
Abstract
Development and progression of many kidney diseases are substantially influenced by aberrant protein acetylation modifications of gene expression crucial for kidney functions. Histone deacetylase (HDAC) expression alterations are detected from renal samples of patients and animal models of various kidney diseases, and the administrations of HDAC inhibitors display impressive renal protective effects in vitro and in vivo. However, when the expression alterations of multiple HDACs occur, not all the HDACs causally affect the disease onset or progression. Identification of a single HDAC as a disease-causing factor will allow subtype-targeted intervention with less side effect. HDAC3 is a unique HDAC with distinct structural and subcellular distribution features and co-repressor dependency. HDAC3 is required for kidney development and its aberrations actively participate in many pathological processes, such as cancer, cardiovascular diseases, diabetes, and neurodegenerative disorders, and contribute significantly to the pathogenesis of kidney diseases. This review will discuss the recent studies that investigate the critical roles of HDAC3 aberrations in kidney development, renal aging, renal cell carcinoma, renal fibrosis, chronic kidney disease, polycystic kidney disease, glomerular podocyte injury, and diabetic nephropathy. These studies reveal the distinct characters of HDAC3 aberrations that act on different molecules/signaling pathways under various renal pathological conditions, which might shed lights into the epigenetic mechanisms of renal diseases and the potentially therapeutic strategies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Kidney performs important biophysiological functions of human body, including removing extra water from the blood (as urine), keeping the balance of ions through filtration, reabsorption, and secretion, and generating hormones that regulate the functions of extrarenal organs. Kidney diseases are emerging as serious health problems with increasing morbidities [1]. In addition to the original etiologies, development and progression of many kidney diseases are substantially associated with epigenetic modifications, such as DNA methylation, protein/histone modification, or small interfering RNA interference, of gene expressions [2]. In particular, protein/histone acetylation alterations seemly play a mechanistic role in the onset or progression of kidney diseases. Histone deacetylase (HDAC) alterations are detected from renal samples of clinical patients and experimental animals of various kidney disease models, and HDAC inhibitors exhibit impressive renoprotections in cell and animal studies [3, 4], suggesting that the increased HDAC expressions or activities potentiate or exacerbate the pathological processes that lead to kidney diseases.
Human HDACs include 18 members subdivided into four major classes, namely class I (1, 2, 3, 8), class II (4, 5, 6, 7, 9, 10), class III HDACs (Sirt1–7), and class IV (11) HDACs, based on their homology to yeast HDACs, subcellular localization, and enzymatic activities [5]. Class I, II, and IV HDACs require Zn2 as cofactor and are sensitive to so-called pan- or respective class-selective HDAC inhibitors, whereas class III HDACs (Sirt1–7) are NAD (nicotinamide adenine dinucleotide)-dependent and irresponsive to the classic HDAC inhibitors [6]. HDACs can modify histone or non-histone proteins and HDAC-incurred lysine deacetylation of histone cores around the gene promoter causes chromatin condense, blocks the access of transcription factors, and inhibits gene transcription [7, 8].
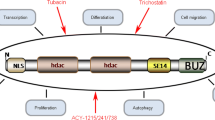
HDAC3 belongs to class I HDAC family and consists of 428 amino acids (aa) with a highly conserved N-terminal deacetylase catalytic domain. Intriguingly, it contains a variable C terminus with no apparent similarity with other HDACs that is required for both deacetylase and transcriptional repression activity [9, 10] (Fig. 1).

Structural characters of class I HDACs
Unlike the other family members mainly appearing in cell nucleus, HDAC3 can be found in the nucleus, the cytoplasm, and at the plasma membrane [11, 12] and exists exclusively in multi-subunit complexes [13] often containing transcriptional repressor NCoR (nuclear receptor co-repressor), SMRT (silencing mediator of retinoic and thyroid receptors) [14], or Ski [15], which separates it from other known HDACs and suggests that HDAC3 may have some unique properties that are not completely redundant with other HDACs [10, 14]. Moreover, a protein–protein interaction network analysis by STRING (https://string-db.org) indicates that HDAC3 directly or indirectly interacts with a number of transcriptional regulatory proteins (Fig. 2), suggesting that HDAC3 acts on multiple signaling pathways in a broad spectrum of cellular processes.

A schematic of HDAC3-protein interaction network analyzed by STRING. BFD4 (bromodomain-containing protein 4), NFKB1 (NF-kappa-B inhibitor alpha),CREBBP (CREB binding protein), polycomb protein EED, EP300 (histone acetyltransferase p300), HDAC4, HIST1H4F (histone cluster 1 H4 family member f), histone acetyltransferase KAT2A, Myc proto-oncogene protein, NCOR1 (nuclear receptor co-repressor 1), NCOR2, NR1D1 (nuclear receptor subfamily 1 group D member 1), NFKB1 (NF-kappa-B p105), RELA (NF-kappa-B p65), PPARG (peroxisome proliferator-activated receptor gamma), histone-binding protein RBBP4, TBL1XR1 (F-box-like/WD repeat-containing protein receptor 1), and YY1 (transcriptional repressor protein Yin Yang 1)
HDAC3 is critical for mammalian embryonic development and aging [16], and its aberrant activities potentially relate or directly lead to human diseases, including cancer [17, 18], epithelial-mesenchymal transition (EMT) [19], macrophage function/inflammation [20,21,22], type I and type II diabetes mellitus [23, 24], cardiovascular disease [25], bone [26,27,28], and different neurodegenerative disorders [29]. The evidences that HDAC3 aberrations contribute to other cellular processes and diseases are also rapidly emerging. HDAC3 is proposed as a drug target to treat patients with various diseases [13]. This review will discuss accumulating studies that investigate the critical roles of HDAC3 in the renal physiology and pathogenesis of kidney diseases (Fig. 3).

HDAC3 regulates many physiopathological processes, including embryonic development, aging, cancer, macrophage/inflammation, diabetes, cardiovascular, neurodegenerative, and bone disorders
HDAC3 is required for kidney development
Kidney development is a sophisticated morphogenetic process involving interaction between nephric duct and the surrounding mesenchyme and formation of pronephros (E8 in mice), mesonephros (E9 in mice), and metanephros (E10.5 in mice), which are tightly controlled by gene transcription regulations and epigenetic modifications [30]. Accumulating data have demonstrated that HDACs regulate fundamental biological processes, such as cell proliferation, differentiation, and apoptosis during kidney development, likely via genomic and nongenomic effects [31]. However, little is known about the role that individual HDAC plays during the process.
Expressions of several class I (1–3) and class II (4, 7, and 9) HDACs are developmentally regulated and HDAC1–3 are highly expressed in nephron precursors, and in addition, HDAC3 is also localized to the glomerular podocytes [32]. Early studies have shown that germline global HDAC3 knockout in mice is embryonically lethal by E9.5 due to a delay in cell cycle progression and developmental defects [8, 33]. Cux1 (Cut Like Homeobox 1) is a transcription repressor and a developmentally regulated protein, and capable of preventing binding of positively activing CCAAT factors to gene promoters to promote cell proliferation [34]. During kidney development, Cux1 is highly expressed in the nephrogenic region. Chromatin immunoprecipitation (ChIP) and promoter reporter analysis showed that Cux1 inhibited the transcription of p27kip1 by forming a transcriptional repressor complex with Grg4 (Groucho 4), HDAC1, and HDAC3 [35]. Protein p27kip1 is a critical cell cycling kinase inhibitor which binds to cyclinE/cdk2, blocking the G1/S transition step required for cell cycle progression. Therefore, p27kip1 inhibition by HDAC3 will negatively affect the renal cell proliferation and kidney development.
Increased HDAC3 accelerates renal aging
Renal aging is a slow process manifesting as declined renal functions and increased susceptibility to various acute or chronic kidney diseases [36], and its development is greatly affected by epigenetic modifications of numerous gene expressions. It has become increasingly clear that protein/histone acetylation plays a crucial role in aging and aging-associated diseases [37, 38]. HDAC inhibitors beneficially regulate aging-related processes likely by targeting non-histone proteins, activating pro-longevity proteins, and/or de-activating anti-longevity proteins [5, 39].
NM_026333 is a newly identified anti-aging gene (no gene name yet) and down-regulated in the kidneys of coupling factor 6 (CF6) transgenic and high salt-fed mice, which had sustained intracellular acidosis due to reduced proton export through Na + -K + ATPase inhibition and displayed shortened lifespan and early senescence-associated phenotypes, such as signs of hair greying and alopecia, weight loss, and/or reduced organ mass [40]. NM_026333 protein directly bound plasma membrane Na + -Ca2 + exchanger 1 (NCX1) to suppress its reverse mode. Endogenous induction or exogenous supplementation of NM_026333 rescued CF6 transgenic cells or CF6-treated human cells from aging by inhibiting the HDAC3-associated suppression of autophagy protein Atg7 and the impaired autophagy, supporting that HDAC3 dysregulation of autophagy promotes renal aging.
Moreover, several known anti-aging proteins, such as telomerase, Klotho, and Nrf2, are also down-regulated in aging kidneys [41], and Klotho suppressions in fibrotic and CKD kidneys are mediated by HDAC3 aberration [42, 43]. Whether their suppressions in renal aging involve HDAC3-associated transcriptional inhibition are currently under investigation.
Aberrant HDAC3 induces renal cell carcinoma
It has been well established that HDACs play critical roles in renal cancer progression, since the expression changes in HDACs, especially the class I HDACs, or inappropriate recruitment of these enzymes has been observed in a number of human renal cancers [13, 17], and HDAC inhibitors have been shown to induce growth arrest, apoptosis, and differentiation in a number of kidney tumor cell lines as well as in mouse tumor models.
Overexpression of HDAC3 is correlated with poor prognosis in various cancers [44]. Inhibition of HDAC3 by I-7ab induces the acetylation process of p53 and p21 expression that results in G1 phase arrest in triple-negative breast cancer [45], and inhibition of HDAC3 may lead to the enhancement of apoptosis of human maxillary cancer [46], suggesting that controlling of cancer cell growth and apoptosis might be the anti-cancer functions of HDAC3 inhibition.
Renal cell carcinoma (RCC), a cancerous disease arising from malignant epithelial cells in the kidneys, is responsible for about 90% of kidney cancers in adults and appears to arise from both genetic defects and environmental influences [47]. HDAC3 is overexpressed in the human RCC tumor tissues [48]. A class I histone deacetylase inhibitor (HDACi) LBH589 displayed antitumor effect on RCC cell lines partially by inhibiting HDAC3 and inducing the degradation of Aurora A and B kinase, leading to G2-M phase arrest and tumor cell apoptosis [48].
BRM (Brahma) is a key subunit of SWI/SNF (switch/sucrose non-fermenting) chromatin remodeling complex and an important tumor suppressor silenced in many tumor types [49,50,51]. BRM knockdown promoted RCC cell proliferation, migration, and invasion. HDAC3-selective inhibitor RGFP966 inhibited the tumor progression of clear cell RCC by restoring BRM expression in vivo and in vitro [52]. Thus, HDAC3 dysregulation of chromatin remodeling facilitates renal cancer progression.
Coactivator activator (CoAA) not only regulates steroid receptor-mediated transcription and alternative RNA splicing but is also a potential tumor suppressor in RCC, which inhibits G1-S transition of human renal cells and suppresses anchorage-independent growth and xenograft tumor formation. The suppression occurs in part by down-regulating C-myc and causing accumulation of p27Kip1 protein. In this cellular setting, CoAA directly represses the proto-oncogene C-myc by recruiting HDAC3 protein and decreasing both the acetylation of histone H3 and the presence of RNA polymerase II on the C-myc promoter [53].
The developments of renal cancer are complex and might be associated with aberrations of multiple HDACs. Several pan- or class-selective HDAC inhibitors, namely Vorinostat, Romidepsin, Belinostat, and Panobinostat, are approved for treatment of patients with hematological malignancies [54]. Moreover, Vorinostat (SAHA, a pan-HDAC inhibitor) [55] and Entinostat (a strong inhibitor of HDAC1 and HDAC3) [56] have been in combination therapy in phase I and II clinical trials for renal cell carcinoma patients. These studies set the stage for possible future application of HDAC3-selective inhibitors for treating patients with renal cancers.
HDAC3 is a key epigenetic player of renal fibrosis
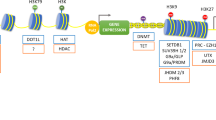
Renal fibrosis is a histological hallmark of renal aging and chronic kidney diseases and characterized by renal cell injury-incurred inflammatory macrophage infiltration, activation of TGF-β signaling, and subsequent myofibroblast trans-differentiation (MTD) with excessive extracellular matrix protein deposition in renal interstitium [57]. Previous studies have established that MTD and renal fibrosis are significantly affected by HDAC aberrations [2, 3]. Mice with HDAC3 knockout resist renal fibrosis induced by UUO (unilateral ureteral obstruction), suggesting that HDAC3 is a pro-fibrogenic factor [42]. HDAC3 among other class I HDAC members was preferentially up-regulated in renal tubular epithelial cells of mouse fibrotic kidneys incurred by UUO [42]. HDAC3 promoter contains a functional Smad3 binding motif and its up-regulation in fibrotic kidney is promoted by TGF-β/Smad signaling, since inhibitors of TGF-β receptor I SB431542 and Smad3 phosphorylation SIS3 (specific inhibitor of SMAD3) block the fibrotic HDAC3 up-regulation in both UUO kidney and TGF-β-treated renal epithelial cells. HDAC3 promotes renal fibrosis, at least in a significant part, by inhibiting the transcription of a kidney-enriched anti-aging and anti-fibrotic protein Klotho, which is markedly suppressed in fibrotic kidney. The up-regulated HDAC3 forms a transcriptional repressive complex with NCoR and NF-κB that inducibly binds to Klotho promoter, resulting in Klotho transcriptional inhibition. Consistently, RGFP966, a selective HDAC3 inhibitor, relieves the Klotho suppression and Klotho restoration dependently alleviates the renal fibrotic injury in UUO and aristolochic acid nephropathy [42]. Since renal fibrosis is an indispensible histopathological character of chronic kidney diseases, these observations have broad implications.
HDAC3 might also regulate macrophage functions during renal fibrogenesis. Renal fibrosis is featured by activation of TGF-β and the associated macrophage M2 polarization [58, 59], which supposedly promote kidney repair and remodeling after renal injury, but these processes are dysregulated during renal fibrosis. TIMAP (TGF-β-inhibited membrane-associated protein) is originally identified from TGF-β-treated renal vascular endothelial cells [60]. Later, TIMAP is also found highly expressed in macrophages and mediates macrophage M2 polarization upon TGF-β stimulation [61]. TIMAP is a subunit of myosin phosphatase that presumably regulates myosin filament contraction and cell motility by dephosphorylating myosin light chain [62]. TIMAP is repressed in UUO kidney and specific HDAC3 inhibition can reverse the repression and inhibit macrophage M2 polarization and the associated phagocytic activities [61], suggesting that HDAC3 inhibition might improve renal fibrosis by targeting macrophage functions.
HDAC3 elevation promotes chronic kidney diseases (CKDs)
CKD is a slow process of gradual loss of kidney function over time incurred by a variety of different causes, including diabetes, hypertension, polycystic kidney disease, obstructive uropathy, glomerular nephrotic, and nephritic syndromes such as focal segmental glomerulosclerosis, membranous nephropathy, and lupus nephritis, and potentially leads to renal failure [63].
Renal protective proteins play essential roles in maintaining kidney homeostasis. PPARγ (peroxisome proliferation-activated receptor gamma) is a nuclear hormone transcriptional factor with eminent renal protective properties. PPARγ protects renal cells and kidney likely via beneficially regulating transcription of genes involved in insulin action, lipid/protein metabolisms, oxidative stress, and inflammation [64]. HDAC3 regulation of CKD seemly involves PPARγ/Klotho pathway. Klotho is not only a kidney-enriched anti-aging protein and a PPARγ target but also a biomarker and proposed therapeutic target of CKD [65]. Klotho is markedly suppressed during CKD development and its levels inversely correlate with the severity and progression of CKD [66]. The lysine residues K240 and K265 on PPARγ are HDAC3-sensitive sites. Administration of a pan-HDAC inhibitor trichostatin A (TSA) and a HDAC3-selective inhibitor RGFP966 extent similarly increases PPARγ acetylation and reverses Klotho suppression in adenine-induced CKD mouse kidney, resulting in improved CKD pathologies in wild-type, but not siRNA-mediated Klotho, knockdown mice [43]. Thus, targeting HDAC3/Klotho axis by HDAC3-selective inhibition might bring clinical benefits.
EMT contributes to the development of peritoneal fibrosis after long-term peritoneal dialysis in patients of end-stage CKD [67,68,69]. Administration of 1,25-dihydroxy vitamin D3 can reduce the EMT of cultured peritoneal mesothelial cells by down-regulating HDAC3 and up-regulating vitamin D receptor (VDR) [70], suggesting a potential role of HDAC3 in this process.
Up-regulated HDAC3 potentiates diabetic nephropathy
Diabetic nephropathy (DN) is a major cause of CKD. In high-fat diet (HFD) combined with low-dose streptozotocin (STZ)-fed mice mimicking DN, HDAC3 is up-regulated in the mouse kidney accompanied by suppression of miR-10a, which inhibits kidney diabetic alterations by targeting CREB1 (cAMP response element binding protein 1) and its downstream extracellular matrix protein fibronectin (FN). Knockdown (KD) of HDAC3 by siRNA significantly increases miR-10a, resulting in decrease of CREB1 and FN expression in kidney of HFD/STZ mice, while HDAC3 overexpression mediated by lentivirus decreases miR-10a and induces diabetic alterations in naïve mice [71]. In another study of similar DN mouse model, Juglanin, a natural compound extracted from crude polygonumaviculare, exhibits marked anti-DN effects via inhibiting NF-κB/HDAC3 signaling accompanied by increased nephrin and podocin expression levels, and the associated inflammation and dyslipidemia [72]. These studies suggest that HDAC3 aberrations likely worsen the development of DN through multiple signaling pathways.
HDAC3 aberration associates with polycystic kidney disease (PKD)
PKD is a kidney disease involving both genetic and epigenetic mechanisms [73]. The multiple cysts filled with fluid in the kidney dilate and impair renal function that might eventually lead to renal failure. HDAC3 is seemly involved in PKD development; however, its exact roles are not conclusive. P27Kip1 is known to be down-regulated by Cux1-associated HDAC3 activities during kidney development [35]. One study treated Pkd1-targeted pregnant mice with TSA or vehicle beginning at E10.5 until E18.5. Newborn Pkd1 mutant mice receiving vehicle exhibited extensive collecting duct cysts, while the mice receiving TSA showed a significant reduction in cysts. The expression of p27Kip1 was up-regulated in TSA-treated Pkd1 mice [74], suggesting that HDAC3 promotes the cyst formation. On the other hand, Prothymosin α (ProT) is an important regulator of CKD and its overexpression induces PKD [75]. ProT is up-regulated in cyst-lining epithelial cells and the suppression of ProT is sufficient to reduce cyst formation [76]. ProT activation involves its interaction with STAT3, a transcription factor normally associated with HDAC3 [77]. The interaction between ProT and STAT3 causes the deprivation of HDAC3 from STAT3 [76], resulting in STAT3 acetylation and activation. This is a good example how HDAC3 is passively due to a competitive mechanism involved in an important signal transduction pathway. Notably, this study was performed with neither pharmacological nor genetic HDAC3 inhibition. Future studies are necessary to determine the exact role of HDAC3 in PKD.
HDAC3 aberration mediates glomerular podocyte injury
Podocytes are integrated parts of glomeruli that maintain the glomerular structural integration and functionality [78]. HDAC3 aberration seems to cause glomerular podocyte injury by down-regulating microRNA-30 family members (miR-30a-30e), which are abundantly expressed in podocytes and crucial for podocyte homeostasis [79]. Down-regulations of miR-30s in podocytes are observed from rats treated with puromycin aminonucleoside and from patients with focal segmental glomerulosclerosis (FSGS) that causes podocyte foot process effacement and proteinuria, like via calcium/calcineurin signaling, in the animal model [80]. Excessive and persistent TGF-β elevations after kidney injury are deleterious for podocytes [81]. HDAC3 is up-regulated in cultured podocytes upon TGF-β stimulation and assists TGF-β-incurred suppression of miR-30s. TSA and HDAC3-selective inhibitor RGFP966 significantly alleviates the miR-30d suppression and reduces podocyte injury [82]. Thus, HDAC3 inhibition of miR-30s likely contributes to podocyte injury-associated renal diseases.
Conclusions
We have discussed the studies demonstrating HDAC3 as an important epigenetic regulator in kidney development, renal aging, renal cell carcinoma, renal fibrosis, CKD, diabetic nephropathy, PKD, and glomerular podocyte injury. These studies reveal the distinct characters of HDAC3 aberrations that not only act on different molecules/signaling pathways under various pathological conditions (Table 1) but also raise many important questions. For example, in addition to renal tubular epithelial cells, what are the other cell types in which HDAC3 is aberrantly up-regulated and exerts its adverse functions? TGF-β seems to be one upstream factor that up-regulates HDAC3 in fibrotic mouse kidney [42, 43, 61, 82]: but under different renal pathological conditions, what are the other common or specific upstream initiators that lead to the HDAC3 aberrations? What are the co-repressors or co-regulators that assist HDAC3-sssociated gene transcriptional inhibition? And how does HDAC3 achieve its target selectivity and specificity? These intriguing questions await future studies to answer.
In addition, kidney diseases are often accompanied by altered expressions of multiple HDACs. Under any circumstance, the results from pan- or class-selective HDAC inhibitors should be validated by genetic gene knockout approach to identify the causal HDAC subtype, as HDAC inhibitors tend to have non-enzymatic or off-target effects. Identification of a single HDAC subtype as a key causal HDAC critically involved in a particular renal disease setting will allow HDAC subtype-targeted therapeutic design and avoid the side effects associated with pan- or class-HDAC inhibitors. RGFP966, reportedly inhibiting HDAC3 with IC50 value of 80 nM while having no effective inhibition on other HDACs up to 15 μM [83], is potent and highly selective for HDAC3, but not in clinical uses. Lately, examination of HDAC3 inhibition as potential therapeutic targets and development of effective HDAC3 inhibitors have received increasing attentions [84, 85]. Future exploration of the precise mechanisms of HDAC3 aberrations in disease pathogenesis and the effective HDAC3-selective inhibitors with tolerable side effects might benefit patients with various diseases of kidney or extrarenal organs.
References
Koye DN, Magliano DJ, Nelson RG, Pavkov ME (2018) The global epidemiology of diabetes and kidney disease. Adv Chronic Kidney Dis 25:121–132. https://doi.org/10.1053/j.ackd.2017.10.011
Wanner N, Bechtel-Walz W (2017) Epigenetics of kidney disease. Cell Tissue Res 369:75–92. https://doi.org/10.1007/s00441-017-2588-x
Tampe B, Zeisberg M (2013) Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol Dial Transplant 29:iv72-iv79. https://doi.org/10.1093/ndt/gft025
Xia J, Cao W (2021) Epigenetic modifications of Klotho expression in kidney diseases. J Mol Med. https://doi.org/10.1007/s00109-021-02044-8
Pasyukova EG, Vaiserman AM (2017) HDAC inhibitors: a new promising drug class in anti-aging research. Mech Ageing Dev 166:6–15. https://doi.org/10.1016/j.mad.2017.08.008
Gregoretti IV, Lee YM, Goodson HV (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 338:17–31. https://doi.org/10.1016/j.jmb.2004.02.006
Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM (2013) The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol 9:672. https://doi.org/10.1038/msb.2013.26
Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW (2008) Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 30:61–72. https://doi.org/10.1016/j.molcel.2008.02.030
Yang W-M, Tsai S-C, Wen Y-D, Fejér G, Seto E (2002) Functional domains of histone deacetylase-3. J Biol Chem 277:9447–9454. https://doi.org/10.1074/jbc.M105993200
Yang WM, Yao YL, Sun JM, Davie JR, Seto E (1997) Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem 272:28001–28007. https://doi.org/10.1074/jbc.272.44.28001
Takami Y, Nakayama T (2000) N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J Biol Chem 275:16191–16201. https://doi.org/10.1074/jbc.M908066199
Grégoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ (2007) Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol 27:1280–1295. https://doi.org/10.1128/mcb.00882-06
Sarkar R, Banerjee S, Amin SA, Adhikari N, Jha T (2020) Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: a review. Eur J Med Chem 192:112171. https://doi.org/10.1016/j.ejmech.2020.112171
Wen YD, Perissi V, Staszewski LM, Yang WM, Krones A, Glass CK, Rosenfeld MG, Seto E (2000) The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci USA 97:7202–7207. https://doi.org/10.1073/pnas.97.13.7202
Tabata T, Kokura K, ten Dijke P, Ishii S (2009) Ski co-repressor complexes maintain the basal repressed state of the TGF-β target gene, SMAD7, via HDAC3 and PRMT5. Genes Cells 14:17–28. https://doi.org/10.1111/j.1365-2443.2008.01246.x
Emmett MJ, Lazar MA (2018) Integrative regulation of physiology by histone deacetylase 3. Nat Rev Mol Cell Biol 20:102–115. https://doi.org/10.1038/s41580-018-0076-0
Adhikari N, Amin SA, Trivedi P, Jha T, Ghosh B (2018) HDAC3 is a potential validated target for cancer: an overview on the benzamide-based selective HDAC3 inhibitors through comparative SAR/QSAR/QAAR approaches. Eur J Med Chem 157:1127–1142. https://doi.org/10.1016/j.ejmech.2018.08.081
Mariadason JM (2014) Dissecting HDAC3-mediated tumor progression. Cancer Biol Ther 7:1581–1583. https://doi.org/10.4161/cbt.7.10.6863
Wu M-Z, Tsai Y-P, Yang M-H, Huang C-H, Chang S-Y, Chang C-C, Teng S-C, Wu K-J (2011) Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell 43:811–822. https://doi.org/10.1016/j.molcel.2011.07.012
Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G (2012) Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci 109:E2865–E2874. https://doi.org/10.1073/pnas.1121131109
Yao Y, Liu Q, Adrianto I, Wu X, Glassbrook J, Khalasawi N, Yin C, Yi Q, Dong Z, Geissmann F, Zhou L (2020) Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat Commun 11. https://doi.org/10.1038/s41467-020-17630-6
Nguyen HCB, Adlanmerini M, Hauck AK, Lazar MA (2020) Dichotomous engagement of HDAC3 activity governs inflammatory responses. Nature 584:286–290. https://doi.org/10.1038/s41586-020-2576-2
Sathishkumar C, Prabu, Balakumar M, Lenin R, Prabhu, Anjana RM, Mohan V, Balasubramanyam M (2016) Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenetics 8. https://doi.org/10.1186/s13148-016-0293-3
Meier BC, Wagner BK (2014) Inhibition of HDAC3 as a strategy for developing novel diabetes therapeutics. Epigenomics 6:209–214
Hoeksema MA, Gijbels MJJ, Van den Bossche J, Velden S, Sijm A, Neele AE, Seijkens T, Stöger JL, Meiler S, Boshuizen MCS, Dallinga-Thie GM, Levels JHM, Boon L, Mullican SE, Spann NJ, Cleutjens JP, Glass CK, Lazar MA, Vries CJM, Biessen EAL, Daemen MJAP, Lutgens E, Winther MPJ (2014) Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med 6:1124–1132. https://doi.org/10.15252/emmm.201404170
Molstad DHH, Mattson AM, Begun DL, Westendorf JJ, Bradley EW (2020) Hdac3 regulates bone modeling by suppressing osteoclast responsiveness to RANKL. J Biol Chem 295:17713–17723. https://doi.org/10.1074/jbc.RA120.013573
Molstad DHH, Zars E, Norton A, Mansky KC, Westendorf JJ, Bradley EW (2020) Hdac3 deletion in myeloid progenitor cells enhances bone healing in females and limits osteoclast fusion via Pmepa1. Sci Rep 10:21804. https://doi.org/10.1038/s41598-020-78364-5
Feigenson M, Shull LC, Taylor EL, Camilleri ET, Riester SM, van Wijnen AJ, Bradley EW, Westendorf JJ (2017) Histone deacetylase 3 deletion in mesenchymal progenitor cells hinders long bone development. J Bone Miner Res 32:2453–2465. https://doi.org/10.1002/jbmr.3236
D’Mello SR (2020) Histone deacetylase-3: friend and foe of the brain. Exp Biol Med (Maywood) 245:1130–1141. https://doi.org/10.1177/1535370220928278
Chen S, El-Dahr SS (2012) Histone deacetylases in kidney development: implications for disease and therapy. Pediatr Nephrol 28:689–698. https://doi.org/10.1007/s00467-012-2223-8
Mason K, Liu Z, Aguirre-Lavin T, Beaujean N (2012) Chromatin and epigenetic modifications during early mammalian development. Anim Reprod Sci 134:45–55. https://doi.org/10.1016/j.anireprosci.2012.08.010
Chen S, Bellew C, Yao X, Stefkova J, Dipp S, Saifudeen Z, Bachvarov D, El-Dahr SS (2011) Histone deacetylase (HDAC) activity is critical for embryonic kidney gene expression, growth, and differentiation. J Biol Chem 286:32775–32789. https://doi.org/10.1074/jbc.M111.248278
You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA (2013) Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol 20:182–187. https://doi.org/10.1038/nsmb.2476
Liu N, Sun Q, Wan L, Wang X, Feng Y, Luo J, Wu H. (2020) CUX1, a controversial player in tumor development. Frontiers in oncology. Front Oncol 10. https://doi.org/10.3389/fonc.2020.00738
Sharma M, Brantley JG, Vassmer D, Chaturvedi G, Baas J, Vanden Heuvel GB (2009) The homeodomain protein Cux1 interacts with Grg4 to repress p27 kip1 expression during kidney development. Gene 439:87–94. https://doi.org/10.1016/j.gene.2009.03.014
Nitta K, Okada K, Yanai M, Takahashi S (2013) Aging and chronic kidney disease. Kidney Blood Press Res 38:109–120. https://doi.org/10.1159/000355760
Wei SY, Pan SY, Li B, Chen YM, Lin SL (2020) Rejuvenation: turning back the clock of aging kidney. J Formos Med Assoc 119:898–906. https://doi.org/10.1016/j.jfma.2019.05.020
Fang Y, Gong AY, Haller ST, Dworkin LD, Liu Z, Gong R (2020) The ageing kidney: molecular mechanisms and clinical implications. Ageing Res Rev 63:101151. https://doi.org/10.1016/j.arr.2020.101151
McIntyre RL, Daniels EG, Molenaars M, Houtkooper RH, Janssens GE (2019) From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol Med 11. https://doi.org/10.15252/emmm.201809854
Osanai T, Tanaka M, Mikami K, Kitajima M, Tomisawa T, Magota K, Tomita H, Okumura K (2018) Novel anti-aging gene NM_026333 contributes to proton-induced aging via NCX1-pathway. J Mol Cell Cardiol 125:174–184. https://doi.org/10.1016/j.yjmcc.2018.10.021
Shiels PG, McGuinness D, Eriksson M, Kooman JP, Stenvinkel P (2017) The role of epigenetics in renal ageing. Nat Rev Nephrol 13:471–482. https://doi.org/10.1038/nrneph.2017.78
Chen F, Gao Q, Wei A, Chen X, Shi Y, Wang H (2020) Histone deacetylase 3 aberration inhibits Klotho transcription and promotes renal fibrosis. DOI. https://doi.org/10.1038/s41418-020-00631-9
Lin W, Zhang Q, Liu L, Yin S, Liu Z, Cao W (2017) Klotho restoration via acetylation of peroxisome proliferation-activated receptor γ reduces the progression of chronic kidney disease. Kidney Int 92:669–679. https://doi.org/10.1016/j.kint.2017.02.023
Weichert W, Röske A, Gekeler V, Beckers T, Ebert MPA, Pross M, Dietel M, Denkert C, Röcken C (2008) Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol 9:139–148. https://doi.org/10.1016/s1470-2045(08)70004-4
Yang M, Dang X, Tan Y, Wang M, Li X, Li G (2018) I-7ab inhibited the growth of TNBC cells via targeting HDAC3 and promoting the acetylation of p53. Biomed Pharmacother 99:220–226. https://doi.org/10.1016/j.biopha.2018.01.063
Narita N, Fujieda S, Tokuriki M, Takahashi N, Tsuzuki H, Ohtsubo T, Matsumoto H (2005) Inhibition of histone deacetylase 3 stimulates apoptosis induced by heat shock under acidic conditions in human maxillary cancer. Oncogene 24:7346–7354. https://doi.org/10.1038/sj.onc.1208879
Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V (2017) Renal cell carcinoma. Nat Rev Dis Primers 3:17009. https://doi.org/10.1038/nrdp.2017.9
Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, Huang SM, Huan SK, Cheng TC, Chen TT, Fan PL, Hsiao PW (2009) Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clinical cancer research: an official Journal of the American Association for Cancer Research 15:840–850. https://doi.org/10.1158/1078-0432.ccr-08-1918
Jancewicz I, Siedlecki JA, Sarnowski TJ, Sarnowska E (2019) BRM: the core ATPase subunit of SWI/SNF chromatin-remodelling complex—a tumour suppressor or tumour-promoting factor? Epigenetics Chromatin 12:68. https://doi.org/10.1186/s13072-019-0315-4
Zhang Z, Li J, Guo H, Wang F, Ma L, Du C, Wang Y, Wang Q, Kornmann M, Tian X, Yang Y (2019) BRM transcriptionally regulates miR-302a-3p to target SOCS5/STAT3 signaling axis to potentiate pancreatic cancer metastasis. Cancer Lett 449:215–225. https://doi.org/10.1016/j.canlet.2019.02.031
Yang Y, Liu L, Fang M, Bai H, Xu Y (2019) The chromatin remodeling protein BRM regulates the transcription of tight junction proteins: implication in breast cancer metastasis. Biochim Biophys Acta Gene Regul Mech 1862:547–556. https://doi.org/10.1016/j.bbagrm.2019.03.002
Fang R, Pan R, Wang X, Liang Y, Wang X, Ma H, Zhou X, Xia Q, Rao Q (2020) Inactivation of BRM/SMARCA2 sensitizes clear cell renal cell carcinoma to histone deacetylase complex inhibitors. Pathol Res Pract 216:152867. https://doi.org/10.1016/j.prp.2020.152867
Kang YK, Schiff R, Ko L, Wang T, Tsai SY, Tsai MJ, O’Malley BW (2008) Dual roles for coactivator activator and its counterbalancing isoform coactivator modulator in human kidney cell tumorigenesis. Can Res 68:7887–7896. https://doi.org/10.1158/0008-5472.can-08-1734
Shah RR (2019) Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf 42:235–245. https://doi.org/10.1007/s40264-018-0773-9
Pili R, Liu G, Chintala S, Verheul H, Rehman S, Attwood K, Lodge MA, Wahl R, Martin JI, Miles KM et al (2017) Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: a multicentre, single-arm phase I/II clinical trial. Br J Cancer 116:874–883. https://doi.org/10.1038/bjc.2017.33
Pili R, Quinn DI, Hammers HJ, Monk P, George S, Dorff TB, Olencki T, Shen L, Orillion A, Lamonica D et al (2017) Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: a multicenter, single-arm, phase I/II trial (NCI-CTEP#7870). Clin Cancer Res 23:7199–7208. https://doi.org/10.1158/1078-0432.ccr-17-1178
Zeisberg M, Neilson EG (2010) Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 21:1819–1834. https://doi.org/10.1681/asn.2010080793
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8:958–969. https://doi.org/10.1038/nri2448
Casalena G, Daehn I, Bottinger E (2012) Transforming growth factor-β, bioenergetics, and mitochondria in renal disease. Semin Nephrol 32:295–303. https://doi.org/10.1016/j.semnephrol.2012.04.009
Cao W, Mattagajasingh SN, Xu H, Kim K, Fierlbeck W, Deng J, Lowenstein CJ, Ballermann BJ (2002) TIMAP, a novel CAAX box protein regulated by TGF-β1 and expressed in endothelial cells. Am J Physiol Cell Physiol 283:C327–C337. https://doi.org/10.1152/ajpcell.00442.2001
Yang J, Yin S, Bi F, Liu L, Qin T, Wang H, Cao W (2016) TIMAP repression by TGFβ and HDAC3-associated Smad signaling regulates macrophage M2 phenotypic phagocytosis. J Mol Med 95:273–285. https://doi.org/10.1007/s00109-016-1479-z
Hartshorne DJ, Ito M, Erdödi F (2004) Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J Biol Chem 279:37211–37214. https://doi.org/10.1074/jbc.R400018200
Wing MR, Ramezani A, Gill HS, Devaney JM, Raj DS (2013) Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin Nephrol 33:363–374. https://doi.org/10.1016/j.semnephrol.2013.05.008
Corrales P, Izquierdo-Lahuerta A, Medina-Gómez G (2018) Maintenance of kidney metabolic homeostasis by PPAR gamma. Int J Mol Sci 19:2063. https://doi.org/10.3390/ijms19072063
Neyra JA, Hu MC (2016) αKlotho and chronic kidney disease. 101: 257–310. https://doi.org/10.1016/bs.vh.2016.02.007
Neyra JA, Hu MC (2017) Potential application of Klotho in human chronic kidney disease. Bone 100:41–49. https://doi.org/10.1016/j.bone.2017.01.017
Choi D, Kim CL, Kim JE, Mo JS, Jeong HS (2020) Hesperetin inhibit EMT in TGF-beta treated podocyte by regulation of mTOR pathway. Biochem Biophys Res Commun 528:154–159. https://doi.org/10.1016/j.bbrc.2020.05.087
Huang S, Susztak K (2016) Epithelial plasticity versus EMT in kidney fibrosis. Trends Mol Med 22:4–6. https://doi.org/10.1016/j.molmed.2015.11.009
Lovisa S, Zeisberg M, Kalluri R (2016) Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol Metab 27:681–695. https://doi.org/10.1016/j.tem.2016.06.004
Liu KH, Fu J, Zhou N, Yin W, Yang YY, Ouyang SX, Liang YM (2019) 1,25-Dihydroxyvitamin D3 prevents epithelial-mesenchymal transition of HMrSV5 human peritoneal mesothelial cells by inhibiting histone deacetylase 3 (HDAC3) and increasing vitamin D receptor (VDR) expression through the Wnt/β-catenin signaling pathway. Med Sci Monit 25:5892–5902. https://doi.org/10.12659/msm.916313
Shan Q, Zheng G, Zhu A, Cao L, Lu J, Wu D, Zhang Z, Fan S, Sun C, Hu B, Zheng Y (2016) Epigenetic modification of miR-10a regulates renal damage by targeting CREB1 in type 2 diabetes mellitus. Toxicol Appl Pharmacol 306:134–143. https://doi.org/10.1016/j.taap.2016.06.010
Li Q, Ge C, Tan J, Sun Y, Kuang Q, Dai X, Zhong S, Yi C, Hu L, Lou D, Xu M (2021) Juglanin protects against high fat diet-induced renal injury by suppressing inflammation and dyslipidemia via regulating NF-kappaB/HDAC3 signaling. Int Immunopharmacol 95:107340. https://doi.org/10.1016/j.intimp.2020.107340
Foo JN, Xia Y (2019) Polycystic kidney disease: new knowledge and future promises. Curr Opin Genet Dev 56:69–75. https://doi.org/10.1016/j.gde.2019.06.007
Livingston S, Carlton C, Sharma M, Kearns D, Baybutt R, Vanden Heuvel GB (2019) Cux1 regulation of the cyclin kinase inhibitor p27(kip1) in polycystic kidney disease is attenuated by HDAC inhibitors. Gene: X 2. https://doi.org/10.1016/j.gene.2019.100007
Li K-J, Shiau A-L, Chiou Y-Y, Yo Y-T, Wu C-L (2005) Transgenic overexpression of prothymosin α induces development of polycystic kidney disease11See Editorial by Gattone, p. 2063. Kidney Int 67:1710–1722. https://doi.org/10.1111/j.1523-1755.2005.00268.x
Chen YC, Su YC, Shieh GS, Su BH, Su WC, Huang PH, Jiang ST, Shiau AL, Wu CL (2019) Prothymosin α promotes STAT3 acetylation to induce cystogenesis in Pkd1-deficient mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 33:13051–13061. https://doi.org/10.1096/fj.201900504R
Yuan ZL, Guan YJ, Chatterjee D, Chin YE (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science (New York, NY) 307:269–273. https://doi.org/10.1126/science.1105166
Braun F, Becker JU, Brinkkoetter PT (2016) Live or let die: is there any cell death in podocytes? Semin Nephrol 36:208–219. https://doi.org/10.1016/j.semnephrol.2016.03.008
Wu J, Zheng C, Fan Y, Zeng C, Chen Z, Qin W, Zhang C, Zhang W, Wang X, Zhu X, Zhang M, Zen K, Liu Z (2014) Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorticoids. J Am Soc Nephrol 25:92–104. https://doi.org/10.1681/asn.2012111101
Wu J, Zheng C, Wang X, Yun S, Zhao Y, Liu L, Lu Y, Ye Y, Zhu X, Zhang C, Shi S, Liu Z (2015) MicroRNA-30 family members regulate calcium/calcineurin signaling in podocytes. J Clin Investig 125:4091–4106. https://doi.org/10.1172/jci81061
Bhati M, D. Prabhu Y, Renu K, Vellingiri B, Thiagarajan P, Panda A, Chakraborty R, Myakala H, Valsala Gopalakrishnan A, (2020) Role of TGF-β signalling in PCOS associated focal segmental glomerulosclerosis. Clin Chim Acta 510:244–251. https://doi.org/10.1016/j.cca.2020.07.032
Liu L, Lin W, Zhang Q, Cao W, Liu Z (2015) TGF-β induces miR-30d down-regulation and podocyte injury through Smad2/3 and HDAC3-associated transcriptional repression. J Mol Med 94:291–300. https://doi.org/10.1007/s00109-015-1340-9
Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA (2013) HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci 110:2647–2652. https://doi.org/10.1073/pnas.1213364110
Zhang L, Chen Y, Jiang Q, Song W, Zhang L (2019) Therapeutic potential of selective histone deacetylase 3 inhibition. Eur J Med Chem 162:534–542. https://doi.org/10.1016/j.ejmech.2018.10.072
Cao F, Zwinderman MRH, Dekker FJ (2018) The process and strategy for developing selective histone deacetylase 3 inhibitors. Molecules 23:551. https://doi.org/10.3390/molecules23030551
Acknowledgements
The authors thank formal and current lab members Dr. Lin Liu, Wenjun Lin, Jun Yang, and Fang Chen for their contributions to the publications cited in this article.
Funding
This study is supported by research grants from National Nature Science Foundation of China General Program 81970577 and 81670762.
Author information
Authors and Affiliations
Contributions
Conceptual design, WC; data mining and collection, LZ; manuscript preparation, LZ and WC; manuscript editing and writing, WC. Both authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Availability of supporting data
Not applicable.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, L., Cao, W. Histone deacetylase 3 (HDAC3) as an important epigenetic regulator of kidney diseases. J Mol Med 100, 43–51 (2022). https://doi.org/10.1007/s00109-021-02141-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-021-02141-8