
Abstract
Small ubiquitin-like modifier (SUMO) plays a key regulatory role in cardiovascular diseases, such as cardiac hypertrophy, hypertension, atherosclerosis, and cardiac ischemia–reperfusion injury. As a multifunctional posttranslational modification molecule in eukaryotic cells, SUMOylation is essentially associated with the regulation of mitochondrial dynamics, especially mitophagy, which is involved in the progression and development of cardiovascular diseases. SUMOylation targeting mitochondrial-associated proteins is admittedly considered to regulate mitophagy activation and mitochondrial functions and dynamics, including mitochondrial fusion and fission. SUMOylation triggers mitochondrial fusion to promote mitochondrial dysfunction by modifying Fis1, OPA1, MFN1/2, and DRP1. The interaction between SUMO and DRP1 induces SUMOylation and inhibits lysosomal degradation of DRP1, which is further involved in the regulation of mitochondrial fission. Both SUMOylation and deSUMOylation contribute to the initiation and activation of mitophagy by regulating the conjugation of MFN1/2 SERCA2a, HIF1α, and PINK1. SUMOylation mediated by the SUMO molecule has attracted much attention due to its dual roles in the development of cardiovascular diseases. In this review, we systemically summarize the current understanding underlying the expression, regulation, and structure of SUMO molecules; explore the biochemical functions of SUMOylation in the initiation and activation of mitophagy; discuss the biological roles and mechanisms of SUMOylation in cardiovascular diseases; and further provide a wider explanation of SUMOylation and deSUMOylation research to provide a possible therapeutic strategy for cardiovascular diseases. Considering the precise functions and exact mechanisms of SUMOylation in mitochondrial dysfunction and mitophagy will provide evidence for future experimental research and may serve as an effective approach in the development of novel therapeutic strategies for cardiovascular diseases.
Graphical abstract
Regulation and effect of SUMOylation in cardiovascular diseases via mitophagy. SUMOylation is involved in multiple cardiovascular diseases, including cardiac hypertrophy, hypertension, atherosclerosis, and cardiac ischemia–reperfusion injury. Since it is expressed in multiple cells associated with cardiovascular disease, SUMOylation can be regulated by numerous ligases, including the SENP family proteins PIAS1, PIASy/4, UBC9, and MAPL. SUMOylation regulates the activation and degradation of PINK1, SERCA2a, PPARγ, ERK5, and DRP1 to mediate mitochondrial dynamics, especially mitophagy activation. Mitophagy activation regulated by SUMOylation further promotes or inhibits ventricular diastolic dysfunction, perfusion injury, ventricular remodelling and ventricular noncompaction, which contribute to the development of cardiovascular diseases

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Small ubiquitin-like modifier (SUMO) is demonstrated to modify multiple proteins by its covalent conjugation to these proteins [1]. Similar to ubiquitin (Ub) in molecular structure, the amino sequence homology of SUMO consists of 18% well-known ubiquitin molecules. A typical ββαββαβ fold and a double glycerin C-terminal are both contained in the three-dimensional structure of SUMO and Ub [2]. The SUMO molecule is expressed from yeast to mammalian cells, and only one gene, Smt3, conservatively possessed SUMO in Saccharomyces cerevisiae, which shares 48% identity and 75% similarity with the mammalian SUMO [3,4]. In mammals, 5 SUMO isoforms display critical regulatory roles in pathophysiological processes, including SUMO1, SUMO2, SUMO3, SUMO4, and SUMO5 [5]. Given sharing 97% identity, SUMO2 and SUMO3 are usually identified as the same protein SUMO2/3; however, SUMO1 is different from SUMO2/3 by its 48% identity [6]. Distinct abilities to form SUMO chains promote the different substrates conjugated by SUMO1 and SUMO2/3. SUMO2/3 normally contains lysine (K) residues near its amino terminus, which is identified as a SUMO conjugation site but not shown in SUMO1. However, SUMO1 and SUMO2/3 can both bind to target substrates by promoting proteolytic maturation, which refers to exposing a carboxy-terminal diglycine (Gly-Gly) motif by sentrin-specific peptidases (SENPs) [2,7]. SUMO4 is similar to SUMO2/3 and is primarily expressed in the kidneys, lymph nodes, and spleen. The amino acid sequence of SUMO4 is reported to consist of a proline (Pro 90) instead of a glutamine that leads to inert maturation by SENPs [8]. SUMO5, a novel SUMO variant, is reported to be involved in the promyelocytic leukemia nuclear body (PML-NB) life cycle in human cells and can also promote the polyconjugation of SUMO2/3 [9].
SUMOylation is a dynamic and reversible process that is balanced for steady-state deSUMOylation [2]. SUMO conjugation is involved in multiple signalling pathways, which are dependent mainly on SUMO-specific proteases. The SUMO E1 activating enzyme, E2 conjugating enzyme, and E3 ligase play crucial regulatory roles in the conjugation and removal of SUMO molecules from target substrates (Table 1) [10]. The SUMO E1 activating enzyme is a heterodimer composed of SUMO activating enzyme subunit 1 (SAE1) and SAE2, which forms the C-terminal of the SUMO molecule [11]. The SUMO E2 conjugating enzyme can promote the formation of an E2-SUMO thioester bond via an isopeptide linkage to a target lysine residue [11]. The SUMO E3 ligase catalyzes the process of SUMOylation by stimulating SUMO transfer to the lysine of target substrates, including protein inhibitors of the activated STAT (PIAS) family [12]. As a multifunctional posttranslational modification (PTM), SUMOylation is involved in a variety of pathophysiological processes, including apoptosis, metabolic disorders, and mitophagy [1,13].
Mitochondrial degradation via a selective form of autophagy induces mitophagy, which is an essential mechanism conserved from yeast to humans that regulates mitochondrial quality and quantity control [14]. Specific outer mitochondrial membrane (OMM) receptors lead to the formation of autophagosomes surrounding mitochondria and promote the elimination of mitochondria, which play an important role in many processes, including early embryonic development, cell differentiation, inflammation, and apoptosis [15]. However, the regulatory mechanisms of potential molecules in mediating mitophagy are still unclear, and how multiple mitophagy pathways modulate cardiovascular diseases in a coordinated manner and its physiological implications determine the importance of mitophagy in regulating cardiovascular diseases [16]. Growing evidence suggests that SUMO-mediated SUMOylation is functionally associated with mitochondrial dysfunction and the initiation and activation of mitophagy [13,17]. Multiple studies have reported that mitochondrial fission is regulated by the mitochondrial membrane-bound E3 ligases MARCH5 and MULAN (MAPL) through the SUMOylation of dynamin-related protein (DRP1) mediated by SUMO1 [18,19]. Likewise, mitochondrial fusion can be regulated by membrane-bound GTPases, including optic atrophy-1 (OPA1) and mitofusins (MFNs). Posttranslational modification of MFNs plays critical roles in mitochondrial fusion and mitophagy initiation. The interaction of MFNs and SUMO2 has been shown to promote lysosomal degradation of mitochondria through SUMOylation of MFNs [20]. Aberrant SUMO expression or dysfunctional SUMOylation modification occurs during disordered mitochondrial dynamics and mitophagy, which contributes to the progression of associated diseases [13]. Although the regulatory roles of SUMOylation in mitophagy activation are distinct, the biochemical functions of SUMO are fundamentally dependent on its molecular motif [21]. However, the biofunctional mechanisms of SUMO molecules on other structural motifs are still unknown. Current research has reported that SUMOylation is involved in cardiovascular diseases via mitophagy activation [17,22,23]. However, SUMOylation-mediated mitophagy in the prevention of cardiovascular diseases remains elusive.
SUMOylation has attracted much attention due to its dual roles in the development of cardiovascular diseases. To provide an update about recent studies on SUMOylation, we systemically summarize the current results and exploit the mechanisms underlying the expression, regulation, and function of SUMO molecules. Additionally, we explored the regulatory role and precise mechanism of SUMOylation in cardiovascular diseases by targeting mitophagy activation. Although the exact mechanisms are elusive, SUMOylation clearly functions as a promising therapeutic strategy for cardiovascular diseases by regulating mitophagy initiation and activation. In this review, therefore, we summarize the recent understanding of the molecular structure, biological roles and biochemical functions of SUMO molecules and their related enzymes, discuss the roles and mechanisms of SUMOylation in mitophagy, and further provide a wider explanation of SUMOylation and deSUMOylation research to provide a possible therapeutic strategy for cardiovascular diseases.
Identification of SUMOylation and deSUMOylation
Small ubiquitin-like modifiers (SUMOs) belong to the family of ubiquitin-related proteins and are a conserved family of proteins [24]. SUMOs are identified to be covalently conjugated to lysine residues of downstream proteins at a specific consensus motif that is ΨKXD/E (Ψ-Lys-X-Glu/Asp) [25]. Ψ is any hydrophobic amino acid, whereas X is any amino acid [26]. The SUMO molecule family contains SUMO1, SUMO2, SUMO3, SUMO4, and the newly discovered SUMO5 [9,27]. The biological function and expressional regulation of SUMOs differ molecularly, and the regulatory mechanism of each SUMO molecule needs to be further investigated.
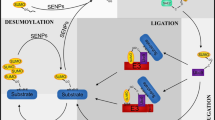
The conjugation and modification of SUMO to target proteins are essentially dependent on enzymatic cascades, including Ubl activating enzyme (E1), Ubl conjugating enzyme (E2), and Ubl protein ligase (E3) (Fig. 1) [28]. The E1 enzyme is a heterodimer that consists of SAE1 and SAE2 subunits, which can catalyze the adenylation of SUMO in the presence of manganese ions (Mg2+) [29]. Both SAE1 and SAE2 initiate the SUMO-mediated modification reaction by catalyzing the formation of SUMO and AMP [30]. During the process of SUMOylation, SUMO molecule is associated with a specific consensus motif recognized by E2 enzymes [25]. SUMO is transferred from SAE2 to cysteine 93 of UBC9, which is identified as an E2 conjugating enzyme. UBC9 can directly recognize target proteins to catalyze the formation between SUMO and the ε-amino group of a lysine in target proteins [31]. In the presence of SAE1, SAE2, and UBC9, SUMO is conjugated to the ΨKXE motif of target proteins. The E3 enzyme has been divided into two major families, including HECT domain-containing E3s and RING domain-containing E3s [32]. Protein inhibitor of activated STAT (PIAS) proteins, including PIAS1, Nse2/Mms21, and Ran-binding protein 2 (RanBP2), are characterized as E3 enzymes that enhance SUMOylation [27]. The cleavage of the isopeptide bond of the SUMO conjugate is involved in SUMO-modified SUMOylation, which is reversible through Ubl-specific proteases (Ulps) and SENPs [10,33]. The regulation of Ulps or SENPs is involved in the removal of a C-terminal sequence following a diglycine motif (Gly-Gly, K-K) in the presence of adenosine triphosphate (ATP) [21]. Research in S. cerevisiae has reported that Ulp1 and Ulp2 can promote SUMOylation of target proteins in the nuclear pore and nucleoplasm, respectively [34,35]. Gao et al. demonstrated that Ulp4 is a positive regulator of the C. elegans mitochondrial UPR (UPRmt) that mediates SUMOylation of DVE-1 and ATFS-1, which promotes the immune response and lifespan extension during mitochondrial stress [36]. SENPs comprise 7 family protein members, including SENP1, SENP2, SENP3, SENP5, SENP6, SENP7, and SENP8 and are further divided into 3 subfamilies [7,37,38]. SENP1 and SENP2 belong to the first subfamily and are specific mainly for 3 mammalian SUMOs, SUMO1, SUMO2, and SUMO3. SENP3 and SENP5 are identified as the second subfamily, which is localized mainly in the nucleolus, favoring SUMO2 and SUMO3. Both SENP6 and SENP7 contain an additional loop inserted in the catalytic domain that targets SUMO2 and SUMO3, which belong to the third subfamily. Notably, SENP8 is a unique family member acting on Nedd8 but not on SUMO [39]. The balance of SUMOylation and deSUMOylation is based mainly on SENP family proteins [40]. DeSUMOylation with the removal of SUMO from target proteins is highly dynamic to SUMOylation, and the regulatory mechanism of SENPs may be involved in processing SUMO precursors [21].

Schematic diagram of the mechanism of the enzymatic cascades leading to the covalent attachment of SUMO to a substrate protein. The SUMO precursor (preSUMO) is initially proteolytically cleaved by SENP family proteins with endopeptidase activity, which exposes the C-terminal diglycine (Gly-Gly) motif. The maturation of SUMO is activated by the SUMO E1 heterodimer SAE1/SAE2 in an ATP-dependent manner. The high-energy thioester bond between SUMO and an internal Cys promotes SUMO loading to the catalytic cysteine of the SUMO E2 enzyme UBC9. Then, UBC9 catalyzes the formation of an isopeptide bond between a lysine residue in the substrate and the GG motif of SUMO, which requires SUMO E3 ligase involvement. The conjugation between SUMO and target proteins is subsequently related to the ΨKXE motif, which further promotes the SUMOylation of target proteins. Under different conditions, SUMOylation of target proteins exhibits multiSUMOylation with 3 SUMO molecules and polySUMOylation. The polySUMOylation of target proteins turns into the ubiquitin–proteasome systems via deSUMOylation-related ligase activation. Eventually, SUMOylation can be reversed to cleave the isopeptide bond and release SUMO molecules by these deSUMOylation-related ligases
As an important dynamic reversible PTM, SUMOylation is involved in regulating the stability, localization, and function of target proteins [41]. Target protein homeostasis mediated by SUMOylation further determines the biological alteration and fate of cells (Table 2) [42]. By directing noncovalent interactions with target proteins through SUMO-interacting motifs (SIMs), SUMO-mediated SUMOylation directly contributes to multiple physiological or pathophysiological mechanisms [5]. SENP1 is reported to be involved in deSUMOylating SUMO-PML but not SUMO-Ran GTPase-activating protein 1 (RanGAP1), suggesting that SENP1 is involved mainly in the inactivation of transcription [43]. However, Lin et al. revealed that blocking SUMOylation by silencing SENP1 enhances the binding of SUMO1 to RanGAP1, which promotes keloid formation by targeting the transforming growth factor-β (TGF-β)/SMAD and hypoxia-inducible factor 1 (HIF1) signalling pathways [44]. In innate immune responses, SENP1 can also target interferon regulatory factor 8 (IRF8) and B-cell lymphoma/leukemia 11B (BCL11B) for deSUMOylation [45,46]. Furthermore, a signal SUMO molecule can be directly bound to an acceptor lysine; however, the polychain of the SUMO molecule (the attachment of multiSUMO)-mediated SUMOylation also interacts with a signal lysine. Rojas-Fernandez et al. have demonstrated that ring finger protein 4 (RNF4) containing an N-terminal domain can bind its polySUMO, which is identified as an E3 ligase without activity [47]. SENP2 deconjugates SUMO from target proteins and plays a critical role in SUMOylation. Chen et al. found that SENP2 deficiency decreased the expression of PLCβ4, and its stability was regulated by SUMO-dependent degradation, which is involved in calcium (Ca2+) homeostasis [48]. As a regulatory ligase responding to deSUMOylation, SUMO2 has broad effects on signal transduction, cell growth, and metabolic regulation [49,50]. Both SENP3 and SENP5 have been established to regulate ribosome biogenesis and are involved in the maturation of 28S rRNA [51,52]. By removal of SUMO, particularly SUMO2 and SUMO3, SENP3 or SENP5 mediates transcriptional processes through demodification of transcriptional coregulators and components of chromatin-modifying complexes. A previous study reported the regulatory role of SENP3 in the nucleolar steps of ribosome biogenesis and further determined that SENP3 can control nucleolar partitioning and ribosome formation by inducing a balance of SUMO conjugation and deconjugation [53,54]. SENP5 is involved mainly in RNA polymerase I-mediated transcription of 47S rRNA, which is also related to ribosome biogenesis [55]. Zunino et al. demonstrated that overexpression of SENP5 inhibited the mitochondrial fragmentation induced by SUMO1-mediated SUMOylation, eventually impacting mitochondrial morphology and metabolism [55]. Additionally, the accumulation of SENP5 promotes the depletion of SUMO in nucleoli, which inhibits defects in ribosome biogenesis [51]. SENP6 is reported to be involved in regulating inflammasome activity by mediating deSUMOylation. Barry et al. found that the inflammasome activation of NLR family pyrin domain containing 3 (NLRP3) is repressed by SUMOylation, and a lack of SENP6 or SENP7 promotes NLRP3-dependent ASC oligomerization and IL-1β release, suggesting that SUMOylation plays a crucial role in inflammatory diseases [56]. However, which SUMO molecule can be precisely regulated by SENP6, such as SUMO2 or SUMO3, remains unclear. Similarly, the deSUMOylation mediated by SENP7 can also be involved in SUMOylation-induced inflammasomes. Li et al. established that knockdown of SENP7 significantly attenuates the NLRP3 inflammasome and NF-κB signalling pathway, suggesting that the balance of SUMOylation is involved in inflammatory regulation through multiple signalling pathways [57].
The importance of SUMOylation has been reported in numerous cellular physiological and pathophysiological processes. Multiple proteins have been identified to covalently associate with SUMO molecules, which is essentially mediated by dynamic reversible SUMOylation/deSUMOylation. Recently, SUMOylation was reported to be involved in the development of cardiovascular diseases, including cardiac hypertrophy, hypertension, atherosclerosis, and ischemia–reperfusion injury of the heart [23]. However, the precise mechanism of SUMOylation in cardiovascular diseases is still unknown. The elimination of dysfunctional mitochondria via mitophagy is essential for the development of cardiovascular diseases [58]. Mitochondrial E3 ubiquitin ligase 1 (Mul1) has been demonstrated to be able to inhibit mitochondrial dynamics and promote mitophagy activation via SUMOylation [59]. However, the association between SUMOylation and mitophagy in regulating the development of cardiovascular diseases is not unclear. Additionally, further investigation of the molecular mechanism by which different SUMO molecules regulate mitophagy is needed.
Regulatory roles and biofunctions of SUMOylation in mitophagy
SUMO1
SUMO1, also known as PIC1, Smt3c, GMP1, SENtrin, or Ubl1, is a sensitive modified protein that regulates the complexity of the proteome in eukaryotic cells. The identification of cDNA coding for humans identified a novel DNA-responding protein SUMO1 that can interact with promyelocytic leukemia (PML) protein and promote DNA allosteric activity in 1996 [60,61]. As one of the most important members, SUMO1 is reversibly and covalently conjugated to RanGAP1 [62]. The association between RanGAP1 and SUMO1 has fundamentally clarified the molecular mechanisms of the covalent attachment of SUMO that can alter the properties of a target protein [63]. In molecular mass of ~ 15 kDa, SUMO1 is a specific unique peptide to RanGAP1, for which SUMO1, by expressed sequence tagged (EST) clones in available cDNA databases, is a predicted 11.5 kDa protein with 18% sequence identity to ubiquitin. Macauley et al. have characterized the complex of SUMO1 with the C-terminal domain of RanGAP1, and the loop containing the consensus SUMOylation site in RanGAP1 and the C-terminus of SUMO-1 are both conformationally flexible by using nuclear magnetic resonance (NMR) spectroscopy [64]. Kaur et al. revealed that the interaction of SUMO1 and metal ions, including cupric (Cu2+) and zinc (Zn2+), can promote SUMOylation-mediated degradation of misfolded proteins by impairing the ubiquitin–proteasome system (UPS) [65]. SUMO1 is expressed in multiple cells and tissues, including cardiac cells, alveolar cells, endothelial cells, liver, brain, and skeletal muscle [66,67,68]. As an important organelle for the balance of metabolism and targets of drugs in these cells, mitochondria are of great significance to cardiovascular diseases due to their physiological function homeostasis [69]. SUMO1-mediated SUMOylation plays a critical role in the regulation and development of cardiovascular diseases [70]. The involvement of SUMO1 in diverse cellular processes of cardiovascular diseases remains to be investigated, and further studies need to focus on the regulatory relationship between SUMO1 and mitophagy in cardiovascular diseases.
The regulatory mechanisms of mitophagy in cardiovascular diseases have been appreciated from in vivo to in vitro studies. The key mediator of mitophagy, PTEN-induced putative kinase 1 (PINK1), leads to cardiac dysfunction and pathological cardiac hypertrophy [71]. Mitochondrial fusion and fission are also involved in mitophagy activation, which is dependent mainly on the outer mitochondrial membrane (OMM) and optic atrophy 1 (OPA1) [72]. In the regulation of mitochondrial fusion and fission, SUMOylation has been shown to mediate mitochondrial dynamics. Neuspiel et al. performed bioinformatics to identify SUMO E3 ligases that contain conserved interesting new gene (RING)-finger motifs, which can further regulate mitochondrial morphology and can be regulated by SUMOylation induced by SUMO1 [73]. Dynamin-related protein (DRP) 1 is a key regulator of mitochondrial fission and is composed of GTP-binding, middle, insert B, and C-terminal GTPase effector (GED) domains. Normally, DRP1 controls mitochondrial dynamics dependent on autophagy via ATG7 activation, indicating that autophagy targets the lysosomal degradation of DRP1 [74]. Ablation of DRP1 in cardiac myocytes has been demonstrated to not only interrupt mitochondrial fission but also to markedly upregulate Parkin, thus provoking mitochondrial depletion that contributes to lethal cardiomyopathy [75]. However, whether SUMOylation is involved in the DRP1-induced upregulation of Parkin and dysfunctional mitochondrial-mediated lethal cardiomyopathy is still unknown [76]. A recent study revealed that DRP1 is a direct target of SUMOylation, and the interaction between the SUMO-conjugating enzyme UBC9 and DRP1 controls the localization of DRP1 and prevents normal mitochondrial fission [77]. Harder et al. further determined that UBC9 and SUMO1 can be specific interaction proteins for dynamin-related protein (DRP1) and further found that the accumulation of SUMO1 seemed to stabilize endogenous DRP1 by suppressing the lysosomal degradation of DRP1; however, an essential role for DRP1 in mitophagy induction is not supported [78]. As a crucial organelle undergoing regulated fusion and fission events, mitochondrial morphology and mitophagy can be regulated by multiple SUMO-related proteins. A previous study demonstrated that the cytosolic pool of SENP5 catalyzes the cleavage of SUMO1 from several mitochondrial substrates [55]. Zunino et al. further revealed that SENP5 deficiency elevated ROS production and promoted mitochondrial fragmentation, which further provoked mitophagy development. Conversely, SUMO1 downregulation by SENP5 overexpression inhibited DRP1 SUMOylation, which rescued mitochondrial fragmentation and suppressed mitophagy activation [55]. Although SUMOylation demonstrates alterations in mitochondrial morphology and mitophagy activation, little is known about the role and the mechanism of SUMO1 in cardiomyocytes and related cardiovascular diseases.
The role of SUMO1-mediated SUMOylation regulated by the transcriptional mechanism in mitophagy has attracted much attention. Long noncoding RNAs (lncRNAs) have been reported to play an important role in mitophagy, yet the role and mechanism of lncRNAs in SUMOylation are unclear [79]. Liu et al. established that lncRNAs promote SUMOylation induced by SUMO1 at K333 and then provoke mitophagy activation, which contributes to the development of medications targeting gliomas [79]. However, the type of lncRNA has not been identified, and further studies are needed to clarify the transcriptional binding mechanism of lncRNAs to SUMO1 for SUMOylation regulation.
SUMO2/3
SUMO1 shares 50% sequence homology with SUMO2/3; nevertheless, SUMO2 is highly homologous to SUMO3 [80]. SUMO2 is 97% identical to SUMO3, in which the SUMO2 precursor consists of 103 amino acids and the SUMO3 precursor contains 95 amino acids [22]. Under normal conditions, SUMO2 and SUMO3 are identified as unconjugated forms, which are rarely involved in the SUMOylation of target proteins [81]. However, SUMO2 and SUMO3 can bind to the lysine site of target proteins for SUMOylation under stress conditions, such as oxidative stress or hypoxia [81]. The morphological alteration of mitochondria is associated mainly with multiple stress conditions, which also activate SUMO2/3-mediated SUMOylation. However, the precise mechanism by which SUMO2/3 regulates mitophagy remains unknown.
SUMOylation of DRP1 is involved in regulating mitochondrial fission and mitophagy activation [55,78]. However, mitochondrial-associated protein ligase (MAPL) can also be a SUMO-conjugation mediator, functioning as an E3 ligase for SUMOylation of DRP1 [82]. Furthermore, SUMOylation may also be involved in mitochondrial biochemical functions, including β-oxidation of fatty acids. Mohanty et al. reported that MAPL, located in the OMM, is the first mitochondrial E3 ligase with a RING-finger domain, of which upregulation promotes mitochondrial fragmentation [83]. Additionally, mitochondrial fragmentation is associated with MAPL-mediated SUMOylation of DRP1. However, in vivo research has revealed that overexpression of SENP5 in murine cardiomyocytes (SENP5 transgenic, SENP5-Tg) leads to a decrease in SUMO2 attachment to DRP1, which further promotes mitochondrial fission; however, whether SENP-mediated deSUMOylation can also regulate mitophagy activation is still unknown. Future investigation should focus on the potential role and mechanism of SENP family proteins in regulating DRP1-induced mitophagy activation [84]. Furthermore, Waters et al. proposed a model in which SENP3-mediated deSUMOylation facilitates Fis1 mitochondrial localization to underpin stress-induced mitophagy. They found that this model promotes the stabilization of SENP3 and leads to Fis1 deSUMOylation, whereas loss of SENP3 abolished mitophagy activation [85]. Possible mechanisms for such different views and contradictions may be associated with differences in the intracellular environment caused by different stages of cell proliferation and growth. Further investigation is necessary to focus on specific roles for the conjugations of SUMO1-DRP1 and SUMO2/3-DRP1 in the process of mitophagy.
The complexity of SUMOylation modification by SUMO2/3 in regulating mitophagy is also achieved by other mitochondrial-related pathways. Dongdem et al. preliminarily identified SUMO2 as a candidate for PINK1-mediated mitophagy. By overexpressing SUMO2 and its C-terminal-GG deletion mutant, carbonyl cyanide m-chlorophenyl hydrazine (CCCP)-induced mitophagy is significantly inhibited in HEK293T cells, suggesting the promotional role of SUMOylation in PINK1/Parkin pathway regulation [13]. Nuclear dot protein 52 (NDP52), also known as CALCOCO2, is associated with mitophagy activation [86]. As an autophagy receptor, NDP52 is required for autophagic engulfment of damaged mitochondria and promotes mitophagy through binding with PINK1 [87]. Recently, Fan et al. reported that classical swine fever virus (CSFV) infection inhibited the expression of NDP52 and the modification of SUMO2 by PINK1/Parkin pathways [88]. Interestingly, Rab35 activation has been reported to be able to contribute to Parkin-mediated mitophagy by recruiting NDP52, which further depolarizes mitochondria [89]. Although there is no direct evidence that SUMO2-mediated NDP52 SUMOylation is involved in PINK1-regulated mitophagy, SUMO2 is believed to be able to target NDP52 or other autophagic receptors located on mitochondria and further promote mitophagy by activating PINK1/Parkin pathways. Future investigations need to follow on the precise modification sites of NDP52 or PINK1 by SUMO2, which contributes to widely discuss the role of SUMOylation in the mitophagy activation. Moreover, the regulatory mechanisms of mitophagy in different diseases remain unclear. SUMOylation in mitochondrial proteins has attracted considerable attention for the formulation of drug intervention strategies for mitochondria-related diseases. Gao et al. revealed a direct relationship between SUMO2 and mitophagy. They demonstrated that the interaction between SUMO2 and the mitochondrial membrane protein SH3GLB1 can promote PINK1-induced mitophagy activation, which further provoked X-ray-induced cardiac hypertrophy [90]. Juncker et al. found that SUMO2-mediated SUMOylation is involved in ataxia telangiectasia (AT), which is a neurodegenerative disease [20]. They demonstrated that SUMO2 interacted with mitofusins (MFNs) and promoted mitochondrial translocation into lysosomes via SUMO2/ubiquitin/LC3 signalling. Furthermore, SUMO2 has been reported to facilitate MFN SUMOylation and attenuate the expression of interferon-stimulated gene 15 (ISG15), which promotes mitophagy activation [20]. Meanwhile, another study showed that MFN1/2 are modified by SUMO2 in HEK293 cells treated with carbonyl cyanide chlorophenylhydrazone (CCCP) and the proteasome inhibitor MG132 and then found that the colocalization of SUMO2 with LC3 and MFN1/2, which protects SUMOylation, protects damaged mitochondria from the heathy mitochondrial network until their removal via mitophagy [13]. Therefore, future studies should focus on the association between SUMO2 and mitochondrial-related proteins, such as the SUMOylation mechanism of SUMO2 in SH3GLB1 or Bnip3-mediated mitophagy fusion and fission [58].
SUMO4
SUMO4 was first discovered in five individuals with type 1 diabetes (T1D) and five controls from European, American, French, and Chinese populations searched for sequence variations in MAP3K7IP2 and SUMO4 [91]. Guo et al. identified SUMO4 as a novel gene sharing 87% amino acid identity with SUMO2 and 90% nucleotide identity [91]. SUMO4 has thus far been identified in humans and has a unique distribution in contrast to SUMO1 and SUMO2/3. Methionine at amino acid position 55 of SUMO4 is evolutionarily conserved among diverse species. The 163A → G single nucleotide polymorphism (SNP) in the CUE domain of SUMO4 has been found to result in an M55 V amino acid substitution [92]. SUMO4 has been reported to be expressed in cardiac, immune, renal, and pancreatic cells [93,94,95]. However, the multiple molecular biological functions of SUMO4 in organelles of these cells are still unclear, and the exact role and precise mechanism of SUMO4 in mitophagy need to be further investigated.
The present studies focused primarily on the transcriptional regulation of SUMO4 in genetic variants, which promotes the establishment of treatment strategies for certain diseases. Neumann identified SUMO4 as a protein modifier by adding the 12B8 tag to the C-terminus of the MHC class-II HLA-DQh chain and attaching the 6D4 epitope to the C-terminus of HLADPh [96]. A study involving 126 liver transplant (LT) patients showed that SUMO4 rs237025 polymorphisms are significantly associated with new-onset diabetes mellitus (NODM) after LT [97]. Baczyk et al. found that the hyper-SUMOylation of SUMO4 is predominantly covalent in nature, resulting in alterations in preeclamptic pregnancies [98]. Furthermore, SUMO4-mediated SUMOylation is found to have a unique spatiotemporal distribution during placental development, and the subcellular localization of SUMO4 can be further modulated by extracellular stressors [99]. Although the role of SUMO4 is constantly being discovered and determined, the regulatory mechanism of SUMO4-mediated SUMOylation is still unclear. The hypothesis that SUMO4 regulates cellular homeostasis may involve its maturation and degradation processes. Under normal conditions, the rapid degradation of SUMO4 is dependent mainly on the cell itself. Under stress conditions, SUMO4 can be matured by stress-induced endogenous hydrolases and covalently conjugated to their substrate proteins. Additionally, SUMO4 can regulate cellular function by SUMOylation of substrate proteins via its C-terminal GG motif, which is not affected by proline-90, which is unique to SUMO4 [8].
Metabolic conditions are involved in the association between genetic regulation and SUMO4-mediated SUMOylation. The upregulation of free fatty acids (FFAs) has been reported to promote lipid metabolism disorder and inflammatory activation, which leads to mitochondrial dysfunction and the development of diabetes mellitus [100]. The datasets of a subnetwork from obesity and diabetes indicate the significant roles of SUMO4 in insulin signalling in diabetes progression. Mitochondria play a critical role in regulating carbohydrate metabolism, in which glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is identified as a significant enzyme [101]. GAPDH has been demonstrated to be able to interact with SUMO4 to induce microvascular complications and faulty insulin signalling, which disrupt insulin sensitivity in diabetic individuals [102]. A number of studies have indicated that reactive oxygen species (ROS), continuously generated by mitochondria, play an important role in stress signalling, which results in a dual role in cardiovascular diseases [103]. Mitochondrial uncoupling proteins (UCPs) regulate the efficiency of oxidative phosphorylation (OXPH) and are involved in GAPDH-mediated mitochondrial ROS production. Other research has shown that both GAPDH and epidermal growth factor receptor (EGFR) are hub genes, and SUMO4 can interact with them directly, which further impairs insulin signalling [102]. However, it is still not possible to infer whether the interaction between GAPDH and SUMO4 could induce SUMO4-glycosylation of GAPDH. Although SUMOylation-targeting processes contribute to the upregulation of mitochondrial ROS production and provocation of mitochondrial dysfunction and mitochondrial fission, the SUMO4-mediated SUMOylation of target substrates in the regulation of mitophagy activation is still unclear.
The association between mitophagy and SUMO4 has rarely been reported, and the posttranslational modification of SUMO4 in mitochondria needs further investigation. The regulatory role of SUMOylation induced by SUMO4 in altering mitochondrial dynamics is the main direction of exploration, especially the modification of DRP1 and MFN1/2 by SUMO4. In addition, the distinct roles and unique modifications among SUMO4, SUMO1, and SUMO2/3 need to be further studied. More details of the SUMOylation sites on mitophagy-related proteins, such as Bnip3, OPA1, and Fis1, should be further investigated (Fig. 2).

Regulatory mechanism of SUMOylation in mitophagy activation. SUMO molecule-mediated SUMOylation is involved in mitochondrial dynamics through multiple signalling pathways, especially in mitophagy activation. The interaction between SUMO and PINK1/Parkin is inhibited by SENP family proteins, which further suppresses mitochondrial oxidation induced by complex I. SUMOylation of DJ-1 inhibits ROS production, whereas SUMOylation of PGC-1α further promotes the expression of SIRT3, which leads to mitochondrial fission and fusion. SUMOylation of DRP1 induced by FADD is critical for mitochondrial fission and mitophagy activation. Accumulation of DRP1 SUMOylation promotes the release of mitochondrial cytochrome C, leading to mitophagy. Other substrates in mitochondria, including OPA1 and MFN1/2, can also be SUMOylated and induce autophagic vesicle engulfment of mitochondria for lysosomal degradation. The dashed line indicates that mitochondria are not necessarily the subcellular location of DRP1 SUMOylation
Potential role of SUMOylation in cardiac hypertrophy
Cardiac hypertrophy is characterized by the enlargement of individual cardiomyocytes, including an increase in cardiac mass and abnormal pathophysiological growth of cardiomyocytes in both width and length [104]. The involvement of mechanical stresses induced by either extrinsic factors or intrinsic factors in the growth response of cardiac cells is significantly associated with the development of cardiac hypertrophy, including increased pressure, volume overload, and ischemia-induced cardiac remodelling [105,106,107]. These pathophysiological factors suggest that prevention of cardiomyocyte alteration serves as a potential strategy for therapeutic intervention of cardiac hypertrophy. Mitochondrial quality control has been fully considered an adaptive response to cardiac hypertrophy [108]. Due to the essential role of mitochondria in mediating the alteration of cardiac cells, mitochondrial dysfunction, and even mitophagy are demonstrated to play a critical role in regulating cardiac hypertrophy. However, the mechanism underlying pathological alteration of cardiac cells and the potential functions of mitophagy in cardiac pathophysiology need to be clarified.
Growing evidence has supported that mitophagy is associated with the process of dysfunctional cardiac cells and the development of cardiac hypertrophy [58]. The strong relationship between mitophagy and cardiac hypertrophy is first involved in Bnip3 expression and regulation [109]. Du et al. reported that doxorubicin (Dox) promotes cardiac hypertrophy by downregulating sirtuin-3 (Sirt3), whereas Bnip3 overexpression completely provoked cardiac toxicity via mitophagy [110]. MST1, a serine/threonine-protein kinase, is considered an upstream regulator in mediating the progression of cardiac hypertrophy. Excessive activation of MST1 significantly suppresses cardiac proliferation and hypertrophy through inhibition of mitophagy induced by Beclin1, which further leads to dilatation of the heart [111]. Recent research has shown that SUMOylation and its related ligases are involved in mediating the mitophagy process, suggesting a central role of SUMOs in regulating cardiac hypertrophy via mitophagy activation [69,112]. Dilated cardiomyopathy (DCM) has been found to be associated with gene variations of lamin A/C (LMNA); however, the precise mechanism of LMNA in cardiac hypertrophy is poorly understood [113]. Sylvius et al. have shown that either dramatic ultrastructural changes of the cardiomyocyte nucleus (D192G) or nonspecific changes (R541S) are involved in end-stage DCM patients carrying LMNA mutations [114]. Further assessment revealed that D192G lamin C expression impaired SUMO1 distribution and functions, suggesting that SUMO1-mediated SUMOylation contributes significantly to DCM via its transcription, chromosome organization, and nuclear trafficking [114]. Moriuchi et al. further identified two lamin A mutations, E203G and K201R, that can be SUMOylated by SUMO2, which contributes to the development of laminopathies [115]. In this research, they found that the SUMOylation-defective E203G mutant inhibited normal localization of WT lamin A in a dominant-negative manner but exhibited abnormal subnuclear distribution, which is associated with familial dilated cardiomyopathy. However, K201R at the amino acid residue of SUMO2 has no effect on the phenotypes of cardiac cells; however, it is not enough to explain why SUMOylation itself is not involved in the pathogenesis of cardiac hypertrophy [115]. Under the transcription and chromosome organization levels, the stable expression of homeobox genes is critical for normal heart growth and development [116,117]. Kim et al. identified Nkx2.5 as a potential target of SUMOylation, which can be further involved in a variety of cellular activities during the development of heart growth [118]. Although the regulatory mechanism of SUMO1 in mediating Nkx2.5 is still unclear, SUMO1 can covalently attach to lysine residue 51 to enhance Nkx2.5 activity. In addition, the association between Nkx2.5 mutants and aberrant SUMOylation has also demonstrated that modification alteration of SUMOylation may underlie the development of cardiac hypertrophy associated with Nkx2.5 mutants [118].
Research has shown that SUMOylation plays a key regulatory role in mediating mitochondrial fusion and fission by modifying Fis1, OPA1, MFN1/2, and DRP1 (Fig. 3) [119]. However, whether SUMOylation-regulated mitophagy can be involved in cardiac hypertrophy remains unknown; in addition, the mechanisms by which SUMOs induce cardiac hypertrophy via mitophagy activation need to be further investigated. Given the critical role of SUMOylation in regulating cardiomyocyte alterations, a recent study has shown that the modification of myomesin-1 by SUMO1 from the nucleus to the cytoplasm regulates cardiac hypertrophy [120]. Wang et al. found that the addition of a SUMO1 mimic to primary cultures of cardiac cardiomyocytes from 1-day-old Sprague–Dawley (SD) rats significantly promoted the nuclear localization of myomesin-1 to the cytoplasm, leading to sarcomere organization-induced cardiac hypertrophy [120]. As a multifunctional post-translational protein, the SUMO molecule contains 101 amino acid residues and modifies target proteins through SUMOylation. However, whether SUMO1-mediated SUMOylation of myomesin-1 can also occur in adult cardiomyocytes is still unclear. Additionally, the SUMO-conjugating machinery to other SUMOylated peptides in cardiomyocytes needs to be further understood. SUMOylation is highly relevant in signal transduction, especially in response to cellular stress-related pathways [5,50]. To further investigate the role of SUMOylation in cardiac hypertrophy, Lee et al. revealed that SUMO1 in mice undergoing transverse aortic constriction (TAC) significantly inhibits cardiac hypertrophy development by promoting SUMOylation of the sarcoplasmic reticulum calcium ATPase (SERCA2a) pump. However, under conditions of cardiac hypertrophy, whether SUMOylation of SERCA2a induced by SUMO1 can regulate mitophagy activation needs further investigation [121]. In TAC animals, SUMO1 overexpression by adeno-associated vector type 9 (AAV9) elevates the level of SERCA2a SUMOylation and inhibits the MEK/ERK signalling pathway but has no effect on JNK/c-JUN signalling. However, a previous study demonstrated that JNK activation contributes to SUMO1 deficiency, which can further promote cardiac hypertrophy [122]. Furthermore, Chaanine et al. showed that overexpression of JNK, which is activated by the transcription factor FOXO3a, increases Bnip3 expression in response to pressure overload and induces mitophagy activation, which further promotes the heart failure process [123]. Therefore, whether SUMO1-mediated SERCA2a SUMOylation is involved in the JNK/c-JUN signalling pathway needs to be further studied, and the mechanism of SUMOylation-regulated mitophagy activation in mediating cardiac hypertrophy development should be the focus of future investigation. Transcriptional regulation plays an important role in the modification of SUMOylation, particularly certain transcription factors that regulate the expression of SUMO molecules. Although SUMOs and their SUMOylation play a crucial role in the development of cardiac hypertrophy, the precise molecular mechanism regulating the stability and trans-activity of target proteins in cardiomyocytes remains unclear. A recent report established that nuclear factor kappa-B (NF-κB) may attenuate the transcriptional activity of myocardin by inhibiting myocardin SUMOylation by SUMO1/PIAS1, thus impairing the development of cardiac hypertrophy. Further investigation is involved in a feedback loop in which miR-1 is upregulated by NF-κB, which also represses SUMO1 expression and inhibits SUMOylation of myocardin [124]. Furthermore, another study also found a mediatory role of SUMOylation in regulating SERCA2a expression; however, the underlying regulatory mechanism remains elusive. By targeting the transfer of AAV9, Oh et al. supported a strong association between miR-146a and SUMO1. In vivo and in vitro research has both revealed that miR-146a downregulates the expression of SUMO1 and then inhibits SUMOylation of SERCA2a, which further attenuates cardiac contractile function and promotes cardiac hypertrophy [66].

Potential role of SUMOylation in cardiac hypertrophy. SUMOylation is associated with the expression and regulation of multiple cardiac proteins, which are involved in the development of cardiac hypertrophy. SUMOylation of Desmin in sarcomeres is dependent on the α-DAG/DMD/ACTG1 signalling pathway, which further promotes LaminA/C expression. Additionally, the release of calcium via Ryr-induced ER stress leads to sensitivity of ATPase activity. HSF2 SUMOylation by PCGF2/MEL18 signalling induces IGF-IIR SUMOylation, resulting in the SUMOylation of PINK1/Parkin and activating mitophagy. Additionally, SUMOylation of IGF-IIR may promote myocardin expression, and its SUMOylation can be inhibited by NF-κB. UBC9 and MAPL promote the interaction between SUMO and ZAK and then inhibit the activity of PI3K/ERβ signalling, which promotes the upregulation of TNF-α and IL-6 induced by PINK1/Parkin. Furthermore, the SUMO molecule is involved in SERCA2a SUMOylation mediated by PIASy through JNK/c-JUN; conversely, the SUMOylation of MEF-2C induced by NFAT3/SENP1/PGC-1α promotes PINK1-mediated IGF1, ACE and TGF-β upregulation. Finally, SUMOylation promotes the development of cardiac hypertrophy via ventricular diastolic dysfunction, perfusion injury, ventricular remodelling, and ventricular noncompaction
Given the critical role of mitochondrial biogenesis and mitophagy in cardiovascular diseases, studies have further explored the function and mechanism of SUMO-related proteins in the development of cardiac hypertrophy [53,55,84]. Cai et al. have found that SENP1 may be a key protein in regulating mitochondrial function and the pathogenesis of cardiomyopathy. By infecting TAC mice with cardiotropic recombinant adeno-associated virus serotype 9 (rAAV9) to achieve targeted SENP1 overexpression, mitochondrial biogenesis is significantly promoted, and partial amelioration of cardiac hypertrophy is observed [125]. Furthermore, they also found that SENP1 was induced by hypertrophic stimuli through calcium/calcineurin-nuclear factor of activated T cells 3 (NFAT3), and deSUMOylation of myocyte enhancer factor-2C (MEF-2C) mediated by SENP1 enhanced peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) transcription, which inhibited mitophagy activation and cardiac hypertrophy [125]. Gupta et al. have shown that SUMOylation, as an integral part of the UPS, is associated with cardiac protein quality control through autophagy. By using gain- and loss-of-function assays in neonatal rat ventricular cardiomyocytes, UBC9 overexpression, a SUMO E2 enzyme, could directly change the autophagic flux. Additionally, the elevated level of SUMOylation increases cardiac autophagy, which protects against cardiomyocyte-specific expression of a mutant α-B-crystallin and mutant CryAB (CryAB(R120G))-induced proteotoxic cardiac disease [126]. These results indicate that overexpression of UBC9 may find a protective protein in regulating cardiac hypertrophy by upregulating autophagic flux. However, the role and exact mechanism of UBC9 and SUMOylation in mitophagy are still unclear, and further investigation is needed to focus on the regulatory function and interacting amino acid sites of SUMO molecules in mitochondrial target proteins in response to the prevention of cardiac hypertrophy. Zhao et al. have also identified the relationship between SUMOylation and mitochondrial alteration in regulating cardiac hypertrophy [127]. They have considered mitochondrial anchored protein ligase (MAPL) as a special SUMO E3 ligase, which can contribute to mitochondrial fission. By silencing MAPL, phenylephrine (PE)-induced upregulation of MFN2 is significantly decreased, accompanied by the inhibition of mitochondrial fission and cardiac hypertrophy. Further investigation focused on the feedback loop in which the miR-485-5p/MAPL/MFN2 axis is involved in mitochondrial machinery and cardiac hypertrophy development. Nevertheless, the underlying mechanism by which SUMO enzymes regulate mitophagy is still unclear. Similar to the E3 SUMO enzyme, muscle RING-finger protein-1 (MuRF1) plays a critical regulatory role in cardiac muscle tissues [128]. Heras et al. discovered that MuRF1 can target SUMO1 for SUMOylation by mediating UBC9 and the E3 PIASy/4, which contributes to cardiac muscle disorders [129]. A new study reported that SUMOylation may play a protective role in cardiac hypertrophy. As a critical protein for hypertensive angiotensin II (Ang-II)-induced cardiac hypertrophy, insulin-like growth factor receptor II (IGF-IIR) signalling can be regulated by heat shock transcription factors (HSFs) during the progression of heart failure [130,131]. However, recent research has demonstrated that HSF2 appears to be SUMOylated by SUMO1 and is reversely inhibited by PCGF2/MEL18. The inhibition of HSF2 SUMOylation by IGF-IIR-mediated signalling suggests that the PCGF2/MEL18/SUMO1/HSF2/IGF-IIR signalling pathway plays a critical role in promoting cardiac hypertrophy [132]. Pai et al. have shown that aberrant expression of leucine zipper- and sterile ɑ motif-containing kinase (ZAK) by SUMO1 modification alters the interaction of phosphoinositide 3-kinase (PI3K) with estrogen receptor β (ERβ) to inhibit the degradation of PI3K in H9C2 cells, which further suppresses myocardial damage and cardiac hypertrophy [133].
As a dynamic organelle, mitochondria undergo a constant cycle of fusion and division to maintain their functions [13]. Therefore, the modification of mitochondrial target proteins seems to play an important role in regulating mitochondrial morphology and biofunctions. SUMOylation is identified as a multifunctional modification for target proteins that is necessary for containing a diglycine motif at the C-terminal sequence [5]. However, other mechanisms of SUMO-related proteins, including SENPs, UBC9, and MAPL, can also be involved in mediating mitochondrial dysfunction and mitophagy activation. The functional role and exact mechanism of mitophagy in the development of cardiac hypertrophy have been widely reported [108,134]. SUMOylation-mediated mitophagy is believed to be involved in multiple processes of cardiac hypertrophy. The mechanisms underlying SUMOylation in cardiac hypertrophy seem to be dependent on PINK1/Parkin, Bnip3 or sestrin2 signalling; however, the exact regulatory mechanism of the interaction of SUMO molecules and these proteins remains unclear [58,135,136]. Although numerous studies have demonstrated that SUMOylation activation by SUMO molecules ultimately maintains mitochondrial function, the precise mechanisms underlying the mitophagy actions of SUMOylation remain elusive. Further studies are needed to explore whether SUMOylation and its related proteins, such as SUMO4, SUMO5, SENPs, and MuRF1, are significantly sufficient to affect mitophagy in response to cardiac hypertrophy.
Involvement of SUMOylation in hypertension
Hypertension has been identified as the most preventable risk factor for cardiovascular diseases and is characterized by persistently high blood pressure in the systemic arteries [137]. Hypertension has been discovered as a serious cardiovascular event with changes in metabolic substrate utilization, dysfunction of the electron transport chain, and adenosine triphosphate (ATP) synthesis [138]. Therefore, mitochondria have been considered crucial in the pathogenesis of hypertension. The phenotype and functionality of cardiomyocytes are regulated by mitochondrial energetic metabolism, of which dysfunction may lead to drastic changes in blood pressure [138]. Multiple studies have revealed that mitochondrial dynamics, especially mitophagy, can properly maintain cellular functions, which are involved in the development of hypertension [139,140]. The mechanisms associated with mitophagy in hypertension are still unknown, and little research has recently focused on PINK1/Parkin signalling or Bnip3-mediated autophagosomes, which fuse with lysosomes for mitochondrial degradation.
Modification of mitochondrial dynamics has been considered to play a critical regulatory role in mitophagy, particularly SUMOylation in mitochondrial fission and fusion (Fig. 4) [132]. The voltage-gated potassium (Kv) channel Kv1.5 is identified as a key protein to repolarize the I (Kur) current in human atrial myocytes, which regulates vascular tone [141]. Benson et al. demonstrated that Kv1.5 can be the target protein for interacting with the SUMO-conjugating enzyme UBC9 and is a target for SUMOylation by SUMO1, SUMO2, and SUMO3. As a SUMOylated substrate, Kv1.5 can be deSUMOylated by SENP2 and Ulp1, and this disruption of conjugation leads to a selective approximately 15-mV hyperpolarizing shift in the voltage dependence of steady-state inactivation [142]. Indirect evidence from Tian’s research has also demonstrated that deSUMOylation mediated by SENP1 is implicated in the development of hypertension [143]. Hypoxic pulmonary hypertension (HPH) in an animal model was established by normobaric intermittent hypoxia, and the expression of SENP1 was found to be downregulated as well as the high level of SUMOylation was found to lead to the enhancement of pulmonary arterial pressure (mPAP). However, whether SENP1-mediated deSUMOylation is involved in mitophagy under chronic hypoxic conditions needs to be further studied. A further mechanism of SUMOylation in HPH was preliminarily clarified by Jiang et al., who showed that SUMO1 promotes SUMOylation upregulation in rat pulmonary arterial smooth muscle cells (PASMCs) by directly interacting with HIF1α [144]. Additionally, almost the same research has clarified that deSUMOylation induced by SENP1 promotes the stability of HIF‑1α, which enhances the proliferative ability of PASMCs and the development of HPH [145]. A recent study has identified Bnip3 as an upstream mediator for HIF‑1α to promote mitophagy activation and is accompanied by abnormal alteration of mitochondrial DNA (mtDNA) and ROS production and aberrant expression of cytochrome c oxidase IV (COX IV) and translocase of outer mitochondrial membrane 20 (TOMM20), which is further involved in regulating hypertension [146]. Future hypotheses should focus on the association between SUMOylation and mitophagy-related proteins, especially Bnip3, which may be a promising therapeutic strategy for the prevention of hypertension in PSMCs. Activating transcription factor 3 (ATF3) functions as an adaptive response protein for inducing hypertension, and a functional experiment has shown that Ang II incubation elevates the levels of SUMO1 and ATF3 [147]. SUMO1 promotes the SUMOylation of ATF3 at position K42, inducing the expression of inflammatory molecules such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-8 and provoking the production of nitric oxide (NO), eventually promoting dysfunction of vascular endothelial cells (ECs) and the development of hypertension [147].

Involvement of SUMOylation in hypertension via mitophagy. SUMOylation regulates numerous signalling pathways to mediate the development of hypertension. The critical role of SUMOylation in hypertension involves the interaction between SUMO and the Kv1.5 channel. Initially, UBC9 promotes Kv1.5 channel SUMOylation, which is further inhibited by SENP2. Additionally, the interaction between MAPK/p38 and SUMO promotes the SUMOylation of MAPK under HIF1α SUMOylation and then promotes the phosphorylation of MK2. The phosphorylation of MK2 induces Bnip3/Bax activation and the upregulation of mitochondrial ROS production and AIF expression, which leads to cytochrome C release. However, both SUMOylation of HIF1α and phosphorylation of MK2 can regulate the expression of inflammatory factors, such as TNF-α, IL-6, and IL-8, and downregulated PCNA inhibited by SENP3 suppresses pulmonary hypertension
Bioinformatics has prospectively predicted that K265 and K402 in dopamine receptor D1 can be SUMOylated by SUMO1, which is also associated with the development of hypertension. The mechanism targets SUMOylation to impede the dephosphorylation of the D1 receptor mediated by the interaction of G protein kinase 4 (GRK4) with protein phosphatase 2A (PP2A), which may provide new insights for the treatment and prevention of hypertension [148]. However, Yao et al. further established a mouse model of HPH and found upregulated expression levels of LC3 and SUMO1. In contrast, the overexpression of SUMO1 promotes vacuolar protein sorting 34 (VPS34) SUMOylation at position K840 and further induces the assembly of the Beclin1/VPS34/ATG14 L complex, which activates autophagic flux and the development of hypertension [149]. Cai et al. have further highlighted the critical role of SENP3 in mediating vascular remodelling and hypertension. By generating an animal model of hypertension in SENP3+/− mice, they found that the interaction between SENP3 and β-catenin inhibited SUMO2/3-mediated SUMOylation and lysosomal degradation of β-catenin, suggesting the important function of SENP3-induced deSUMOylation in the development of hypertension [150]. However, the exact role and precise mechanism of SUMOylation in regulating the hypertension process via mitophagy activation remain unclear. Mitophagy-related signalling pathways have been strongly implicated in the development of hypertension [151]. Multiple physiological stimuli, including hypoxia, Ang-II and NO, can promote mitophagy activation in vascular cells, which is involved in hypertension processes [138]. As a multifunctional modification in mitochondrial dynamics, SUMOylation is considered to play a dual role in mediating hypertension via mitophagy. Therefore, future research should determine the accurate role of SUMOylation in modifying mitochondrial dynamics and mitophagy for the occurrence and development of hypertension. Additionally, targeting SUMOylation to mitochondrial proteins has been confirmed to be exploited to develop therapeutic strategies to prevent hypertension.
SUMOylation targets atherosclerosis via mitophagy
Atherosclerosis is a progressive chronic inflammatory cardiovascular disorder that is characterized by the formation of lipid-filled plaques within arteries [152]. Numerous studies have demonstrated that the accumulation of lipids in the walls of medium- and large-sized arteries can lead to atherosclerotic plaque development [153]. However, the mechanism of lipid accumulation and the progression of atherosclerotic plaque formation are both unknown. Mitophagy, an autophagic response that specifically targets mitochondria, is reported to be involved in pathological processes of atherosclerosis [154]. Mitochondrial dysfunction and mitophagy are potentially linked to the degradation of atherosclerotic plaques, which further provokes the development of atherosclerosis [155]. Numerous studies have shown that lipid metabolism alterations, inflammation, and oxidative stress are significantly associated with mitophagy activation, suggesting that mitophagy may play a protective role in preventing atherosclerosis by stabilizing atherosclerotic plaques [156,157]. Although mitophagy is essential for oxidative metabolism, in which lipid disorder is a crucial event in atherosclerosis, the proatherogenic molecular mechanism of mitophagy is less clarified [158].
SUMOylation is accomplished with activating enzyme E1 (SAE1/SAE2), conjugating enzyme E2 (UBC9) and ligating enzyme E3, which contributes to maturation, conjugation and ligation [41]. Atherosclerosis has been demonstrated to be associated with dyslipidemia that involves increased serum triglycerides and LDL and decreased high-density lipoprotein (HDL) [152]. SUMOylation regulates multiple cellular processes to maintain metabolic homeostasis, including cholesterol metabolism, synthesis of steroid hormones, and bile acid metabolism (Fig. 5) [159]. Dehnavi et al. found that the lysine residue on PPARα can be modified by the SUMO molecule [160]. Furthermore, Kim et al. reported that the expression of SUMO2 is elevated in the aortic endothelium of hypercholesterolemic LDL receptor-deficient (LDLR−/−) mice and is responsible for the impairment of endothelium-dependent vasorelaxation, suggesting that SUMO2-mediated SUMOylation may promote the development of atherosclerosis [161]. SUMOylation can also be associated with reverse cholesterol transport (RCT), which is characterized as an antiatherogenic process. The SUMOylation of liver receptor homologue 1 (LRH-1) at position K289 promotes its interaction with prospero homeobox protein 1 (PROX1), thereby altering the RCT process and promoting atherosclerosis [162]. Additionally, both UBC9 and PIASy can induce the SUMOylation of PPARα at position K185 to suppress the transcriptional activity of PPARα, which further promotes dyslipidemia [163]. Lim et al. have shown that SUMOylation of PPARα at position K107 is involved in vascular smooth muscle cell (VSMC) proliferation and migration; conversely, the deSUMOylation of PPARα might play an important role against atherosclerosis [164]. In contrast, SUMOylation may be involved in a protective role in inhibiting VSMC proliferation. The regulatory role of Rho-specific guanine nucleotide dissociation inhibitor (RhoGDI) is widely reported in vascular remodelling [165]. Dai et al. demonstrated that Ang-II can promote RhoGDI degradation by inducing RhoGDI SUMOylation; conversely, the competitive binding of SUMO to RhoGDI regulates RhoGDI stability and VSMC proliferation [166]. McPherson and Gauthier have shown that SUMOylation of sterol regulatory element binding proteins (SREBPs) plays a unique and fundamental role in both cholesterol and fatty acid metabolism, and the regulation of SREBP-1c and SREBP-2 by SUMO ultimately mediates dyslipidemia and insulin resistance [167]. The mechanism of SUMO in lipid metabolism disorders of atherosclerosis may be associated with estrogen receptor α (ERα) modification. Kobayashi et al. found that UBC9 and PIAS1 can interact with ERα in a manner dependent on the presence of 17β-estradiol by using a yeast two-hybrid system from a human heart cDNA library, which plays important roles in the pathophysiology of atherosclerosis [168]. Endothelial dysfunction is reported to be involved in atherosclerosis development [169]. The maintenance of endothelial function is dependent mainly on shear stress-induced ERK5 activation, the regulation of Kruppel-like Factor 2 (KLF2) and endothelial NO synthase expression [170,171]. Woo et al. further clarified that H2O2 and advanced glycation end-products (AGEs) induce SUMOylation of ERK5 mediated by PIAS1 and UBC9 at positions K6 and K22, respectively, subsequently inducing the expression of KLF2 and endothelial NO synthase (eNOS), which further promotes endothelial dysfunction [172]. Moreover, PIAS1-mediated SUMOylation of NF-κB also initiates inflammatory responses, including TNFα and lipopolysaccharide (LPS), that contribute to atherosclerosis [173]. The mechanism of SUMOylation of NF-κB in proatherosclerosis may involve conjugation between SUMO and IκBα, thereby promoting the interaction of adipocyte enhancer-binding protein 1 (AEBP1) with NF-κB and facilitating inflammatory conditions [174]. Other oxidative products, such as peroxynitrite (ONOO−), are associated with the binding of PIASy to protein kinase C ζ (PKCζ), leading to SUMOylation of p53 and inducing disturbed blood flow, which causes atherosclerosis [175]. However, the mechanism of PKCζ in regulating atherosclerosis is still unclear, and future studies need to focus on the regulatory role and molecular mechanism of SUMO-mediated conjugation in downstream mediators. Surprisingly, Heo et al. concisely demonstrated that ONOO− upregulates the activation of PKCζ and induces the interaction of p53 with Bcl-2 by SUMOylating p53. Subsequently, ONOO enhances the interaction between PKCζ and PIASy via the Siz/PIAS-RING domain at amino acid position 301–410 of PIASy, for which p53 SUMOylation mediated by PKCζ promotes endothelial dysfunction and the development of atherosclerosis [176]. Overall, these results reveal the promoting role of disturbed blood flow in the regulation of endothelial dysfunction via ONOO− production-dependent PKCζ. Then, the interaction of PKCζ with PIASy enhances the SUMOylation of p53 by decreasing the expression of KLF2 and eNOS, eventually promoting inflammation and atherosclerosis.

Participation of SUMOylation in molecular signalling pathways involved in atherosclerosis. The regulation of lipid metabolism by SUMOylation is involved in the formation of atherosclerotic plaques. On the one hand, LPS promotes PPARα expression, and both UBC9 and PIASy induce PPARα SUMOylation, leading to the interaction between SUMO and RhoGDI, which further promotes the lysosomal degradation of RhoGDI. On the other hand, TNF-α signalling induces UBC9 activation and promotes the SUMOylation of phosphorylated IκBα, and modified IκBα promotes the release of RE1A and p50 from IκBα, which induces the expression of downstream substrates, including ABCA1, LXR, IFN-γ, and AEBP1. Moreover, UBC9 can also promote SUMOylation of ERK5, which induces the interaction between LRH-1 and PROX1. ERK5 SUMOylation provokes ROS production through KLF2 SUMOylation, induces PIASy activation and promotes the nuclear translocation of p53 via PKCζ. Disturbed flow remains a critical role in the development of atherosclerosis through ER stress via SUMOylation. ER stress activates UBC9 and inhibits SENP2, which promotes SUMOylation of p90RSK and MAGI1, eventually inducing ABCA1, ABCG1, and PML expression and provoking the formation of atherosclerotic plaques
DeSUMOylation is also involved in disturbed blood flow-induced inflammatory responses in endothelial cells, further causing atherosclerosis. Heo et al. further revealed that SENP2-mediated deSUMOylation induces the SUMOylation of p53 and ERK5 in regulating atherosclerosis in SENP2+/−/LDLR−/− mice [177]. Although the elucidation of the precise mechanisms regulating SUMOylation in atherosclerosis is needed to better understand the proatherogenic molecular mechanism, the exact role and regulatory mechanism of SUMO conjugation in mitophagy remain unclear. Moreover, Xiao et al. reported that SENP2 in atherosclerosis is significantly associated with SUMOylation-mediated keratinocyte migration. Mechanistically, SENP2 targets nuclear Dbf2-related 1 for SUMOylation at position K465 to promote p38/ERK1/2 activation by attenuating the interaction between MEKK1/2 and NDR1 [178]. Other modifications, such as phosphorylation, may cooperate with SUMOylation to play a regulatory role in atherosclerosis. Abe et al. have shown that the interaction of p90 ribosomal S6 kinase (p90RSK) with membrane-associated guanylate kinase with inverted domain structure-1 (MAGI1) leads to the phosphorylation of SENP2 at position T368 and subsequently induces deSUMOylation of MAGI1 at position K931. The deSUMOylation of MAGI1 further enhances nuclear translocation of p90RSK-MAGI1 and formation of the ATF6-MAGI1 complex, which accelerates endothelial dysfunction and atherosclerosis [179]. Dysregulated miRNA-181b is reported to mediate the polarization of M1 macrophages via inhibition of PIAS1-induced SUMOylation of KLF4, eventually causing atherosclerosis [180]. However, whether SUMOylation can be involved in macrophage lipid metabolism and foam cell formation is still unclear, and the regulatory role of SUMOylation in the cholesterol efflux of macrophages needs to be further investigated. ABCG1 plays an important role in regulating macrophage cholesterol metabolism, which contributes to the development of atherosclerosis [181]. Dong et al. recently found that interferon-γ (IFN-γ) promotes the degradation of liver X receptor α (LXRα) via SUMOylation at K22 and K326 by elevating the expression of UBC9 and PIAS1, and the interaction of LXRα with SUMO1 and SUMO2/3 further enhances the inhibition of ABCG1 expression. Suppression of ABCG1 expression decreases cholesterol efflux from macrophages to blood flow, which further promotes the development of atherosclerosis [182]. IFN-γ can also regulate the stability of atherosclerotic plaques by promoting SUMOylation of PML; however, the precise modification of PML and the exact downstream mediator in vascular smooth muscle cells (VSMCs) are still unclear [183].
Multiple studies have demonstrated that SUMOylation of atherosclerotic proteins contributes to the initiation and development of atherosclerosis [163]. SUMO molecules, especially SUMO1 and SUMO2/3, play important roles in the pathogenesis of atherosclerosis. DeSUMOylation was recently found to be involved in an inhibitory role in maintaining endothelial function and the stability of atherosclerotic plaques [178,179]. However, some studies have reported a contradictory conclusion that SUMOylation may play dual roles in the prevention of atherosclerosis [164,165]. Future research should focus on the regulatory role of SUMOylation in mitophagy activation, which may provide a possible explanation of SUMOylation in atherosclerosis development.
Regulation of SUMOylation in mediating cardiac ischemia reperfusion injury
Cardiac ischemia/reperfusion (I/R) injury is identified as one of the acute coronary occlusions that leads to myocardium death, expands the size of myocardial infarction, damages cardiac systolic function, and provokes the development of heart failure [184]. The mechanism of cardiac I/R has been demonstrated to involve mitochondrial dysfunction and mitophagy impairment [185,186]. The dual roles of mitophagy and how mitochondrial dysfunction regulates interrupted cardiac homeostasis remain elusive. PINK1, Bnip3, and Beclin1 are reported to be localized on the OMM to regulate mitophagy activation, which contributes to the development of cardiac I/R. These mitophagy-related proteins are fundamentally anchored at the OMM by their transmembrane domains and attach mitochondria to autophagosomes through the LC3 interacting region (LIR) motif. Driven by the LIR motif, these supervising proteins are capable of recruiting autophagic proteins LC3 and gamma-aminobutyric acid receptor associated protein (GABARAP) to the mitochondrial membrane, further inducing mitophagy initiation independent of the ubiquitin pathway [187]. However, the mechanism of mitophagy in regulating cardiac I/R is still unclear, even though mitophagy-related proteins may play a critical role in the development of cardiac I/R. SUMOylation by SUMO in mitophagy-related proteins has been suggested to alleviate or exacerbate cardiomyocyte damage and is involved in the pathological development of cardiac I/R (Fig. 6). Previous studies have shown that deSUMOylation of DRP1 by SENP3 is essential for physiological and pathophysiological mitochondrial fission and promotes cell death during reperfusion after ischemia by enhancing DRP1 partitioning to the OMM, which causes cytochrome c release. Guo et al. further investigated the mechanism by which deSUMOylation recruits DRP1 to the OMM and found that deSUMOylation selectively promotes DRP1 binding to mitochondrial fission factor (MFF) [188,189].

The regulatory role and function of SUMOylation in mediating cardiac ischemia–reperfusion injury. Cardiac ischemia induces mitochondrial dynamics disorder, which is dependent mainly on SUMOylation activation. The dashed box remains the possible mechanism by which the inhibition of SENP family proteins by cardiac ischemia suppresses the interaction between SUMO and DRP1, and zinc-induced DRP1 SUMOylation promotes mitophagy activation. Upregulation of PIAS1 promotes SUMOylation of PPARγ and then promotes SUMOylation of Sirtuin1 via NF-κB/ERK5/eEF2 signalling. Additionally, PIAS1 promotes SUMOylation of HDAC4 and HIF1α by repressing SENP1-induced deSUMOylation of DJ-7 and further promotes SUMOylation of PML, leading to cardiac injury. UBC9 induces SERCA2a SUMOylation in the endoplasmic reticulum to release calcium, which disrupts the mitochondrial potential and facilitates mitophagy activation
Recent studies have implied that abundant expression of microRNA-499 (miR-499) can prevent cardiac I/R injury by regulating the calcineurin-mediated activation of DRP1 [190]. A possible mechanism may be involved in the regulation of mitochondrial dynamics through miR-499 and the SUMOylation-mediated ubiquitin–proteasome system (UPS). Further investigation has identified that p53 regulates miR-499 transcription and inhibits mitochondrial fission by repressing the SUMOylation-UPS axis, which decreases I/R-induced cardiac hypertrophy and collagen deposition in transgenic mice [190]. These results suggest that SUMOylation-related UPS may mediate the key regulatory progression of target proteins in mitophagy, which is further associated with the development of cardiac I/R. McCrink et al. further found that the interaction between β-arrestin-2 (βarr2) and SERCA2a inhibits postmyocardial infarction (MI) remodelling by promoting acute SUMOylation of SERCA2a, which further protects cardiac functions from ischemic injury [191]. Natural products have also been demonstrated to protect cardiac I/R injury through SUMOylation. Luteolin is reported to alter cardiomyocyte contractility and Ca2+ transients by promoting the expression and stability of SERCA2a. Luteolin upregulates SUMO1-mediated SUMOylation of SERCA2a via PI3K/Akt signalling, which further ameliorates myocardial fibrosis and inhibits cardiac I/R injury [192]. Another study further clarified the regulatory mechanism of SUMOylation in attenuating cardiac I/R injury under luteolin invention. Du et al. found that luteolin increases the expression of SUMO1 to improve mitochondrial membrane potential (ΔΨm) and reduce apoptotic cells by enhancing SUMOylation of SERCA2a at position K585, which further protected cardiac myocardial I/R injury [193]. Moreover, puerarin has been identified as a cardioprotective natural product. Puerarin significantly increases the expression of SUMO2 and promotes SUMOylation in H9C2 cells, which inhibits cardiac I/R injury by attenuating the release of lactate dehydrogenase (LDH) through an estrogen receptor (ER)/ERK/SUMO2-dependent mechanism [194]. Shishido et al. established a myocardial infarction (MI) mouse model by using cardiac-specific CA-MEK5α transgenic mice (CA-MEK5α-Tg) with left coronary artery ligation and found that SUMOylation by UBC9 or PIAS1 inhibited the transcriptional activity of ERK5 and promoted H2O2 production, suggesting the destructive role of SUMOylation in cardiac I/R injury [195]. However, different conclusions from Xie et al. have revealed a protective role of PIAS1-mediated SUMOylation in ameliorating cardiac I/R injury. SUMOylation of peroxisome proliferator-activated receptor γ (PPARγ) by PIAS1 inhibits inflammation by repressing NF-κB activity. However, whether PIAS1 can regulate mitophagy via PPARγ or SERCA2a in mitochondria is still unclear. Additionally, another study reported the underlying therapeutic potential of SUMOylation in cardiac I/R injury. Tilemann et al. found that SUMO1 mediates SUMOylation of SERCA2a and promotes stabilized LV volumes, which further protects cardiac functions in a swine model of ischemic heart failure [196]. DeSUMOylation is reported to also be involved in cardiac I/R injury. Gu et al have demonstrated that deconjugation of SUMO from HIF1α by SENP1 regulates stable expression of HIF1α, indicating that deSUMOylation by SENP1 plays important protective roles in cardiac I/R injury [197]. The paradoxical conclusions indicate that both SUMOylation and deSUMOylation of HIF1α are beneficial to the prevention of cardiac I/R injury. Liu et al. have shown that a high level of SUMOylation of HIF1α promotes lysosomal degradation of HIF1α and downregulates vascular endothelial growth factor (VEGF) expression, suggesting the therapeutic potential of SUMOylation in the prevention of cardiac I/R injury [194]. Previous data have shown that deSUMOylation enzymes are involved in regulating mitochondrial fission in response to cardiac I/R injury. In knockout of Parkinson’s disease autosomal recessive, early onset 7 (DJ-7) mice, accumulation of SUMO1 and reduction of SUMO2/3 leads to mitochondrial fission; furthermore, downregulation of SENP1 and upregulation of SENP5 also promote mitochondrial dysfunction and mitophagy initiation, eventually provoking cardiac I/R injury [198]. Considering the physiological function of mitophagy, we hypothesized that alleviating the expression of PINK1 or Bnip3 may be a potential therapeutic strategy for cardioprotection, and future studies need to verify the mechanism of mitophagy in regulating cardiac I/R injury via SENP1 and SENP5.
As a reversible PTM pathway, SUMOylation catalyzes the conjugation of SUMO proteins to lysine residues of distinct target proteins. SUMOylation modifies multiple cellular regulators, thereby affecting a multitude of key processes in a highly dynamic manner. SUMOylation undergoes dynamic alterations during cardiac I/R, suggesting the involvement of SUMO in the quality control and expression stability of target proteins in the ischemic heart [199]. The recovery of cardiac function during cardiac I/R injury is significantly associated with eukaryotic elongation factor 2 (eEF2) [200]. Phosphorylated eEF2 can also be SUMOylated, and the nuclear translocation of eEF2 elevates the expression of Bcl-2, which further promotes cardiac I/R injury [201]. Other studies have further explored the regulatory role of SUMOylation in cardiac I/R injury via histone deacetylase 4 (HDAC4) expression. After the invention of irisin, the protein level of HDAC4 was observed in H9C2 cells. However, the overexpression of HDAC4 provokes mitochondrial dysfunction by increasing the mitochondrial permeability transition pore (mPTP) and attenuating mitochondrial transit peptide (MTP). Furthermore, irisin addition decreases HDAC4 expression by promoting SUMOylation, which displays a protective effect against cardiac I/R injury by reducing LDH leakage and restraining the activation of caspase-3 and annexin V [202]. As a specific inhibitor of SUMOylation, ginkgolic acid (GA) has been reported to be involved in cardiac fibrosis induced by MI. Inhibition of PML and disruption of Pin1 significantly suppress the recruitment of TGF-β1, which is dependent on SUMO1-mediated PML SUMOylation [203].
Less is known about the role of SENPs, including SENP6 and SENP7, in cardiac I/R injury. However, SENP3 was recently identified as a novel potential therapeutic mechanism in protecting cardiac I/R injury [204]. Mitophagy plays a critical role in the development of cardiac I/R injury; however, the association between SUMOylation and mitochondrial dysfunction in cardiac I/R injury is less studied. Recently, DRP1 was shown to be a substrate of SUMO1, and Zn2+-induced SUMOylation of DRP1 can regulate mitophagy through ΔΨm collapse, which further contributes to the protective effect of cardiac I/R injury [205]. RNF4, as a poly-SUMO-specific E3 ubiquitin ligase, is involved in mediating cardiac I/R injury. Knockdown of endogenous RNF4 exacerbates H2O2-induced cardiomyocyte apoptosis and enhances SUMOylation of PML, which promotes ischemia-induced cardiac dysfunction [206]. Whether SENP6 or SENP7 plays regulatory roles in ROS production, especially H2O2, may be a future research direction. Investigations into the emerging role and possible mechanism of SENPs in regulating cardiac I/R injury via mitophagy are challenging tasks. Other SUMO ligases have also been reported to be involved in protecting cardiac I/R injury. A wide range of SUMOylation for proteins induced by UBC9, including Beclin1 and VPS34, promotes the colocalization of LAMP1 and VPS34, and UBC9-mediated SUMOylation also promotes PIK3-III complex assembly and autophagic flux, which further contributes to the protection of cardiac I/R injury [207]. Further investigations into cardiac SUMO-related proteins, such as UBC9, SENP6, and SENP7, mediating SUMOylation in mitophagy are needed to define the precise functional significance in cardiac I/R injury. Additionally, the exact roles and diverse mechanisms of SUMOylation and deSUMOylation in mitophagy-mediated cardiac I/R injury should be clarified in the future.
Perspective and conclusions
The SUMO molecule family, including SUMO1 ~ SUMO5, is a highly conserved PTM form in all eukaryotes [5]. Despite the precise modification of the SUMO molecule in downstream proteins, the exact mechanism of SUMOylation in mitophagy is still unclear. The identification of the modification sites of SUMOylation and deSUMOylation in proteins is fundamental for understanding the biological functions and regulatory mechanisms of SUMO [41]. SUMO-mediated SUMOylation requires that the residue preceding Gly-Gly (diGly) at the C-terminus be replaced with lysine (KGG) [21]. The attachment of an acceptor lysine in the target protein has been reported to be involved mainly in a single SUMO moiety, whereas polySUMO-mediated SUMOylation promotes the superposition of SUMO to form a SUMO polypeptide chain [208]. Although the homology and structure of these five SUMOs are obviously distinguished, the differences in their functions and the deSUMOylation enzymes need to be further explored and subdivided. Multiple studies link the modulation of mitophagy by PTMs, such as phosphorylation and ubiquitination, to altered cardiac cell severity or progression in the development of cardiovascular diseases [14,209,210]. As a multifunctional modification, SUMOylation is involved in multiple pathophysiological processes associated with mitophagy, including cellular oxidative stress, the inflammatory response and hypoxia [80,121]. Ample evidence indicates that activation of SUMOylation or inhibition of deSUMOylation is mechanistically associated with the progression and development of cardiovascular diseases by targeting mitophagy-related proteins, including PINK1, HIF1α, and MFN1/2 [17,127,197].
Mitophagy is involved in the progression and development of cardiovascular diseases [211]. The maintenance of mitochondrial morphology and function is critical for cardiac physiology, especially cellular metabolic homeostasis. Given that mitophagy activation is regulated by mitochondrial dynamic proteins, including OPA1, DRP1, PINK1, and MFN1/2, the modification of these target proteins needs to be further investigated [212]. SUMO molecules, especially SUMO1, promote mitochondrial fusion and fission by binding to mitochondrial-related proteins, which play an important role in regulating mitochondrial dysfunction [112]. Additionally, SUMO conjugation has been implicated in the modulation of the mitochondrial energetic process, which further facilitates the mitochondrial translocation into autophagosomes for lysosomal degradation [13]. In contrast, deSUMOylation is also involved in mitophagy activation through the E2 ligase UBC9 or SUMO proteases SENPs. The mediation of SENPs is dependent mainly on the removal of a C-terminal sequence following a diGly motif with the presence of ATP, and UBC9 catalyzes the formation between SUMO and the ε-amino group of a lysine by directly recognizing target proteins [31,55]. However, the transcriptional regulation of SUMO-conjugated proteins in mediating mitochondrial dysfunction and mitophagy is still unclear, and the regulatory role of miRNAs and transcription factors in inducing SUMOylation needs to be further studied. Mitochondrial dysfunction and mitophagy activation are essential for the development of cardiovascular diseases, and SUMOylation may be a multifunctional regulatory action in mediating mitophagy-induced cardiovascular diseases [17]. Growing evidence has reported that SUMO1- and SUMO2-mediated SUMOylation is involved in regulating cardiac hypertrophy via its transcription, chromosome organization, and nuclear trafficking [114]. Additionally, the SUMOylation of Nkx2.5 may be related to mitochondrial fusion and fission by modifying OPA1, DRP1, or Fis1 [119]. SERCA2a was identified as an important acceptor protein in the development of cardiac hypertrophy. The SUMOylation of SERCA2a by SUMO1 promotes heart failure, which further induces cardiac hypertrophy [122]. Other SUMOylation or deSUMOylation by SENP1, UBC9, and PIASy/4 can also promote cardiac hypertrophy by activating mitophagy [129,145]. SUMOylation of Kv1.5 promotes dysfunction of atrial myocytes, which further provokes hypertension. Additionally, deSUMOylation of Kv1.5 by SENP2 and Ulp1 leads to a 15-mV hyperpolarizing shift in the voltage dependence of steady-state inactivation [142]. HIF1α-mediated mitophagy plays a crucial role in hypertension development. SUMO1 directly interacts with HIF1α to promote the SUMOylation of HIF1α and is accompanied by Bnip3-mediated alteration of mtDNA, which further provokes the development of hypertension [146]. SUMO2-mediated SUMOylation of PPARα is involved in the proliferation and migration of VSMCs, and conversely, the deSUMOylation of PPARα contributes to the prevention of atherosclerosis [164]. In addition, the regulatory expression of SREBPs, eNOS, p90RSK and LXRα and PML by PIASy, SENP2 and IFN-γ via SUMOylation further promotes the development of atherosclerosis [179,182,183]. miR-499 and SUMOylation-mediated UPS are involved in the regulation of mitochondrial dynamics, which further mediates cardiac I/R injury [190]. Additionally, SUMOylation of SERCA2a by SUMO1 via PI3K ameliorates myocardial fibrosis [192]. Inflammatory response-related proteins, such as PPARγ, NF-κB, and LDH, are also SUMOylated by the SUMO molecule UBC9 or PIAS1, promoting H2O2 production and mitophagy, which induces cardiac I/R injury [195]. Moreover, both SUMOylation and deSUMOylation of HIF1α by SENP1 and SUMO1 are involved in vascular VEGF expression, suggesting the therapeutic potential of SUMOylation in the prevention of cardiac I/R injury [194]. Therefore, the precise mechanisms of SUMOylation in cardiovascular diseases should focus on its regulatory actions for mitophagy activation, particularly on its modification of mitochondrial-related proteins, such as SERCA2a, HIF1α, PINK1, and Bnip3.
SUMOylation mediated by SUMO conjugation was recently demonstrated to be an important modification in regulating cardiovascular diseases [23]. Although multiple studies have endeavored to elucidate the mechanisms of SUMOylation in mitophagy-associated cardiovascular diseases, numerous contradictions and perplexities remain regarding SUMO molecule expression and regulation in the protein modification system. A wide range of investigations indicates that the protective role of SUMOylation is involved in the prevention and regulation of cardiovascular diseases. However, some other results have shown that SUMOylation exhibits dual roles in cardiovascular disease, suggesting that an additional mechanism of SUMOylation is associated with cardiovascular diseases [164,165]. In addition, the upstream factors, such as transcription factors and micro/lnc-RNAs, mediating the regulation of SUMO expression are unclear. Future studies need to exhaustively clarify the precise role and exact mechanism of SUMOylation in regulating mitophagy, and the comprehensive evaluation of the effect of SUMO molecules on cardiovascular diseases should be further understood. The complex and multifaceted regulatory mechanisms of SUMOylation in regulating mitochondrial dynamics and mitophagy activation are still unclear, and a few studies have implicated SUMOylation in the development of cardiovascular diseases, especially in the regulation of mitochondrial fusion/fission and mitochondrial-autophagic flux under SUMOylation. Further research should strive to clarify why both SUMOylation and deSUMOylation exhibit the same role in regulating mitophagy and the development of cardiovascular diseases. A possible explanation may be involved in the multiple kinds of proteins that induce mitophagy activation, and their SUMOylation modification pathways may be distinct under different conditions. Since five types of SUMO molecules have been identified, they may exhibit different activities under distinct inducing conditions, or the activity of SUMO-conjugating enzymes and deSUMOylation-related ligases may also be altered by certain factors. Therefore, it is necessary to clarify the explicit biofunctional basis of SUMO molecules and expand the types and specific functions of SUMO-related enzymes, which will summarize the extensive effects of SUMOylation on mitophagy and cardiovascular diseases. Once a deep understanding of the biological, physiological, and pathophysiological roles and mechanisms of SUMOylation in mitophagy is comprehensively clarified, artificial intervention in SUMOylation initiation and activation can precisely bypass the harms of SUMOylation during cardiovascular diseases, which will provide a promising platform for the future development of novel therapeutic targets for cardiovascular diseases.
Availability of data and material
Not applicable.
Abbreviations
- SUMO:
-
Small ubiquitin-likemodifier
- Ub:
-
Ubiquitin
- SENPs:
-
Sentrin-specific peptidases
- PML-NBs:
-
Promyelocytic leukemia nuclear bodies
- SAEs:
-
SUMO activating enzyme subunits
- PIAS:
-
Protein inhibitors of activated STAT
- PTM:
-
Posttranslational modification
- DRP1:
-
Dynamin-related protein
- OPA1:
-
Optic atrophy-1
- MFNs:
-
Mitofusins
- Mg2+ :
-
Manganese ions
- Ca2+ :
-
Calcium
- Cu2+ :
-
Cupric
- Zn2+ :
-
Zinc
- RanBP2:
-
Ran-binding protein 2
- Ulps:
-
Ubl-specific proteases
- ATP:
-
Adenosine triphosphate
- UPRmt :
-
Mitochondrial UPR
- SIMs:
-
SUMO-interacting motifs
- RanGAP1:
-
Ran GTPase-activating protein 1
- TGF-β:
-
Transforming growth factor-β
- HIF1:
-
Hypoxia-inducible factor 1
- IRF8:
-
Interferon regulatory factor 8
- BCL11B:
-
B-cell lymphoma/leukemia 11B
- RNF4:
-
Ring finger protein 4
- NLRP3:
-
NLR family pyrin domain containing 3
- Mul1:
-
Mitochondrial E3 ubiquitin ligase 1
- UPS:
-
Ubiquitin-proteasome system
- PINK1:
-
PTEN-induced putative kinase 1
- OMM:
-
Outer mitochondrial membrane
- LncRNAs:
-
Long noncoding RNAs
- MAPL:
-
Mitochondrial-associated protein ligase
- NDP52:
-
Nuclear dot protein 52
- AT:
-
Ataxia telangiectasia
- ISG15:
-
Interferon-stimulated gene 15
- T1D:
-
Type 1 diabetes
- SNP:
-
Single nucleotide polymorphism
- ROS:
-
Reactive oxygen species
- EGFR:
-
Epidermal growth factor receptor
- SERCA2a:
-
Sarcoplasmic reticulum calcium ATPase
- NF-κB:
-
Nuclear factor kappa-B
- PGC-1α:
-
Proliferator-activated receptor γ coactivator-1α
- HSFs:
-
Heat shock transcription factors
- ERβ:
-
Estrogen receptor β
- Kv:
-
Voltage-gated potassium
- mtDNA:
-
Mitochondrial DNA
- TOMM20:
-
Translocase of outer mitochondrial membrane 20
- KLF2:
-
Kruppel-like factor 2
- eNOS:
-
Endothelial NO synthase
- AEBP1:
-
Adipocyte enhancer-binding protein 1
- p90RSK:
-
P90 ribosomal S6 kinase
- I/R:
-
Ischemia/reperfusion
- GABARAP:
-
Gamma-aminobutyric acid receptor associated protein
- ΔΨm:
-
Mitochondrial membrane potential
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- eEF2:
-
Eukaryotic elongation factor 2
- mPTP:
-
Mitochondrial permeability transition pore
References
Wilson VG (2017) Introduction to sumoylation. Adv Exp Med Biol 963:1–12. https://doi.org/10.1007/978-3-319-50044-7_1
Wang Y, Dasso M (2009) SUMOylation and deSUMOylation at a glance. J Cell Sci 122:4249–4252. https://doi.org/10.1242/jcs.050542
Huang WC, Ko TP, Li SS et al (2004) Crystal structures of the human SUMO-2 protein at 1.6 A and 1.2 A resolution: implication on the functional differences of SUMO proteins. Eur J Biochem 271:4114–4122. https://doi.org/10.1111/j.1432-1033.2004.04349.x
Chen A, Mannen H, Li SS (1998) Characterization of mouse ubiquitin-like SMT3A and SMT3B cDNAs and gene/pseudogenes. Biochem Mol Biol Int 46:1161–1174. https://doi.org/10.1080/15216549800204722
Sheng Z, Zhu J, Deng YN et al (2021) SUMOylation modification-mediated cell death Open Biol 11:210050. https://doi.org/10.1098/rsob.210050
Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11:861–871. https://doi.org/10.1038/nrm3011
Drag M, Salvesen GS (2008) DeSUMOylating enzymes–SENPs. IUBMB Life 60:734–742. https://doi.org/10.1002/iub.113
Wei W, Yang P, Pang J et al (2008) A stress-dependent SUMO4 sumoylation of its substrate proteins. Biochem Biophys Res Commun 375:454–459. https://doi.org/10.1016/j.bbrc.2008.08.028
Liang YC, Lee CC, Yao YL et al (2016) SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci Rep 6:26509. https://doi.org/10.1038/srep26509
Yeh ET (2009) SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem 284:8223–8227. https://doi.org/10.1074/jbc.R800050200
Bossis G, Melchior F (2006) Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell 21:349–357. https://doi.org/10.1016/j.molcel.2005.12.019
Rangrez AY, Borlepawar A, Schmiedel N et al (2020) The E3 ubiquitin ligase HectD3 attenuates cardiac hypertrophy and inflammation in mice. Commun Biol 3:562. https://doi.org/10.1038/s42003-020-01289-2
Kim C, Juncker M, Reed R et al (2021) SUMOylation of mitofusins: a potential mechanism for perinuclear mitochondrial congression in cells treated with mitochondrial stressors. Biochim Biophys Acta Mol Basis Dis 1867:166104. https://doi.org/10.1016/j.bbadis.2021.166104
Onishi M, Yamano K, Sato M et al (2021) Molecular mechanisms and physiological functions of mitophagy. EMBO J 40:e104705. https://doi.org/10.15252/embj.2020104705
Enerback S (2016) Beige communication through gap junctions and adaption by autophagy. Cell Metab 24:370–371. https://doi.org/10.1016/j.cmet.2016.08.024
Al Rawi S, Louvet-Vallee S, Djeddi A et al (2011) Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 334:1144–1147. https://doi.org/10.1126/science.1211878
Teresak P, Lapao A, Subic N et al (2021) Regulation of PRKN-independent mitophagy. Autophagy:1–16. https://doi.org/10.1080/15548627.2021.1888244
Maharjan S, Oku M, Tsuda M et al (2014) Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep 4:5896. https://doi.org/10.1038/srep05896
Okatsu K, Saisho K, Shimanuki M et al (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15:887–900. https://doi.org/10.1111/j.1365-2443.2010.01426.x
Juncker M, Kim C, Reed R et al (2021) ISG15 attenuates post-translational modifications of mitofusins and congression of damaged mitochondria in Ataxia Telangiectasia cells. Biochim Biophys Acta Mol Basis Dis 1867:166102. https://doi.org/10.1016/j.bbadis.2021.166102
Nayak A, Muller S (2014) SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol 15:422. https://doi.org/10.1186/s13059-014-0422-2
Le NT, Martin JF, Fujiwara K et al (2017) Sub-cellular localization specific SUMOylation in the heart. Biochim Biophys Acta Mol Basis Dis 1863:2041–2055. https://doi.org/10.1016/j.bbadis.2017.01.018
Wang J (2009) SUMO conjugation and cardiovascular development. Front Biosci (Landmark Ed) 14:1219–1229. https://doi.org/10.2741/3304
Yang SH, Galanis A, Witty J et al (2006) An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J 25:5083–5093. https://doi.org/10.1038/sj.emboj.7601383
Rodriguez MS, Dargemont C, Hay RT (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem 276:12654–12659. https://doi.org/10.1074/jbc.M009476200
Sampson DA, Wang M, Matunis MJ (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem 276:21664–21669. https://doi.org/10.1074/jbc.M100006200
Flotho A, Melchior F (2013) Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 82:357–385. https://doi.org/10.1146/annurev-biochem-061909-093311
Yang Y, He Y, Wang X et al (2017) Protein SUMOylation modification and its associations with disease. Open Biol. https://doi.org/10.1098/rsob.170167
Schulman BA, Harper JW (2009) Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol 10:319–331. https://doi.org/10.1038/nrm2673
Kessler JD, Kahle KT, Sun T et al (2012) A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335:348–353. https://doi.org/10.1126/science.1212728
Hsieh YL, Kuo HY, Chang CC et al (2013) Ubc9 acetylation modulates distinct SUMO target modification and hypoxia response. EMBO J 32:791–804. https://doi.org/10.1038/emboj.2013.5
Pichler A, Knipscheer P, Saitoh H et al (2004) The RanBP2 SUMO E3 ligase is neither HECT- nor RING-type. Nat Struct Mol Biol 11:984–991. https://doi.org/10.1038/nsmb834
Kumar A, Zhang KY (2015) Advances in the development of SUMO specific protease (SENP) inhibitors. Comput Struct Biotechnol J 13:204–211. https://doi.org/10.1016/j.csbj.2015.03.001
Takahashi Y, Mizoi J, Toh EA et al (2000) Yeast Ulp1, an Smt3-specific protease, associates with nucleoporins. J Biochem 128:723–725. https://doi.org/10.1093/oxfordjournals.jbchem.a022807
Li SJ, Hochstrasser M (2000) The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol 20:2367–2377. https://doi.org/10.1128/MCB.20.7.2367-2377.2000
Gao K, Li Y, Hu S et al (2019) SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. Elife. https://doi.org/10.7554/eLife.41792
Mikolajczyk J, Drag M, Bekes M et al (2007) Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem 282:26217–26224. https://doi.org/10.1074/jbc.M702444200
Chen DV, Suzuki T, Itoh Y et al (2021) Deneddylation by SENP8 restricts hepatitis B virus propagation. Microbiol Immunol 65:125–135. https://doi.org/10.1111/1348-0421.12874
Shin YC, Tang SJ, Chen JH et al (2011) The molecular determinants of NEDD8 specific recognition by human SENP8. PLoS ONE 6:e27742. https://doi.org/10.1371/journal.pone.0027742
Liu K, Guo C, Lao Y et al (2020) A fine-tuning mechanism underlying self-control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy 16:975–990. https://doi.org/10.1080/15548627.2019.1647944
Marston AL (2021) A SUMOylation wave to anchor the genome. J Cell Biol. https://doi.org/10.1083/jcb.202110031
Welch MA, Jansen LR, Baro DJ (2021) SUMOylation of the Kv4.2 ternary complex increases surface expression and current amplitude by reducing internalization in HEK 293 cells. Front Mol Neurosci 14:757278. https://doi.org/10.3389/fnmol.2021.757278
Gong L, Millas S, Maul GG et al (2000) Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J Biol Chem 275:3355–3359. https://doi.org/10.1074/jbc.275.5.3355
Lin X, Wang Y, Jiang Y et al (2020) Sumoylation enhances the activity of the TGF-beta/SMAD and HIF-1 signaling pathways in keloids. Life Sci 255:117859. https://doi.org/10.1016/j.lfs.2020.117859
Chang TH, Xu S, Tailor P et al (2012) The small ubiquitin-like modifier-deconjugating enzyme sentrin-specific peptidase 1 switches IFN regulatory factor 8 from a repressor to an activator during macrophage activation. J Immunol 189:3548–3556. https://doi.org/10.4049/jimmunol.1201104
Zhang LJ, Vogel WK, Liu X et al (2012) Coordinated regulation of transcription factor Bcl11b activity in thymocytes by the mitogen-activated protein kinase (MAPK) pathways and protein sumoylation. J Biol Chem 287:26971–26988. https://doi.org/10.1074/jbc.M112.344176
Rojas-Fernandez A, Plechanovova A, Hattersley N et al (2014) SUMO chain-induced dimerization activates RNF4. Mol Cell 53:880–892. https://doi.org/10.1016/j.molcel.2014.02.031
Chen X, Qin Y, Zhang Y et al (2021) SENP2-PLCbeta4 signaling regulates neurogenesis through the maintenance of calcium homeostasis. Cell Death Differ. https://doi.org/10.1038/s41418-021-00857-1
Chen X, Qin Y, Zhang Z et al (2021) Hyper-SUMOylation of ERG is essential for the progression of acute myeloid leukemia. Front Mol Biosci 8:652284. https://doi.org/10.3389/fmolb.2021.652284
Chen Y, Xu T, Li M et al (2021) Inhibition of SENP2-mediated Akt deSUMOylation promotes cardiac regeneration via activating Akt pathway. Clin Sci (Lond) 135:811–828. https://doi.org/10.1042/CS20201408
Yun C, Wang Y, Mukhopadhyay D et al (2008) Nucleolar protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J Cell Biol 183:589–595. https://doi.org/10.1083/jcb.200807185
Haindl M, Harasim T, Eick D et al (2008) The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep 9:273–279. https://doi.org/10.1038/embor.2008.3
Chen F, Yan H, Guo C et al (2021) Assessment of SENP3-interacting proteins in hepatocytes treated with diethylnitrosamine by BioID assay. Acta Biochim Biophys Sin (Shanghai) 53:1237–1246. https://doi.org/10.1093/abbs/gmab096
Finkbeiner E, Haindl M, Muller S (2011) The SUMO system controls nucleolar partitioning of a novel mammalian ribosome biogenesis complex. EMBO J 30:1067–1078. https://doi.org/10.1038/emboj.2011.33
Zunino R, Schauss A, Rippstein P et al (2007) The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci 120:1178–1188. https://doi.org/10.1242/jcs.03418
Barry R, John SW, Liccardi G et al (2018) SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun 9:3001. https://doi.org/10.1038/s41467-018-05321-2
Li X, Jiao F, Hong J et al (2020) SENP7 knockdown inhibited pyroptosis and NF-kappaB/NLRP3 inflammasome pathway activation in Raw 264.7 cells. Sci Rep 10:16265. https://doi.org/10.1038/s41598-020-73400-w
Gao A, Jiang J, Xie F et al (2020) Bnip3 in mitophagy: novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin Chim Acta 506:72–83. https://doi.org/10.1016/j.cca.2020.02.024
Peng J, Ren KD, Yang J et al (2016) Mitochondrial E3 ubiquitin ligase 1: a key enzyme in regulation of mitochondrial dynamics and functions. Mitochondrion 28:49–53. https://doi.org/10.1016/j.mito.2016.03.007
Shen Z, Pardington-Purtymun PE, Comeaux JC et al (1996) UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics 36:271–279. https://doi.org/10.1006/geno.1996.0462
Boddy MN, Howe K, Etkin LD et al (1996) PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 13:971–982
Werner A, Flotho A, Melchior F (2012) The RanBP2/RanGAP1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase. Mol Cell 46:287–298. https://doi.org/10.1016/j.molcel.2012.02.017
Ritterhoff T, Das H, Hofhaus G et al (2016) The RanBP2/RanGAP1*SUMO1/Ubc9 SUMO E3 ligase is a disassembly machine for Crm1-dependent nuclear export complexes. Nat Commun 7:11482. https://doi.org/10.1038/ncomms11482
Macauley MS, Errington WJ, Okon M et al (2004) Structural and dynamic independence of isopeptide-linked RanGAP1 and SUMO-1. J Biol Chem 279:49131–49137. https://doi.org/10.1074/jbc.M408705200
Kaur A, Jaiswal N, Raj R et al (2020) Characterization of Cu(2+) and Zn(2+) binding sites in SUMO1 and its impact on protein stability. Int J Biol Macromol 151:204–211. https://doi.org/10.1016/j.ijbiomac.2020.02.116
Oh JG, Watanabe S, Lee A et al (2018) miR-146a suppresses SUMO1 expression and induces cardiac dysfunction in maladaptive hypertrophy. Circ Res 123:673–685. https://doi.org/10.1161/CIRCRESAHA.118.312751
Xu H, Wang H, Zhao W et al (2020) SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics 10:5671–5686. https://doi.org/10.7150/thno.42539
Su X, Wan Y, Xie L et al (2019) Expression of SUMO1P3 compared with SUMO1 is an independent predictor of patient outcome in lung adenocarcinoma. Med Sci Monit 25:6691–6701. https://doi.org/10.12659/MSM.916887
Larrick JW, Larrick JW, Mendelsohn AR (2020) SUMO wrestles with mitophagy to extend lifespan. Rejuvenation Res 23:527–532. https://doi.org/10.1089/rej.2020.2406
Li XC, Zeng Y, Sun RR et al (2017) SUMOylation in cardiac disorders - a review. Eur Rev Med Pharmacol Sci 21:1583–1587
Billia F, Hauck L, Konecny F et al (2011) PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 108:9572–9577. https://doi.org/10.1073/pnas.1106291108
Ishihara N, Fujita Y, Oka T et al (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25:2966–2977. https://doi.org/10.1038/sj.emboj.7601184
Neuspiel M, Schauss AC, Braschi E et al (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18:102–108. https://doi.org/10.1016/j.cub.2007.12.038
Purnell PR, Fox HS (2013) Autophagy-mediated turnover of dynamin-related protein 1. BMC Neurosci 14:86. https://doi.org/10.1186/1471-2202-14-86
Song M, Gong G, Burelle Y et al (2015) Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res 117:346–351. https://doi.org/10.1161/CIRCRESAHA.117.306859
Yamashita SI, Jin X, Furukawa K et al (2016) Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol 215:649–665. https://doi.org/10.1083/jcb.201605093
Figueroa-Romero C, Iniguez-Lluhi JA, Stadler J et al (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23:3917–3927. https://doi.org/10.1096/fj.09-136630
Harder Z, Zunino R, McBride H (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14:340–345. https://doi.org/10.1016/j.cub.2004.02.004
Liu C, Peng Z, Li P et al (2020) lncRNA RMST suppressed GBM cell mitophagy through enhancing FUS SUMOylation. Mol Ther Nucleic Acids 19:1198–1208. https://doi.org/10.1016/j.omtn.2020.01.008
Wang J, Schwartz RJ (2010) Sumoylation and regulation of cardiac gene expression. Circ Res 107:19–29. https://doi.org/10.1161/CIRCRESAHA.110.220491
Saitoh H, Hinchey J (2000) Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem 275:6252–6258. https://doi.org/10.1074/jbc.275.9.6252
Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10:748–754. https://doi.org/10.1038/embor.2009.86
Mohanty A, Zunino R, Soubannier V et al (2021) A new functional role of mitochondria-anchored protein ligase in peroxisome morphology in mammalian cells. J Cell Biochem 122:1686–1700. https://doi.org/10.1002/jcb.30114
Kim EY, Zhang Y, Beketaev I et al (2015) SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J Mol Cell Cardiol 78:154–164. https://doi.org/10.1016/j.yjmcc.2014.08.003
Waters E, Wilkinson KA, Harding AL et al (2022) The SUMO protease SENP3 regulates mitochondrial autophagy mediated by Fis1. EMBO Rep 23:e48754. https://doi.org/10.15252/embr.201948754
Sun M, Zhang W, Bi Y et al (2021) NDP52 protects against myocardial infarction-provoked cardiac anomalies through promoting autophagosome-lysosome fusion via recruiting TBK1 and RAB7. Antioxid Redox Signal. https://doi.org/10.1089/ars.2020.8253
Heo JM, Ordureau A, Paulo JA et al (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20. https://doi.org/10.1016/j.molcel.2015.08.016
Fan S, Wu K, Luo C et al (2019) Dual NDP52 function in persistent CSFV infection. Front Microbiol 10:2962. https://doi.org/10.3389/fmicb.2019.02962
Furuya N, Kakuta S, Sumiyoshi K et al (2018) NDP52 interacts with mitochondrial RNA poly(A) polymerase to promote mitophagy. EMBO Rep. https://doi.org/10.15252/embr.201846363
Gao A, Zou J, Mao Z et al (2022) SUMO2-mediated SUMOylation of SH3GLB1 promotes ionizing radiation-induced hypertrophic cardiomyopathy through mitophagy activation. Eur J Pharmacol 924:174980. https://doi.org/10.1016/j.ejphar.2022.174980
Guo D, Li M, Zhang Y et al (2004) A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet 36:837–841. https://doi.org/10.1038/ng1391
Wang CY, She JX (2008) SUMO4 and its role in type 1 diabetes pathogenesis. Diabetes Metab Res Rev 24:93–102. https://doi.org/10.1002/dmrr.797
Chen S, Yang T, Liu F et al (2014) Inflammatory factor-specific sumoylation regulates NF-kappaB signalling in glomerular cells from diabetic rats. Inflamm Res 63:23–31. https://doi.org/10.1007/s00011-013-0675-3
Ma C, Li YJ, Pan CS et al (2014) High resolution diffusion weighted magnetic resonance imaging of the pancreas using reduced field of view single-shot echo-planar imaging at 3 T. Magn Reson Imaging 32:125–131. https://doi.org/10.1016/j.mri.2013.10.005
Li H, Yang Z, Pu LM et al (2016) Adiponectin receptor 1 and small ubiquitin-like modifier 4 polymorphisms are associated with risk of coronary artery disease without diabetes. J Geriatr Cardiol 13:776–782. https://doi.org/10.11909/j.issn.1671-5411.2016.09.001
Neumann J (2005) Novel antibody tags from the rat lysosomal protein RT1.DM for immunodetection of recombinant proteins. J Immunol Methods 301:66–76. https://doi.org/10.1016/j.jim.2005.03.014
Zhang T, Liu Y, Hu Y et al (2017) Association of donor and recipient SUMO4 rs237025 genetic variant with new-onset diabetes mellitus after liver transplantation in a Chinese population. Gene 627:428–433. https://doi.org/10.1016/j.gene.2017.06.060
Baczyk D, Audette MC, Drewlo S et al (2017) SUMO-4: A novel functional candidate in the human placental protein SUMOylation machinery. PLoS ONE 12:e0178056. https://doi.org/10.1371/journal.pone.0178056
Baczyk D, Audette MC, Coyaud E et al (2018) Spatiotemporal distribution of small ubiquitin-like modifiers during human placental development and in response to oxidative and inflammatory stress. J Physiol 596:1587–1600. https://doi.org/10.1113/JP275288
Rocha M, Apostolova N, Diaz-Rua R et al (2020) Mitochondria and T2D: role of autophagy, ER stress, and inflammasome. Trends Endocrinol Metab 31:725–741. https://doi.org/10.1016/j.tem.2020.03.004
Chiche J, Reverso-Meinietti J, Mouchotte A et al (2019) GAPDH expression predicts the response to R-CHOP, the tumor metabolic status, and the response of DLBCL patients to metabolic inhibitors. Cell Metab 29(1243–1257):e1210. https://doi.org/10.1016/j.cmet.2019.02.002
Sengupta U, Ukil S, Dimitrova N et al (2009) Expression-based network biology identifies alteration in key regulatory pathways of type 2 diabetes and associated risk/complications. PLoS ONE 4:e8100. https://doi.org/10.1371/journal.pone.0008100
Cadenas S (2018) Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg 1859:940–950. https://doi.org/10.1016/j.bbabio.2018.05.019
Nakamura M, Sadoshima J (2018) Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15:387–407. https://doi.org/10.1038/s41569-018-0007-y
Ren Z, Yu P, Li D et al (2020) Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation 141:1704–1719. https://doi.org/10.1161/CIRCULATIONAHA.119.043053
Omidkhoda N, Wallace Hayes A, Reiter RJ et al (2019) The role of microRNAs on endoplasmic reticulum stress in myocardial ischemia and cardiac hypertrophy. Pharmacol Res 150:104516. https://doi.org/10.1016/j.phrs.2019.104516
You J, Wu J, Zhang Q et al (2018) Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am J Physiol Heart Circ Physiol 314:H552–H562. https://doi.org/10.1152/ajpheart.00212.2017
Tong M, Saito T, Zhai P et al (2019) Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 124:1360–1371. https://doi.org/10.1161/CIRCRESAHA.118.314607
Dorn GW 2nd (2010) Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3:374–383. https://doi.org/10.1007/s12265-010-9174-x
Du Q, Zhu B, Zhai Q et al (2017) Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am J Transl Res 9:3360–3373
Maejima Y, Kyoi S, Zhai P et al (2013) Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 19:1478–1488. https://doi.org/10.1038/nm.3322
Luan Y, Luan Y, Feng Q et al (2021) Emerging role of mitophagy in the heart: therapeutic potentials to modulate mitophagy in cardiac diseases. Oxid Med Cell Longev 2021:3259963. https://doi.org/10.1155/2021/3259963
Chen SN, Sbaizero O, Taylor MRG et al (2019) Lamin A/C cardiomyopathy: implications for treatment. Curr Cardiol Rep 21:160. https://doi.org/10.1007/s11886-019-1224-7
Sylvius N, Bilinska ZT, Veinot JP et al (2005) In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J Med Genet 42:639–647. https://doi.org/10.1136/jmg.2004.023283
Moriuchi T, Muraoka T, Mio K et al (2014) Long-term expression of the lamin A mutant associated with dilated cardiomyopathy induces senescence. Genes Cells 19:901–918. https://doi.org/10.1111/gtc.12189
Gardner LD, Peck KA, Goetz GW et al (2019) Cardiac remodeling in response to embryonic crude oil exposure involves unconventional NKX family members and innate immunity genes. J Exp Biol. https://doi.org/10.1242/jeb.205567
Schwartz RJ, Schneider MD (2006) CAMTA in cardiac hypertrophy. Cell 125:427–429. https://doi.org/10.1016/j.cell.2006.04.015
Kim EY, Chen L, Ma Y et al (2011) Expression of sumoylation deficient Nkx2.5 mutant in Nkx2.5 haploinsufficient mice leads to congenital heart defects. PLoS One 6:e20803. https://doi.org/10.1371/journal.pone.0020803
Zungu M, Schisler J, Willis MS (2011) All the little pieces. -Regulation of mitochondrial fusion and fission by ubiquitin and small ubiquitin-like modifer and their potential relevance in the heart. Circ J 75:2513–2521. https://doi.org/10.1253/circj.cj-11-0967
Wang X, Liu X, Wang S et al (2012) Myofibrillogenesis regulator 1 induces hypertrophy by promoting sarcomere organization in neonatal rat cardiomyocytes. Hypertens Res 35:597–603. https://doi.org/10.1038/hr.2011.228
Lee A, Jeong D, Mitsuyama S et al (2014) The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid Redox Signal 21:1986–2001. https://doi.org/10.1089/ars.2014.5983
Buccarello L, Dragotto J, Iorio F et al (2020) The pivotal role of SUMO-1-JNK-Tau axis in an in vitro model of oxidative stress counteracted by the protective effect of curcumin. Biochem Pharmacol 178:114066. https://doi.org/10.1016/j.bcp.2020.114066
Chaanine AH, Joyce LD, Stulak JM et al (2019) Mitochondrial morphology, dynamics, and function in human pressure overload or ischemic heart disease with preserved or reduced ejection fraction. Circ Heart Fail 12:e005131. https://doi.org/10.1161/CIRCHEARTFAILURE.118.005131
Liao XH, Wang N, Zhao DW et al (2014) NF-kappaB (p65) negatively regulates myocardin-induced cardiomyocyte hypertrophy through multiple mechanisms. Cell Signal 26:2738–2748. https://doi.org/10.1016/j.cellsig.2014.08.006
Cai R, Gu J, Sun H et al (2015) Induction of SENP1 in myocardium contributes to abnormities of mitochondria and cardiomyopathy. J Mol Cell Cardiol 79:115–122. https://doi.org/10.1016/j.yjmcc.2014.11.014
Gupta MK, McLendon PM, Gulick J et al (2016) UBC9-mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ Res 118:1894–1905. https://doi.org/10.1161/CIRCRESAHA.115.308268
Zhao Y, Ponnusamy M, Liu C et al (2017) MiR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim Biophys Acta Mol Basis Dis 1863:2871–2881. https://doi.org/10.1016/j.bbadis.2017.07.034
Peris-Moreno D, Taillandier D, Polge C (2020) MuRF1/TRIM63, master regulator of muscle mass. Int J Mol Sci. https://doi.org/10.3390/ijms21186663
Heras G, Namuduri AV, Traini L et al (2019) Muscle RING-finger protein-1 (MuRF1) functions and cellular localization are regulated by SUMO1 post-translational modification. J Mol Cell Biol 11:356–370. https://doi.org/10.1093/jmcb/mjy036
Lin YM, Badrealam KF, Kuo CH et al (2021) Small molecule compound nerolidol attenuates hypertension induced hypertrophy in spontaneously hypertensive rats through modulation of Mel-18-IGF-IIR signalling. Phytomedicine 84:153450. https://doi.org/10.1016/j.phymed.2020.153450
Chang RL, Chang CF, Ju DT et al (2018) Short-term hypoxia upregulated Mas receptor expression to repress the AT1 R signaling pathway and attenuate Ang II-induced cardiomyocyte apoptosis. J Cell Biochem 119:2742–2749. https://doi.org/10.1002/jcb.26440
Huang CY, Kuo CH, Pai PY et al (2018) Inhibition of HSF2 SUMOylation via MEL18 upregulates IGF-IIR and leads to hypertension-induced cardiac hypertrophy. Int J Cardiol 257:283–290. https://doi.org/10.1016/j.ijcard.2017.10.102
Pai P, Shibu MA, Chang RL et al (2018) ERbeta targets ZAK and attenuates cellular hypertrophy via SUMO-1 modification in H9c2 cells. J Cell Biochem 119:7855–7864. https://doi.org/10.1002/jcb.27199
Lin HB, Naito K, Oh Y et al (2020) Innate immune Nod1/RIP2 signaling is essential for cardiac hypertrophy but requires mitochondrial antiviral signaling protein for signal transductions and energy balance. Circulation 142:2240–2258. https://doi.org/10.1161/CIRCULATIONAHA.119.041213
Gao A, Li F, Zhou Q et al (2020) Sestrin2 as a potential therapeutic target for cardiovascular diseases. Pharmacol Res 159:104990. https://doi.org/10.1016/j.phrs.2020.104990
Zhou J, Li XY, Liu YJ et al (2021) Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy:1–16. https://doi.org/10.1080/15548627.2021.1984656
Poulter NR, Prabhakaran D, Caulfield M (2015) Hypertension Lancet 386:801–812. https://doi.org/10.1016/S0140-6736(14)61468-9
Oparil S, Acelajado MC, Bakris GL et al (2018) Hypertension Nat Rev Dis Primers 4:18014. https://doi.org/10.1038/nrdp.2018.14
Chen G, Lin Y, Chen L et al (2020) Role of DRAM1 in mitophagy contributes to preeclampsia regulation in mice. Mol Med Rep 22:1847–1858. https://doi.org/10.3892/mmr.2020.11269
Haslip M, Dostanic I, Huang Y et al (2015) Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol 35:1166–1178. https://doi.org/10.1161/ATVBAHA.114.304865
Du Y, Wang T, Guo J et al (2021) Kv1.5 channels are regulated by PKC-mediated endocytic degradation. J Biol Chem 296:100514. https://doi.org/10.1016/j.jbc.2021.100514
Benson MD, Li QJ, Kieckhafer K et al (2007) SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci USA 104:1805–1810. https://doi.org/10.1073/pnas.0606702104
Tian H, Dai AG, Fu DY et al (2012) The expression and possible role of SENP1 in the pulmonary vascular wall of rat during the development of hypoxic pulmonary hypertension. Zhongguo Ying Yong Sheng Li Xue Za Zhi 28:123–127
Jiang Y, Wang J, Tian H et al (2015) Increased SUMO-1 expression in response to hypoxia: Interaction with HIF-1alpha in hypoxic pulmonary hypertension. Int J Mol Med 36:271–281. https://doi.org/10.3892/ijmm.2015.2209
Zhou F, Dai A, Jiang Y et al (2016) SENP1 enhances hypoxiainduced proliferation of rat pulmonary artery smooth muscle cells by regulating hypoxiainducible factor1alpha. Mol Med Rep 13:3482–3490. https://doi.org/10.3892/mmr.2016.4969
Fu ZJ, Wang ZY, Xu L et al (2020) HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 36:101671. https://doi.org/10.1016/j.redox.2020.101671
Zhang ZB, Ruan CC, Chen DR et al (2016) Activating transcription factor 3 SUMOylation is involved in angiotensin II-induced endothelial cell inflammation and dysfunction. J Mol Cell Cardiol 92:149–157. https://doi.org/10.1016/j.yjmcc.2016.02.001
Yu CQ, Yin LQ, Tu ZT et al (2017) The regulatory role of dopamine receptor D1 on PP2A via SUMO-1 modification. Eur Rev Med Pharmacol Sci 21:3270–3276
Yao Y, Li H, Da X et al (2019) SUMOylation of Vps34 by SUMO1 promotes phenotypic switching of vascular smooth muscle cells by activating autophagy in pulmonary arterial hypertension. Pulm Pharmacol Ther 55:38–49. https://doi.org/10.1016/j.pupt.2019.01.007
Cai Z, Wang Z, Yuan R et al (2021) Redox-sensitive enzyme SENP3 mediates vascular remodeling via de-SUMOylation of beta-catenin and regulation of its stability. EBioMedicine 67:103386. https://doi.org/10.1016/j.ebiom.2021.103386
Schreckenberger ZJ, Wenceslau CF, Joe B et al (2020) Mitophagy in hypertension-associated premature vascular aging. Am J Hypertens 33:804–812. https://doi.org/10.1093/ajh/hpaa058
Gao A, Cayabyab FS, Chen X et al (2017) Implications of sortilin in lipid metabolism and lipid disorder diseases. DNA Cell Biol 36:1050–1061. https://doi.org/10.1089/dna.2017.3853
Ahotupa M (2017) Oxidized lipoprotein lipids and atherosclerosis. Free Radic Res 51:439–447. https://doi.org/10.1080/10715762.2017.1319944
Huynh DTN, Heo KS (2021) Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res. https://doi.org/10.1007/s12272-021-01360-4
Foote K, Reinhold J, Yu EPK et al (2018) Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell 17:e12773. https://doi.org/10.1111/acel.12773
Orekhov AN, Poznyak AV, Sobenin IA et al (2020) Mitochondrion as a selective target for the treatment of atherosclerosis: role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr Neuropharmacol 18:1064–1075. https://doi.org/10.2174/1570159X17666191118125018
Kattoor AJ, Pothineni NVK, Palagiri D et al (2017) Oxidative stress in atherosclerosis. Curr Atheroscler Rep 19:42. https://doi.org/10.1007/s11883-017-0678-6
Poznyak AV, Nikiforov NG, Wu WK et al (2021) Autophagy and mitophagy as essential components of atherosclerosis. Cells. https://doi.org/10.3390/cells10020443
Talamillo A, Ajuria L, Grillo M et al (2020) SUMOylation in the control of cholesterol homeostasis. Open Biol 10:200054. https://doi.org/10.1098/rsob.200054
Dehnavi S, Sadeghi M, Penson PE et al (2019) The role of protein SUMOylation in the pathogenesis of atherosclerosis. J Clin Med. https://doi.org/10.3390/jcm8111856
Kim YR, Jacobs JS, Li Q et al (2019) SUMO2 regulates vascular endothelial function and oxidative stress in mice. Am J Physiol Heart Circ Physiol 317:H1292–H1300. https://doi.org/10.1152/ajpheart.00530.2019
Stein S, Oosterveer MH, Mataki C et al (2014) SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab 20:603–613. https://doi.org/10.1016/j.cmet.2014.07.023
Liu YZ, Xiao X, Hu CT et al (2020) SUMOylation in atherosclerosis. Clin Chim Acta 508:228–233. https://doi.org/10.1016/j.cca.2020.05.033
Lim S, Ahn BY, Chung SS et al (2009) Effect of a peroxisome proliferator-activated receptor gamma sumoylation mutant on neointimal formation after balloon injury in rats. Atherosclerosis 206:411–417. https://doi.org/10.1016/j.atherosclerosis.2009.02.031
Zhang J, Tang L, Dai F et al (2019) ROCK inhibitors alleviate myofibroblast transdifferentiation and vascular remodeling via decreasing TGFbeta1-mediated RhoGDI expression. Gen Physiol Biophys 38:271–280. https://doi.org/10.4149/gpb_2019017
Dai F, Qi Y, Guan W et al (2019) RhoGDI stability is regulated by SUMOylation and ubiquitination via the AT1 receptor and participates in Ang II-induced smooth muscle proliferation and vascular remodeling. Atherosclerosis 288:124–136. https://doi.org/10.1016/j.atherosclerosis.2019.07.010
McPherson R, Gauthier A (2004) Molecular regulation of SREBP function: the Insig-SCAP connection and isoform-specific modulation of lipid synthesis. Biochem Cell Biol 82:201–211. https://doi.org/10.1139/o03-090
Kobayashi S, Shibata H, Yokota K et al (2004) FHL2, UBC9, and PIAS1 are novel estrogen receptor alpha-interacting proteins. Endocr Res 30:617–621. https://doi.org/10.1081/erc-200043789
Gimbrone MA Jr, Garcia-Cardena G (2016) Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118:620–636. https://doi.org/10.1161/CIRCRESAHA.115.306301
Jha P, Das H (2017) KLF2 in Regulation of NF-kappaB-mediated immune cell function and inflammation. Int J Mol Sci. https://doi.org/10.3390/ijms18112383
Komaravolu RK, Adam C, Moonen JR et al (2015) Erk5 inhibits endothelial migration via KLF2-dependent down-regulation of PAK1. Cardiovasc Res 105:86–95. https://doi.org/10.1093/cvr/cvu236
Woo CH, Shishido T, McClain C et al (2008) Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res 102:538–545. https://doi.org/10.1161/CIRCRESAHA.107.156877
Liu B, Shuai K (2008) Targeting the PIAS1 SUMO ligase pathway to control inflammation. Trends Pharmacol Sci 29:505–509. https://doi.org/10.1016/j.tips.2008.07.008
Majdalawieh A, Ro HS (2010) Regulation of IkappaBalpha function and NF-kappaB signaling: AEBP1 is a novel proinflammatory mediator in macrophages. Mediators Inflamm 2010:823821. https://doi.org/10.1155/2010/823821
Takabe W, Alberts-Grill N, Jo H (2011) Disturbed flow: p53 SUMOylation in the turnover of endothelial cells. J Cell Biol 193:805–807. https://doi.org/10.1083/jcb.201104140
Heo KS, Lee H, Nigro P et al (2011) PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol 193:867–884. https://doi.org/10.1083/jcb.201010051
Heo KS, Chang E, Le NT et al (2013) De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res 112:911–923. https://doi.org/10.1161/CIRCRESAHA.111.300179
Xiao N, Li H, Yu W et al (2019) SUMO-specific protease 2 (SENP2) suppresses keratinocyte migration by targeting NDR1 for de-SUMOylation. FASEB J 33:163–174. https://doi.org/10.1096/fj.201800353R
Abe JI, Ko KA, Kotla S et al (2019) MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight. https://doi.org/10.1172/jci.insight.125570
Wang Z, Li C, Sun X et al (2020) Hypermethylation of miR-181b in monocytes is associated with coronary artery disease and promotes M1 polarized phenotype via PIAS1-KLF4 axis. Cardiovasc Diagn Ther 10:738–751. https://doi.org/10.21037/cdt-20-407
Frambach S, de Haas R, Smeitink JAM et al (2020) Brothers in arms: ABCA1- and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacol Rev 72:152–190. https://doi.org/10.1124/pr.119.017897
Dong M, Zhang Y, Xu C et al (2020) Interferon-gamma decreases ATP-binding cassette subfamily G member 1-mediated cholesterol efflux through small ubiquitin-like modifier/ubiquitin-dependent liver X receptor-alpha degradation in macrophages. Biotechnol Appl Biochem. https://doi.org/10.1002/bab.2063
Karle W, Becker S, Stenzel P et al (2021) Promyelocytic leukemia protein promotes the phenotypic switch of smooth muscle cells in atherosclerotic plaques of human coronary arteries. Clin Sci (Lond) 135:887–905. https://doi.org/10.1042/CS20201399
Du J, Li Y, Zhao W (2020) Autophagy and myocardial ischemia. Adv Exp Med Biol 1207:217–222. https://doi.org/10.1007/978-981-15-4272-5_15
Lee TL, Lee MH, Chen YC et al (2020) Vitamin D attenuates ischemia/reperfusion-induced cardiac injury by reducing mitochondrial fission and mitophagy. Front Pharmacol 11:604700. https://doi.org/10.3389/fphar.2020.604700
Jin Q, Li R, Hu N et al (2018) DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol 14:576–587. https://doi.org/10.1016/j.redox.2017.11.004
Zhang W, Chen C, Wang J et al (2018) Mitophagy in cardiomyocytes and in platelets: a major mechanism of cardioprotection against ischemia/reperfusion injury. Physiology (Bethesda) 33:86–98. https://doi.org/10.1152/physiol.00030.2017
Guo C, Hildick KL, Luo J et al (2013) SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J 32:1514–1528. https://doi.org/10.1038/emboj.2013.65
Guo C, Wilkinson KA, Evans AJ et al (2017) SENP3-mediated deSUMOylation of Drp1 facilitates interaction with Mff to promote cell death. Sci Rep 7:43811. https://doi.org/10.1038/srep43811
Wang JX, Jiao JQ, Li Q et al (2011) miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med 17:71–78. https://doi.org/10.1038/nm.2282
McCrink KA, Maning J, Vu A et al (2017) beta-Arrestin2 improves post-myocardial infarction heart failure via sarco(endo)plasmic reticulum Ca(2+)-ATPase-dependent positive inotropy in cardiomyocytes. Hypertension 70:972–981. https://doi.org/10.1161/HYPERTENSIONAHA.117.09817
Hu W, Xu T, Wu P et al (2017) Luteolin improves cardiac dysfunction in heart failure rats by regulating sarcoplasmic reticulum Ca(2+)-ATPase 2a. Sci Rep 7:41017. https://doi.org/10.1038/srep41017
Du Y, Liu P, Xu T et al (2018) Luteolin modulates SERCA2a leading to attenuation of myocardial ischemia/ reperfusion injury via sumoylation at lysine 585 in mice. Cell Physiol Biochem 45:883–898. https://doi.org/10.1159/000487283
Liu H, Bian X, Xu M et al (2021) HIF1alpha protein SUMOylation is an important protective mechanism of action of hypothermia in hypoxic cardiomyocytes. Mol Med Rep. https://doi.org/10.3892/mmr.2021.12115
Shishido T, Woo CH, Ding B et al (2008) Effects of MEK5/ERK5 association on small ubiquitin-related modification of ERK5: implications for diabetic ventricular dysfunction after myocardial infarction. Circ Res 102:1416–1425. https://doi.org/10.1161/CIRCRESAHA.107.168138
Tilemann L, Lee A, Ishikawa K et al (2013) SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med 5:211ra159. https://doi.org/10.1126/scitranslmed.3006487
Gu J, Fan Y, Liu X et al (2014) SENP1 protects against myocardial ischaemia/reperfusion injury via a HIF1alpha-dependent pathway. Cardiovasc Res 104:83–92. https://doi.org/10.1093/cvr/cvu177
Shimizu Y, Lambert JP, Nicholson CK et al (2016) DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol 97:56–66. https://doi.org/10.1016/j.yjmcc.2016.04.008
Hotz PW, Wiesnet M, Tascher G et al (2020) Profiling the murine SUMO proteome in response to cardiac ischemia and reperfusion injury. Molecules. https://doi.org/10.3390/molecules25235571
Crozier SJ, Vary TC, Kimball SR et al (2005) Cellular energy status modulates translational control mechanisms in ischemic-reperfused rat hearts. Am J Physiol Heart Circ Physiol 289:H1242-1250. https://doi.org/10.1152/ajpheart.00859.2004
Zhang C, Liu X, Zhang C et al (2017) Phosphorylated eEF2 is SUMOylated and induces cardiomyocyte apoptosis during myocardial ischemia reperfusion. J Cardiol 69:689–698. https://doi.org/10.1016/j.jjcc.2016.05.020
Zhao YT, Wang H, Zhang S et al (2016) Irisin ameliorates hypoxia/reoxygenation-induced injury through modulation of histone deacetylase 4. PLoS ONE 11:e0166182. https://doi.org/10.1371/journal.pone.0166182
Qiu F, Dong C, Liu Y et al (2018) Pharmacological inhibition of SUMO-1 with ginkgolic acid alleviates cardiac fibrosis induced by myocardial infarction in mice. Toxicol Appl Pharmacol 345:1–9. https://doi.org/10.1016/j.taap.2018.03.006
Rawlings N, Lee L, Nakamura Y et al (2019) Protective role of the deSUMOylating enzyme SENP3 in myocardial ischemia-reperfusion injury. PLoS ONE 14:e0213331. https://doi.org/10.1371/journal.pone.0213331
Bian X, Xu J, Zhao H et al (2019) Zinc-induced SUMOylation of dynamin-related protein 1 protects the heart against ischemia-reperfusion injury. Oxid Med Cell Longev 2019:1232146. https://doi.org/10.1155/2019/1232146
Qiu F, Han Y, Shao X et al (2020) Knockdown of endogenous RNF4 exacerbates ischaemia-induced cardiomyocyte apoptosis in mice. J Cell Mol Med 24:9545–9559. https://doi.org/10.1111/jcmm.15363
Xiao Q, Chen XH, Jiang RC et al (2020) Ubc9 attenuates myocardial ischemic injury through accelerating autophagic flux. Front Pharmacol 11:561306. https://doi.org/10.3389/fphar.2020.561306
Keiten-Schmitz J, Schunck K, Muller S (2019) SUMO chains rule on chromatin occupancy. Front Cell Dev Biol 7:343. https://doi.org/10.3389/fcell.2019.00343
Wang B, Nie J, Wu L et al (2018) AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res 122:712–729. https://doi.org/10.1161/CIRCRESAHA.117.312317
Sliter DA, Martinez J, Hao L et al (2018) Parkin and PINK1 mitigate STING-induced inflammation. Nature 561:258–262. https://doi.org/10.1038/s41586-018-0448-9
Zheng H, Zhu H, Liu X et al (2021) Mitophagy in diabetic cardiomyopathy: roles and mechanisms. Front Cell Dev Biol 9:750382. https://doi.org/10.3389/fcell.2021.750382
Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C et al (2016) Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol 594:509–525. https://doi.org/10.1113/JP271301
Serbyn N, Bagdiul I, Noireterre A et al (2021) SUMO orchestrates multiple alternative DNA-protein crosslink repair pathways. Cell Rep 37:110034. https://doi.org/10.1016/j.celrep.2021.110034
Ryu HY, Su D, Wilson-Eisele NR et al (2019) The Ulp2 SUMO protease promotes transcription elongation through regulation of histone sumoylation. EMBO J 38:e102003. https://doi.org/10.15252/embj.2019102003
Shin EJ, Shin HM, Nam E et al (2012) DeSUMOylating isopeptidase: a second class of SUMO protease. EMBO Rep 13:339–346. https://doi.org/10.1038/embor.2012.3
Schulz S, Chachami G, Kozaczkiewicz L et al (2012) Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep 13:930–938. https://doi.org/10.1038/embor.2012.125
Wang T, Cao Y, Zheng Q et al (2019) SENP1-Sirt3 signaling controls mitochondrial protein acetylation and metabolism. Mol Cell 75(823–834):e825. https://doi.org/10.1016/j.molcel.2019.06.008
Selby TL, Biel N, Varn M et al (2019) The Epstein-Barr virus oncoprotein, LMP1, regulates the function of SENP2, a SUMO-protease. Sci Rep 9:9523. https://doi.org/10.1038/s41598-019-45825-5
Hu Z, Teng XL, Zhang T et al (2021) SENP3 senses oxidative stress to facilitate STING-dependent dendritic cell antitumor function. Mol Cell 81(940–952):e945. https://doi.org/10.1016/j.molcel.2020.12.024
Li J, Lu D, Dou H et al (2018) Desumoylase SENP6 maintains osteochondroprogenitor homeostasis by suppressing the p53 pathway. Nat Commun 9:143. https://doi.org/10.1038/s41467-017-02413-3
Coleman KE, Bekes M, Chapman JR et al (2017) SENP8 limits aberrant neddylation of NEDD8 pathway components to promote cullin-RING ubiquitin ligase function. Elife. https://doi.org/10.7554/eLife.24325
Morozko EL, Smith-Geater C, Monteys AM et al (2021) PIAS1 modulates striatal transcription, DNA damage repair, and SUMOylation with relevance to Huntington’s disease. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.2021836118
Ran M, Chen H, Liang B et al (2018) Alcohol-induced autophagy via upregulation of PIASy promotes HCV replication in human hepatoma cells. Cell Death Dis 9:898. https://doi.org/10.1038/s41419-018-0845-x
Huang J, Xie P, Dong Y et al (2021) Inhibition of Drp1 SUMOylation by ALR protects the liver from ischemia-reperfusion injury. Cell Death Differ 28:1174–1192. https://doi.org/10.1038/s41418-020-00641-7
Anderson CA, Blackstone C (2013) SUMO wrestling with Drp1 at mitochondria. EMBO J 32:1496–1498. https://doi.org/10.1038/emboj.2013.103
Cai R, Yu T, Huang C et al (2012) SUMO-specific protease 1 regulates mitochondrial biogenesis through PGC-1alpha. J Biol Chem 287:44464–44470. https://doi.org/10.1074/jbc.M112.422626
Zhong N, Xu J (2008) Synergistic activation of the human MnSOD promoter by DJ-1 and PGC-1alpha: regulation by SUMOylation and oxidation. Hum Mol Genet 17:3357–3367. https://doi.org/10.1093/hmg/ddn230
Shinbo Y, Niki T, Taira T et al (2006) Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ 13:96–108. https://doi.org/10.1038/sj.cdd.4401704
Katafuchi T, Holland WL, Kollipara RK et al (2018) PPARgamma-K107 SUMOylation regulates insulin sensitivity but not adiposity in mice. Proc Natl Acad Sci USA 115:12102–12111. https://doi.org/10.1073/pnas.1814522115
Funding
This work is supported by grants from the Scientific Research Project of Hunan Provincial Health Commission of China (No. 202203013975), the Natural Science Foundation of Hunan Province (2022JJ40386), the Ph.D. Research Start-up Fund of the Second Affiliated Hospital of Hengyang Medical College of University of South China (No. 2021B02), and the scientific research project of Hunan Provincial Department of Education (No. 21B0407).
Author information
Authors and Affiliations
Contributions
Hong Xiao, Hong Zhou, and Anbo Gao wrote the manuscript. Hong Zhou created the figures and tables. Gaofeng Zeng contributed to the conceptualization and formatting of the manuscript. Junfa Zeng and Anbo Gao edited and revised the manuscript and were responsible for funding acquisition. All authors approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xiao, H., Zhou, H., Zeng, G. et al. SUMOylation targeting mitophagy in cardiovascular diseases. J Mol Med 100, 1511–1538 (2022). https://doi.org/10.1007/s00109-022-02258-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-022-02258-4