the single nucleotide polymorphism rs2335052 (A164P) in GATA2 significantly correlates with increased risk of colorectal cancer recurrence [171]. Overexpression of GATA2 promotes the progression of colorectal cancer by upregulating genes for proliferation, invasion, epithelial-mesenchymal transition (EMT), and stemness [172]. Located next to the GATA2 gene is a region coding for GATA2 antisense RNA 1 (GATA2-AS1), which when expressed, promotes GATA2 expression in colorectal cancer cells. Here, GATA2 directly induces GATA2-AS1 expression by binding to its promoter, with GATA2-AS1 then recruiting DEAD-box helicase 3 X-linked to the 3′UTR of GATA2 where it stabilizes the GATA2 mRNA, thus enhancing GATA2 expression [172].
Other cancers
GATA2 expression is significantly decreased in renal tumor tissue compared to normal tissue, where decreased GATA2 expression correlates with renal tumor development and aggressiveness [173]. GATA2 can also indirectly contribute to tumor growth by aiding in the formation of an immunosuppressive environment. In glioblastoma, overexpression of GATA2 induced expression of PD-L1 and PD-L2 in glioma cells. These then inhibit T cell production of IFNγ, thus worsening the immunological control of glioma [174]. Aberrant expression of GATA2 is not always deleterious in the context of cancer. For example, higher GATA2 expression in ovarian cancer is correlated with better patient survival [175].
GATA2 in lung disease
In humans, GATA2 has been found to be expressed in alveolar cells, bronchial epithelium, and endothelial cells of the lung, while in rats GATA2 expression has been reported in bronchial epithelial cells, alveolar macrophages, and resident lung monocytes [176]. This expression pattern may be unique to rats, as the alveolar macrophages of mice express minimal levels of GATA2 unless CISH—a negative regulator of GM-CSF signaling—is deleted [177]. At this time there are no published studies analyzing GATA2 expression in human alveolar macrophages or their monocytic progenitors, but unpublished data from our lab shows that human monocytes do not express GATA2 at a level detectable by flow cytometry (data not shown). Patients with germline GATA2 mutations experience frequent lung infections, driven in-part by the loss of myeloid immune cells, resulting in susceptibility to a range of viral, fungal, and mycobacterial infections [105]. These recurrent infections can lead to lung remodeling and a loss of elasticity due to depletion of elastin [178, 179]. Therefore, identification of GATA2 deficiency is important to prevent the recurrent infection and concordant lung damage that can occurs in these patients (Fig. 3) [178].
Lung cancer
Lung cancer is one of the leading causes of death worldwide. Repressed GATA2 expression is associated with human lung cancer development with a similar phenotype observed in mouse models [180]. Repression of GATA2 in humans occurs through GATA2 promoter CpG hypermethylation that develops early in transformation, with similar epigenic silencing observed in mouse models [180]. Zhang et al. found that GATA2-AS1 also represses GATA2 expression in lung cancer cells, thus promoting cancer cell growth [181]. Comprehensive analysis of lung cancers found lower expression of GATA2 in lung adenocarcinoma, squamous cell carcinoma, and large cell lung carcinoma. However, no significant relationship between GATA2 expression and patient survival was found [182]. Overexpression of GATA2 inhibits A549 and H460 lung cancer cell line proliferation, invasion, migration, and EMT through upregulation of long non-coding RNA LINC00891. GATA2 directly binds to the promoter of LINC00891 to induce its expression, with LINC00891 inhibiting malignant behaviors by supressing the RhoA pathway required for invasion and migration [183]. While suppression of GATA2 generally promotes lung cancer formation, this is not always the case. Kumar et al. found that deletion of GATA2 decreased KRAS-mutated non-small cell lung cancer cell viability and increased apoptosis [184], with in vivo delivery of a GATA2 siRNA suppressing proliferation and inducing apoptosis in a mouse model of lung cancer [185]. Thus, whether GATA2 expression is pro- or anti-tumorigenic in lung cancer likely depends on the gene expression profile and presence or absence of other tumorigenic mutations within the cancerous cell.
Pulmonary alveolar proteinosis
Pulmonary alveolar proteinosis (PAP) is a rare lung disorder characterized by the abnormal accumulation of alveolar surfactant in the alveoli of the lung and dysfunction of alveolar macrophages, which together lead to hypoxemic respiratory failure and even death [186]. Pulmonary surfactant is a mixture of phospholipids and proteins which coat the alveoli, reducing their surface tension at the air-cell interface, thereby promoting alveoli inflation and gas exchange [187]. However, abnormal accumulation of surfactant can block lung airways and impair the transfer of oxygen to the blood. Most cases of PAP are caused by autoantibodies against GM-CSF, but the subset of PAP patients who lack these autoantibodies often have mutations in GATA2 [188]. For example, Matias et al. identified an otherwise benign variant of GATA2 (rs1573858) in a 43-year-old PAP patient who was anti-GM-CSF antibody negative [189]. Pilarski et al. identified a frameshift mutation (exon 5 C.1020_1029dup: p.R344GfsX43) in the GATA2 gene of a PAP patient who had severe interstitial lung fibrosis [190]. In autoantibody-mediated PAP, much of the lung pathology is due to a loss of GM-CSF signaling in alveolar macrophages. This results in the formation of abnormal alveolar macrophages with a poor ability to phagocytose and process surfactant, thus allowing for surfactant to accumulate [191]. How GATA2 mutations contribute to PAP is unclear. One possibility is that there may be depletion of alveolar macrophages due to decreasing levels of the circulating monocytes which give rise to alveolar macrophages in adults [192]. Alternatively, an inability to upregulate GATA2 in response to inflammatory stimuli may prevent alveolar macrophages from responding properly to the accumulation of surfactant. While not reported in humans, mouse macrophage cell lines upregulate GATA2 in response to LPS and IL-1β [193], while in rats, infection by Pneumocystis carinii induces GATA2 expression in the lung, with GATA2 knockdown impairing pathogen phagocytosis by alveolar macrophages [176, 194, 195]. A similar loss of GATA2-enhanced phagocytosis may impair the phagocytic function of alveolar macrophages in PAP patients, reducing their ability to catabolize excess surfactant [191]. GATA2 may further modulate macrophage activity and differentiation by modulating the expression of the M-CSF receptor, which is required for macrophage differentiation and survival [196]. Therefore, GATA2 deficiency may induce PAP by either reducing alveolar macrophages number or altering alveolar macrophage function. For patients with GATA2-mediated PAP, treatment with whole lung lavages to reduce surfactant and HSC transplantation improved their symptoms [197].
Pneumocystis pneumonia
While not observed in humans, GATA2 has been shown to be an important mediator of macrophage function during fungal pneumocystis pneumonia. Pneumocystis jirovecii is a yeast-like fungal pathogen which causes severe pneumonia in immunocompromised patients [198, 199]. P. jirovecii selectively infects humans and asymptomatically colonizes the lungs of up to 80% of the human population [200]. In rats, GATA2 mRNA is present at detectable levels in lung homogenates under basal conditions and its expression decreases upon infection with P. carinii – the rat-infecting species of the Pneumocystis genus [176]. Alveolar macrophages of P. carinii infected rats exhibit poor phagocytosis of the fungal invader [201]. A similar phenotype is observed in HIV-patients infected with P. jirovecii, with this loss of phagocytic function attributed to the decreased expression of the phagocytic mannose receptor by alveolar macrophages [202]. siRNA depletion of GATA2 in the rat lung restored the phagocytic capacity of alveolar macrophages to near-normal levels, and improved clearance of P. carinii [194]. While this was interpreted as evidence that GATA2 expressed by alveolar macrophages mediates the suppression of phagocytosis, later microarray analyses of highly purified alveolar macrophages from P. carinii infected rats failed to detect GATA2 expression in either control or infected animals [203]. Moreover, in rats this suppression of alveolar macrophage phagocytosis can be transferred to cultured macrophages via cell/fungus-free bronchoalveolar lavage fluid [204]. Combined, these results indicate that a soluble factor produced normally repressed by GATA2 and expressed by a non-macrophage cell type—not a cell-intrinsic defect of macrophages—mediates this suppression of phagocytosis during pneumocystis pneumonia.
SARS-CoV-2
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection causes Corona Virus Disease 19 (COVID-19). Pneumonia and Acute Respiratory Distress Syndrome are severe outcomes of COVID-19 that are predominantly caused by dysregulated and uncontrolled inflammation. Much of this inflammation is driven by a cytokine storm, with this cytokine storm being one of the main drivers of COVID-19 mortality [205]. Among the cytokines released during the cytokine storm, high levels of IL-1β have been found in severe and critical patients compared to patients with mild or moderate disease [206]. This elevated IL-1β induces expression of transmembrane protease/serine subfamily 2 (TMPRSS2) in lung epithelial cells [207], which can then enhance SARS-CoV-2 entry into host cells by cleaving and activating the viral spike protein (S) following its binding to host angiotensin I Converting Enzyme 2. This cleavage of the S protein by TMPRSS2 is required to induce membrane fusion and viral entry, with increased TMPRSS2 expression enhancing viral entry [208]. GATA2 promotes SARS-CoV-2 virus infection by inducing TMPRSS2 transcription [207]. This is mediated by binding of GATA2 to the -13kb DNA enhancer region of the TMPRSS2 gene, thus promoting TNPRSS2 expression [209]. GATA2 also contributes to the cytokine storm by inducing the expression of several pro-inflammatory cytokines including IL-1β and CXCL2 [210]. Indeed, Islam et al. found 177 differentially expressed genes in lung epithelial cells responding to SARS-CoV-2 infection, with a quintet of transcription factors—GATA2, FOXC1, YY1, FOXL1, and NFKB1—forming a central hub that regulated the differential expression of these genes [211]. To date, these studies have been limited to analyses of infected lung epithelial cells, but given the broad trophism of SARS-CoV-2, it is likely that GATA2 has additional roles in other SARS-CoV-2 infected cell types.
GATA2 in the cardiovascular system
Atherosclerosis
Atherosclerosis is the thickening or hardening of arteries caused by a buildup of plaque beneath the inner lining of an artery. These plaques are unstable structures comprised of fats, cholesterol, and necrotic cell debris. Several single nucleotide polymorphisms in GATA2 are associated with the development of atherosclerosis and coronary artery disease, although the mechanism leading to this susceptibility remains unclear [212]. We identified higher GATA2 expression in macrophages collected from early human aortic plaque, and demonstrated that oxidized low-density lipoprotein (oxLDL)—a major driver of atherosclerosis [213,214,215,216]—induced significant GATA2 expression in cultured macrophages [217]. During atherosclerosis, several distinct macrophage populations arise via polarization of monocyte-derived macrophages which infiltrate the plaque [218]. This includes polarization toward inflammatory (M1 and Mox), pro-resolving (M2), lipid-metabolizing (TREM2), and heme-metabolizing (Mhem) populations [218,219,220,221,222]. Interestingly, the early-lesion macrophages we identified lacked markers of polarization toward any of these phenotypes, and instead were characterized primarily by their high expression of GATA2. In vitro, GATA2 overexpression increased cholesterol efflux from macrophages, suggesting that GATA2 may be induced in response to the cellular stress caused by cholesterol accumulation [223]. However, GATA2 overexpression also decreased the expression of several receptors and signaling molecules required for the removal of apoptotic cells and pathogens, and of genes required for the degradation and processing of materials engulfed by the macrophages [217]. These combined defects lead to poor uptake and processing of apoptotic cells, likely contributing to the accumulation of necrotic debris in the developing plaque [224,225,226,227]. Importantly, knockdown of GATA2 reversed many of these defects, even in the presence of high concentrations of oxLDL [217]. This suggests that GATA2 expression in early-stage plaque may be a maladaptive response to the stress of cholesterol accumulation, with GATA2 expression improving the ability of macrophages to remove intracellular cholesterol, but in doing so, generates a population of macrophages which are unable to engage in the anti-atherogenic removal of dead and dying cells.
Endothelial cell function and repair
Thrombosis occurs in a large portion of patients with germline inherited GATA2 variants, with up to 25% of these patients experiencing one or more strokes, venous thromboembolism, or other thrombotic events [228,229,230]. Using chromatin immunoprecipitation, Purgatoro et al. determined that GATA2 binds to the promoter of endothelial nitric oxide synthase (eNOS) and is required for the expression of eNOS. Consequentially, it was found that both basal nitric oxide production and nitric oxide production following platelet activation were reduced in cells isolated from a patient with MonoMAC syndrome [231]. Nitric oxide promotes endothelial cell migration and, through enhancing migration, drives both angiogenesis and the closure of wounded endothelium [232, 233]. Consistently with this role of nitric oxide, reduced angiogenesis was observed in endothelial cells derived from this MonoMAC patient, with restoration of both nitric oxide production and angiogenesis observed when eNOS expression was increased by treatment with atorvastatin.
Conclusions
GATA2 has a central role in multiple diseases including inherited hematological disorders, a broad range of cancers, and in responses to infectious and inflammatory stimuli. While the role of GATA2 in hematological disorders and cancer is well understood, research into its role in infectious and inflammatory diseases has only begun. Further exploration of GATA2 will improve our understanding of these diseases, and of how GATA2 alters the transcriptional landscape in response to environmental and developmental challenges. While GATA2 may not be a viable therapeutic target due to its central role in hematopoiesis, identification of the transcription factors and signaling pathways which regulate GATA2 expression may allow for the therapeutic targeting of GATA2 in the many diseases where its expression serves a pathological role.
Data availability
This is a review and therefore contains no new data. All materials used to prepare this manuscript can be found in the reference section.
Abbreviations
- AML:
-
Acute myeloid leukemia
- ALL:
-
Acute lymphoblastic leukemia
- AR:
-
Androgen receptor
- CGA:
-
α-Subunit of glycoprotein hormone
- EGF:
-
Epidermal growth factor
- EMT:
-
Epithelial-mesenchymal transition
- GATA2-AS1:
-
GATA2 anti-sense 1
- HPC:
-
Hematopoietic progenitor cells
- HSC:
-
Hematopoietic stem cells
- MonoMAC:
-
Monocytopenia and mycobacterial infection
- NLS:
-
Nuclear localization signal
- NRD:
-
Negative regulatory domain
- oxLDL:
-
Oxidized low-density lipoprotein
- PTEN:
-
Phosphatase and tensin homolog
- PAP:
-
Pulmonary alveolar proteinosis
- TAD:
-
Trans-activation domain
- TMPRSS2:
-
Transmembrane protease/serine subfamily 2
- ZnF:
-
Zinc-finger domain
References
Bresnick EH et al (2010) GATA switches as developmental drivers. J Biol Chem 285(41):31087–31093
Rodrigues NP et al (2012) GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int J Biochem Cell Biol 44(3):457–460
La Ferla K et al (2002) Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-kappaB. Faseb J 16(13):1811–1813
Kitajima K et al (2002) GATA-2 and GATA-2/ER display opposing activities in the development and differentiation of blood progenitors. Embo J 21(12):3060–3069
Tsuzuki S et al (2004) Cross talk between retinoic acid signaling and transcription factor GATA-2. Mol Cell Biol 24(15):6824–6836
Ling KW et al (2004) GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J Exp Med 200(7):871–882
Tsai FY et al (1994) An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 371(6494):221–226
Tsai FY, Orkin SH (1997) Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood 89(10):3636–3643
Minegishi N et al (1999) The mouse GATA-2 gene is expressed in the para-aortic splanchnopleura and aorta-gonads and mesonephros region. Blood 93(12):4196–4207
Nardelli J et al (1999) Expression and genetic interaction of transcription factors GATA-2 and GATA-3 during development of the mouse central nervous system. Dev Biol 210(2):305–321
Ng YK et al (1994) GATA factor activity is required for the trophoblast-specific transcriptional regulation of the mouse placental lactogen I gene. Development 120(11):3257–3266
Dorfman DM et al (1992) Human transcription factor GATA-2. Evidence for regulation of preproendothelin-1 gene expression in endothelial cells. J Biol Chem 267(2):1279–1285
Kornhauser JM et al (1994) Temporal and spatial changes in GATA transcription factor expression are coincident with development of the chicken optic tectum. Brain Res Mol Brain Res 23(1–2):100–110
Lim KC et al (2012) Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J Clin Invest 122(10):3705–3717
Zhou Y, Yamamoto M, Engel JD (2000) GATA2 is required for the generation of V2 interneurons. Development 127(17):3829–3838
Tremblay M, Sanchez-Ferras O, and Bouchard M (2018) GATA transcription factors in development and disease. Development 145(20)
Lee ME et al (1991) Cloning of the GATA-binding protein that regulates endothelin-1 gene expression in endothelial cells. J Biol Chem 266(24):16188–16192
Kobayashi-Osaki M et al (2005) GATA motifs regulate early hematopoietic lineage-specific expression of the Gata2 gene. Mol Cell Biol 25(16):7005–7020
Fleenor DE et al (1996) Comparison of human and Xenopus GATA-2 promoters. Gene 179(2):219–223
Minegishi N et al (1998) Alternative promoters regulate transcription of the mouse GATA-2 gene. J Biol Chem 273(6):3625–3634
Pan X et al (2000) Identification of human GATA-2 gene distal IS exon and its expression in hematopoietic stem cell fractions. J Biochem 127(1):105–112
Tress ML, Abascal F, Valencia A (2017) Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci 42(2):98–110
Shen C et al (2016) The PU.1-Modulated MicroRNA-22 Is a regulator of monocyte/macrophage differentiation and acute myeloid leukemia. PLoS Genet 12(9):e1006259
Vicente C et al (2012) The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit Rev Oncol Hematol 82(1):1–17
Inoue F et al (2017) A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res 27(1):38–52
Lulli V et al (2006) Overexpression of Ets-1 in human hematopoietic progenitor cells blocks erythroid and promotes megakaryocytic differentiation. Cell Death Differ 13(7):1064–1074
Maeno M et al (1996) The role of BMP-4 and GATA-2 in the induction and differentiation of hematopoietic mesoderm in Xenopus laevis. Blood 88(6):1965–1972
Robert-Moreno A et al (2005) RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development 132(5):1117–1126
Kumano K et al (2001) Notch1 inhibits differentiation of hematopoietic cells by sustaining GATA-2 expression. Blood 98(12):3283–3289
Walsh JC et al (2002) Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity 17(5):665–676
Yatsula B et al (2005) Identification of binding sites of EVI1 in mammalian cells. J Biol Chem 280(35):30712–30722
Yuasa H et al (2005) Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J 24(11):1976–1987
Khandekar M et al (2007) A Gata2 intronic enhancer confers its pan-endothelia-specific regulation. Development 134(9):1703–1712
Gao X et al (2013) Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med 210(13):2833–2842
Soukup AA et al (2019) Single-nucleotide human disease mutation inactivates a blood-regenerative GATA2 enhancer. J Clin Invest 129(3):1180–1192
Martowicz ML et al (2005) Dynamic GATA factor interplay at a multicomponent regulatory region of the GATA-2 locus. J Biol Chem 280(3):1724–1732
Grass JA et al (2006) Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol 26(19):7056–7067
Brandt W et al (2008) Defining the functional boundaries of the Gata2 locus by rescue with a linked bacterial artificial chromosome transgene. J Biol Chem 283(14):8976–8983
Sanalkumar R et al (2014) Mechanism governing a stem cell-generating cis-regulatory element. Proc Natl Acad Sci USA 111(12):E1091–E1100
Meng A et al (1997) Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc Natl Acad Sci USA 94(12):6267–6272
Hirahara N et al (2020) Liganded T3 receptor beta2 inhibits the positive feedback autoregulation of the gene for GATA2, a transcription factor critical for thyrotropin production. PLoS ONE 15(1):e0227646
Grass JA et al (2003) GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc Natl Acad Sci USA 100(15):8811–8816
Celton M et al (2014) Epigenetic regulation of GATA2 and its impact on normal karyotype acute myeloid leukemia. Leukemia 28(8):1617–1626
Niimi K et al (2013) GATA2 zinc finger 2 mutation found in acute myeloid leukemia impairs myeloid differentiation. Leuk Res Rep 2(1):21–25
Zhang P et al (1999) Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc Natl Acad Sci USA 96(15):8705–10
Chang AN et al (2002) GATA-factor dependence of the multitype zinc-finger protein FOG-1 for its essential role in megakaryopoiesis. Proc Natl Acad Sci USA 99(14):9237–9242
Chun TH et al (2003) Modification of GATA-2 transcriptional activity in endothelial cells by the SUMO E3 ligase PIASy. Circ Res 92(11):1201–1208
Ozawa Y et al (2001) Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood 98(7):2116–2123
Tsuzuki S, Enver T (2002) Interactions of GATA-2 with the promyelocytic leukemia zinc finger (PLZF) protein, its homologue FAZF, and the t(11;17)-generated PLZF-retinoic acid receptor alpha oncoprotein. Blood 99(9):3404–3410
Tong Q et al (2000) Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 290(5489):134–138
Tong Q et al (2005) Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol Cell Biol 25(2):706–715
Luesink M et al (2012) High GATA2 expression is a poor prognostic marker in pediatric acute myeloid leukemia. Blood 120(10):2064–2075
Leubolt G, Redondo Monte E, and Greif PA (2020) GATA2 mutations in myeloid malignancies: Two zinc fingers in many pies. IUBMB Life 72(1):151–158
Hsu AP et al (2011) Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 118(10):2653–2655
Visvader JE et al (1995) The C-terminal zinc finger of GATA-1 or GATA-2 is sufficient to induce megakaryocytic differentiation of an early myeloid cell line. Mol Cell Biol 15(2):634–641
Nguyen Ba AN et al (2009) NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 10:202
Dingwall C et al (1988) The nucleoplasmin nuclear location sequence is larger and more complex than that of SV-40 large T antigen. J Cell Biol 107(3):841–849
Minegishi N et al (2003) Expression and domain-specific function of GATA-2 during differentiation of the hematopoietic precursor cells in midgestation mouse embryos. Blood 102(3):896–905
Viger RS et al (2008) Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol Endocrinol 22(4):781–798
Yang Z et al (1994) Human GATA-3 trans-activation, DNA-binding, and nuclear localization activities are organized into distinct structural domains. Mol Cell Biol 14(3):2201–2212
Kitajima K et al (2018) Domain-specific biological functions of the transcription factor Gata2 on hematopoietic differentiation of mouse embryonic stem cells. Genes Cells 23(9):753–766
Bates DL et al (2008) Crystal structures of multiple GATA zinc fingers bound to DNA reveal new insights into DNA recognition and self-association by GATA. J Mol Biol 381(5):1292–1306
Minegishi N et al (2005) Rapid turnover of GATA-2 via ubiquitin-proteasome protein degradation pathway. Genes Cells 10(7):693–704
Towatari M et al (1995) Regulation of GATA-2 phosphorylation by mitogen-activated protein kinase and interleukin-3. J Biol Chem 270(8):4101–4107
Hayakawa F et al (2004) Functional regulation of GATA-2 by acetylation. J Leukoc Biol 75(3):529–540
Whetton AD, Dexter TM (1993) Influence of growth factors and substrates on differentiation of haemopoietic stem cells. Curr Opin Cell Biol 5(6):1044–1049
Miyajima A et al (1993) Receptors for granulocyte-macrophage colony-stimulating factor, interleukin-3, and interleukin-5. Blood 82(7):1960–1974
Menghini R et al (2005) Phosphorylation of GATA2 by Akt increases adipose tissue differentiation and reduces adipose tissue-related inflammation: a novel pathway linking obesity to atherosclerosis. Circulation 111(15):1946–1953
Koga S et al (2007) Cell-cycle-dependent oscillation of GATA2 expression in hematopoietic cells. Blood 109(10):4200–4208
Kaplan T et al (2011) Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development. PLoS Genet 7(2):e1001290
Iwafuchi-Doi M, Zaret KS (2014) Pioneer transcription factors in cell reprogramming. Genes Dev 28(24):2679–2692
Zaret KS, Carroll JS (2011) Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25(21):2227–2241
Ye Y, Chen X, Zhang W (2020) Mammalian SWI/SNF chromatin remodeling complexes in embryonic stem cells: regulating the balance between pluripotency and differentiation. Front Cell Dev Biol 8:626383
Wu D et al (2014) Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res 42(6):3607–3622
Jung MM et al (2023) Pathogenic human variant that dislocates GATA2 zinc fingers disrupts hematopoietic gene expression and signaling networks. J Clin Invest 133(7)
Dore LC et al (2012) Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood 119(16):3724–3733
Beck D et al (2013) Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood 122(14):e12-22
May G et al (2013) Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell Stem Cell 13(6):754–768
Wilson NK et al (2010) Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7(4):532–544
Paralkar VR et al (2014) Lineage and species-specific long noncoding RNAs during erythro-megakaryocytic development. Blood 123(12):1927–1937
Man HSJ et al (2023) Long noncoding RNA GATA2-AS1 augments endothelial hypoxia inducible factor 1-alpha induction and regulates hypoxic signaling. J Biol Chem 299(5):103029
Froese N et al (2022) Endothelial cell GATA2 modulates the cardiomyocyte stress response through the regulation of two long non-coding RNAs. Biology (Basel) 11(12)
Tipping AJ et al (2009) High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood 113(12):2661–2672
Thoms JAI et al (2021) Disruption of a GATA2-TAL1-ERG regulatory circuit promotes erythroid transition in healthy and leukemic stem cells. Blood 138(16):1441–1455
Hong W et al (2005) FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J 24(13):2367–2378
Tsai SF et al (1989) Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 339(6224):446–451
Cantor AB et al (2008) Antagonism of FOG-1 and GATA factors in fate choice for the mast cell lineage. J Exp Med 205(3):611–624
Ikonomi P et al (2000) Levels of GATA-1/GATA-2 transcription factors modulate expression of embryonic and fetal hemoglobins. Gene 261(2):277–287
Takahashi S et al (2000) GATA factor transgenes under GATA-1 locus control rescue germline GATA-1 mutant deficiencies. Blood 96(3):910–916
Huang J et al (2016) Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev Cell 36(1):9–23
Gillespie MA et al (2020) Absolute quantification of transcription factors reveals principles of gene regulation in erythropoiesis. Mol Cell 78(5):960–974e11
Bresnick EH et al (2012) Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res 40(13):5819–5831
Fujiwara Y et al (1996) Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci USA 93(22):12355–12358
Orlic D et al (1995) Pluripotent hematopoietic stem cells contain high levels of mRNA for c-kit, GATA-2, p45 NF-E2, and c-myb and low levels or no mRNA for c-fms and the receptors for granulocyte colony-stimulating factor and interleukins 5 and 7. Proc Natl Acad Sci USA 92(10):4601–4605
Zhang SJ et al (2008) Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA 105(6):2076–2081
Ezoe S et al (2002) GATA-2/estrogen receptor chimera regulates cytokine-dependent growth of hematopoietic cells through accumulation of p21(WAF1) and p27(Kip1) proteins. Blood 100(10):3512–3520
de Pater E et al (2013) Gata2 is required for HSC generation and survival. J Exp Med 210(13):2843–2850
Pasquet M et al (2013) High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood 121(5):822–829
Persons DA et al (1999) Enforced expression of the GATA-2 transcription factor blocks normal hematopoiesis. Blood 93(2):488–499
Rodrigues NP et al (2005) Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood 106(2):477–484
Bruzzese A et al (2020) GATA2 Related conditions and predisposition to pediatric myelodysplastic syndromes. Cancers (Basel) 12(10)
Hsu AP, McReynolds LJ, Holland SM (2015) GATA2 deficiency. Curr Opin Allergy Clin Immunol 15(1):104–109
Ostergaard P et al (2011) Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet 43(10):929–931
Bresnick EH, Johnson KD (2019) Blood disease-causing and -suppressing transcriptional enhancers: general principles and GATA2 mechanisms. Blood Adv 3(13):2045–2056
Vinh DC et al (2010) Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 115(8):1519–1529
Dickinson RE et al (2011) Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte. B and NK lymphoid deficiency Blood 118(10):2656–2658
Hahn CN et al (2011) Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 43(10):1012–1017
Mace EM et al (2013) Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood 121(14):2669–2677
Mace EM, Orange JS (2016) Genetic causes of human NK cell deficiency and their effect on NK cell subsets. Front Immunol 7:545
Cascone P et al (1992) Neuromuscular assessment and postural examination in patients with TMJ condylo-meniscal incoordination. Minerva Stomatol 41(3):79–90
Bigley V et al (2011) The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med 208(2):227–234
Ding LW et al (2017) Mutational profiling of a MonoMAC syndrome family with GATA2 deficiency. Leukemia 31(1):244–245
Camargo JF et al (2013) MonoMAC syndrome in a patient with a GATA2 mutation: case report and review of the literature. Clin Infect Dis 57(5):697–699
Ishida H et al (2012) GATA-2 anomaly and clinical phenotype of a sporadic case of lymphedema, dendritic cell, monocyte, B- and NK-cell (DCML) deficiency, and myelodysplasia. Eur J Pediatr 171(8):1273–1276
Portich JP, Condino Neto A, Faulhaber GAM (2020) Humoral deficiency in a novel GATA2 mutation: a new clinical presentation successfully treated with hematopoietic stem cell transplantation. Pediatr Blood Cancer 67(9):e28374
Mendes-de-Almeida DP et al (2019) GATA2 mutation in long stand Mycobacterium kansasii infection, myelodysplasia and MonoMAC syndrome: a case-report. BMC Med Genet 20(1):64
Simonis A et al (2018) Allogeneic hematopoietic cell transplantation in patients with GATA2 deficiency-a case report and comprehensive review of the literature. Ann Hematol 97(10):1961–1973
Damian L et al (2018) Pseudo-sarcoidosis revealing MonoMAC syndrome. J Clin Immunol 38(7):739–741
Sologuren I et al (2018) Lethal influenza in two related adults with inherited GATA2 deficiency. J Clin Immunol 38(4):513–526
Yamamoto H et al (2018) MonoMAC syndrome patient developing myelodysplastic syndrome following persistent EBV infection. Rinsho Ketsueki 59(3):315–322
Eguchi K et al (2018) Nontuberculous mycobacteria-associated hemophagocytic lymphohistiocytosis in MonoMAC syndrome. Pediatr Blood Cancer 65(7):e27017
Vila A et al (2017) Multiple opportunistic infections in a woman with GATA2 mutation. Int J Infect Dis 54:89–91
Ganapathi KA et al (2015) GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood 125(1):56–70
Donadieu J et al (2018) Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 103(8):1278–1287
Homan CC et al (2021) GATA2 deficiency syndrome: a decade of discovery. Hum Mutat 42(11):1399–1421
Shen Y et al (2021) Diagnosing MonoMAC syndrome in GATA2 germline mutated myelodysplastic syndrome via next-generation sequencing in a patient with refractory and complex infection: case report and literature review. Infect Drug Resist 14:1311–1317
Cuellar-Rodriguez J et al (2011) Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood 118(13):3715–3720
Chu VH et al (2012) MonoMAC versus idiopathic CD4+ lymphocytopenia. Comment to Haematologica. 2011;96(8):1221–5. Haematologica 97(4):e9–11; author reply e12
Yamamoto JF, Goodman MT (2008) Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control 19(4):379–390
De Kouchkovsky I, Abdul-Hay M (2016) Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J 6(7):e441
Grimwade D (2012) The changing paradigm of prognostic factors in acute myeloid leukaemia. Best Pract Res Clin Haematol 25(4):419–425
Hartmann L, Metzeler KH (2019) Clonal hematopoiesis and preleukemia-genetics, biology, and clinical implications. Genes Chromosomes Cancer 58(12):828–838
Corces MR, Chang HY, Majeti R (2017) Preleukemic hematopoietic stem cells in human acute myeloid leukemia. Front Oncol 7:263
Sill H et al (2011) Therapy-related myeloid neoplasms: pathobiology and clinical characteristics. Br J Pharmacol 162(4):792–805
Lagunas-Rangel FA et al (2017) Acute myeloid leukemia-genetic alterations and their clinical prognosis. Int J Hematol Oncol Stem Cell Res 11(4):328–339
de Jonge HJ, Huls G, de Bont ES (2011) Gene expression profiling in acute myeloid leukaemia. Neth J Med 69(4):167–176
Dohner H et al (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129(4):424–447
Shiba N et al (2014) Mutations of the GATA2 and CEBPA genes in paediatric acute myeloid leukaemia. Br J Haematol 164(1):142–145
Groschel S et al (2014) A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157(2):369–381
Yamazaki H et al (2014) A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25(4):415–427
Theis F et al (2016) Clinical impact of GATA2 mutations in acute myeloid leukemia patients harboring CEBPA mutations: a study of the AML study group. Leukemia 30(11):2248–2250
Mufti GJ (2004) Pathobiology, classification, and diagnosis of myelodysplastic syndrome. Best Pract Res Clin Haematol 17(4):543–557
Vardiman JW (2010) The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: an overview with emphasis on the myeloid neoplasms. Chem Biol Interact 184(1–2):16–20
Vardiman JW et al (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114(5):937–951
Hasle H, Niemeyer CM (2011) Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154(2):185–195
Gohring G et al (2010) Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 116(19):3766–3769
Wlodarski MW et al (2016) Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127(11):387–1397; quiz 1518
Howlader N, Krapcho NA, Miller M, Bishop D, Kosary K, Yu CL, Ruhl M, Tatalovich J, Mariotto Z, Lewis A, Chen DR, Feuer HS, Cronin EJ (eds) (2017, April) SEER Cancer Stat Rev 1975–2014. National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2014/. based on November 2016 SEER data submission, posted to the SEER web site
Xu H et al (2013) Novel susceptibility variants at 10p12.31–12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105(10):733–42
Somasundaram R et al (2015) Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126(2):144–152
Girardi T et al (2017) The genetics and molecular biology of T-ALL. Blood 129(9):1113–1123
Leonard M et al (1993) Dynamics of GATA transcription factor expression during erythroid differentiation. Blood 82(4):1071–1079
Wang H et al (2021) Aberrant GATA2 activation in pediatric B-cell acute lymphoblastic leukemia. Front Pediatr 9:795529
Rodriguez-Bravo V et al (2017) The role of GATA2 in lethal prostate cancer aggressiveness. Nat Rev Urol 14(1):38–48
Kaochar S et al (2021) Inhibition of GATA2 in prostate cancer by a clinically available small molecule. Endocr Relat Cancer 29(1):15–31
Hankey W, Chen Z, Wang Q (2020) Shaping chromatin states in prostate cancer by pioneer transcription factors. Cancer Res 80(12):2427–2436
Wang Q et al (2007) A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27(3):380–392
Vidal SJ et al (2015) A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell 27(2):223–239
Chiang YT et al (2014) GATA2 as a potential metastasis-driving gene in prostate cancer. Oncotarget 5(2):451–461
He B et al (2014) GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc Natl Acad Sci USA 111(51):18261–18266
Wang Q et al (2009) Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138(2):245–256
Wang Z et al (2015) GATA2 promotes glioma progression through EGFR/ERK/Elk-1 pathway. Med Oncol 32(4):87
Wang Y et al (2012) GATA2 negatively regulates PTEN by preventing nuclear translocation of androgen receptor and by androgen-independent suppression of PTEN transcription in breast cancer. Hum Mol Genet 21(3):569–576
Wang Y et al (2011) Differential regulation of PTEN expression by androgen receptor in prostate and breast cancers. Oncogene 30(42):4327–4338
Masuda K et al (2005) Androgen receptor binding sites identified by a GREF_GATA model. J Mol Biol 353(4):763–771
Erceylan OF, Savas A, Gov E (2021) Targeting the tumor stroma: integrative analysis reveal GATA2 and TORYAIP1 as novel prognostic targets in breast and ovarian cancer. Turk J Biol 45(2):127–137
Hamadou WS et al (2017) GATA2 gene analysis in several forms of hematological malignancies including familial aggregations. Ann Hematol 96(10):1635–1639
Song SH et al (2018) Aberrant GATA2 epigenetic dysregulation induces a GATA2/GATA6 switch in human gastric cancer. Oncogene 37(8):993–1004
Cao T et al (2022) A CGA/EGFR/GATA2 positive feedback circuit confers chemoresistance in gastric cancer. J Clin Invest 132(6)
Xu K et al (2016) GATA binding protein 2 overexpression is associated with poor prognosis in KRAS mutant colorectal cancer. Oncol Rep 36(3):1672–1678
Liu X et al (2015) GATA2 rs2335052 Polymorphism predicts the survival of patients with colorectal cancer. PLoS ONE 10(8):e0136020
Pan Y et al (2022) A feedback loop between GATA2-AS1 and GATA2 promotes colorectal cancer cell proliferation, invasion, epithelial-mesenchymal transition and stemness via recruiting DDX3X. J Transl Med 20(1):287
Peters I et al (2015) Decreased mRNA expression of GATA1 and GATA2 is associated with tumor aggressiveness and poor outcome in clear cell renal cell carcinoma. Target Oncol 10(2):267–275
Fu Y et al (2020) GATA2 Regulates constitutive PD-L1 and PD-L2 expression in brain tumors. Sci Rep 10(1):9027
Zhou Q et al (2022) Distinct expression and prognostic values of GATA transcription factor family in human ovarian cancer. J Ovarian Res 15(1):49
Tang X et al (2000) Down-regulation of GATA-2 transcription during Pneumocystis carinii infection. Infect Immun 68(8):4720–4724
Shoger KE et al (2021) CISH attenuates homeostatic cytokine signaling to promote lung-specific macrophage programming and function. Sci Signal 14(698):eabe5137
Svobodova T et al (2015) Diffuse parenchymal lung disease as first clinical manifestation of GATA-2 deficiency in childhood. BMC Pulm Med 15:8
Marciano BE et al (2021) Pulmonary manifestations of GATA2 deficiency. Chest 160(4):1350–1359
Tessema M et al (2014) GATA2 is epigenetically repressed in human and mouse lung tumors and is not requisite for survival of KRAS mutant lung cancer. J Thorac Oncol 9(6):784–793
Zhang L et al (2019) A MYC target long non-coding RNA GATA2-AS1 regulates non-small cell lung cancer growth. Neoplasma 66(6):954–962
Gong C et al (2021) Comprehensive analysis of expression and prognostic value of GATAs in lung cancer. J Cancer 12(13):3862–3876
Zhang Y et al (2022) LINC00891 regulated by miR-128-3p/GATA2 axis impedes lung cancer cell proliferation, invasion and EMT by inhibiting RhoA pathway. Acta Biochim Biophys Sin (Shanghai) 54(3):378–387
Kumar MS et al (2012) The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 149(3):642–655
Shen S et al (2014) Cationic lipid-assisted polymeric nanoparticle mediated GATA2 siRNA delivery for synthetic lethal therapy of KRAS mutant non-small-cell lung carcinoma. Mol Pharm 11(8):2612–2622
Trapnell BC et al (2019) Pulmonary alveolar proteinosis. Nat Rev Dis Primers 5(1):16
Bernhard W (2016) Lung surfactant: function and composition in the context of development and respiratory physiology. Ann Anat 208:146–150
Griese M et al (2015) GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders. BMC Pulm Med 15:87
China N et al (2020) rs1573858 GATA-2 homozygote variant associated with pulmonary alveolar proteinosis, cytopenia and neurologic dysfunction. Pulmonology 26(3):178–180
Ballerie A et al (2016) Association of pulmonary alveolar proteinosis and fibrosis: patient with GATA2 deficiency. Eur Respir J 48(5):1510–1514
Carey B, Trapnell BC (2010) The molecular basis of pulmonary alveolar proteinosis. Clin Immunol 135(2):223–235
Liu Z et al (2019) Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell 178(6):1509–1525 e19
Wu TT et al (2013) GATA-2 transduces LPS-induced il-1beta gene expression in macrophages via a toll-like receptor 4/MD88/MAPK-dependent mechanism. PLoS ONE 8(8):e72404
Lasbury ME et al (2003) Effect of transcription factor GATA-2 on phagocytic activity of alveolar macrophages from Pneumocystis carinii-infected hosts. Infect Immun 71(9):4943–4952
Lasbury ME et al (2001) Effect of the transcription factor GATA-2 on phagocytic activity of alveolar macrophages from Pneumocystis carinii-infected hosts. J Eukaryot Microbiol Suppl:158S-159S
Collin M, Dickinson R, Bigley V (2015) Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169(2):173–187
van Lier YF et al (2020) Allogeneic hematopoietic cell transplantation in the management of GATA2 deficiency and pulmonary alveolar proteinosis. Clin Immunol 218:108522
Aliouat-Denis CM et al (2008) Pneumocystis species, co-evolution and pathogenic power. Infect Genet Evol 8(5):708–726
Allen CM et al (2010) Imaging lung manifestations of HIV/AIDS. Ann Thorac Med 5(4):201–216
Ponce CA et al (2010) Pneumocystis colonization is highly prevalent in the autopsied lungs of the general population. Clin Infect Dis 50(3):347–353
Chen W, Mills JW, Harmsen AG (1992) Development and resolution of Pneumocystis carinii pneumonia in severe combined immunodeficient mice: a morphological study of host inflammatory responses. Int J Exp Pathol 73(6):709–720
Koziel H et al (1998) Reduced binding and phagocytosis of Pneumocystis carinii by alveolar macrophages from persons infected with HIV-1 correlates with mannose receptor downregulation. J Clin Invest 102(7):1332–1344
Cheng BH et al (2010) Microarray studies on effects of Pneumocystis carinii infection on global gene expression in alveolar macrophages. BMC Microbiol 10:103
Lasbury ME, Durant PJ, and Lee CH (2001) Bronchoalveolar lavage fluid from Pneumocystis carinii-infected rats inhibits phagocytosis in normal alveolar macrophages. J Eukaryot Microbiol Suppl:163S-164S
Angioni R et al (2020) Age-severity matched cytokine profiling reveals specific signatures in Covid-19 patients. Cell Death Dis 11(11):957
McElvaney OJ et al (2020) Characterization of the inflammatory response to severe COVID-19 illness. Am J Respir Crit Care Med 202(6):812–821
Cioccarelli C et al (2021) IL1beta promotes TMPRSS2 expression and SARS-CoV-2 cell entry through the p38 MAPK-GATA2 axis. Front Immunol 12:781352
Hoffmann M et al (2020) SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–280 e8
Clinckemalie L et al (2013) Androgen regulation of the TMPRSS2 gene and the effect of a SNP in an androgen response element. Mol Endocrinol 27(12):2028–2040
Katsumura KR et al (2016) GATA Factor-dependent positive-feedback circuit in acute myeloid leukemia cells. Cell Rep 16(9):2428–2441
Islam T et al (2020) Integrative transcriptomics analysis of lung epithelial cells and identification of repurposable drug candidates for COVID-19. Eur J Pharmacol 887:173594
Muiya NP et al (2014) A study of the role of GATA2 gene polymorphism in coronary artery disease risk traits. Gene 544(2):152–158
Brown AJ et al (2000) Cholesterol and oxysterol metabolism and subcellular distribution in macrophage foam cells. Accumulation of oxidized esters in lysosomes. J Lipid Res 41(2):226–37
Goo YH, Yechoor VK, Paul A (2016) Transcriptional profiling of foam cells in response to hypercholesterolemia. Genom Data 9:37–39
Kellner-Weibel G et al (1998) Effects of intracellular free cholesterol accumulation on macrophage viability: a model for foam cell death. Arterioscler Thromb Vasc Biol 18(3):423–431
Tangirala RK et al (1994) Formation of cholesterol monohydrate crystals in macrophage-derived foam cells. J Lipid Res 35(1):93–104
Yin C et al (2020) Efferocytic defects in early atherosclerosis are driven by GATA2 overexpression in macrophages. Front Immunol 11:594136
Cochain C et al (2018) Single-cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 122(12):1661–1674
Stoger JL et al (2012) Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 225(2):461–468
Boyle JJ et al (2009) Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol 174(3):1097–1108
Boyle JJ et al (2012) Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res 110(1):20–33
Kadl A et al (2010) Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res 107(6):737–746
Koller D et al (2014) Effects of oxidized phospholipids on gene expression in RAW 264.7 macrophages: a microarray study. PLoS One 9(10):e110486
Linton MF et al (2016) Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J 80(11):2259–2268
Thorp E, Subramanian M, Tabas I (2011) The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol 41(9):2515–2518
Thorp EB (2010) Mechanisms of failed apoptotic cell clearance by phagocyte subsets in cardiovascular disease. Apoptosis 15(9):1124–1136
Thorp E, Tabas I (2009) Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol 86(5):1089–1095
Spinner MA et al (2014) GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123(6):809–821
Zolton JR et al (2018) Oocyte cryopreservation for women with GATA2 deficiency. J Assist Reprod Genet 35(7):1201–1207
Berry D, Fekrat S (2019) Central retinal vein occlusion in Gata2 deficiency. Retin Cases Brief Rep 13(2):181–184
Purgatorio G et al (2022) Germline GATA2 variant disrupting endothelial eNOS function and angiogenesis can be restored by c-Jun/AP-1 upregulation. Haematologica 107(5):1072–1085
Ziche M et al (1994) Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest 94(5):2036–2044
Noiri E et al (1997) Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. J Biol Chem 272(3):1747–1752
Funding
This work was funded by a Grant-In-Aid from the Heart and Stroke Foundation of Canada to BH. AA was funded by Ontario Graduate Scholarship and RGE Murray Scholarship from Western University. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AA authored most of the manuscript. BH contributed to the writing and editing of the manuscript and oversaw its preparation. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics and informed consent
Not applicable.
Conflict of interest
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aktar, A., Heit, B. Role of the pioneer transcription factor GATA2 in health and disease. J Mol Med 101, 1191–1208 (2023). https://doi.org/10.1007/s00109-023-02359-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-023-02359-8