
Abstract
Arginase-1 (ARG1) deficiency is a rare autosomal recessive disorder that affects the liver-based urea cycle, leading to impaired ureagenesis. This genetic disorder is caused by 40+ mutations found fairly uniformly spread throughout the ARG1 gene, resulting in partial or complete loss of enzyme function, which catalyzes the hydrolysis of arginine to ornithine and urea. ARG1-deficient patients exhibit hyperargininemia with spastic paraparesis, progressive neurological and intellectual impairment, persistent growth retardation, and infrequent episodes of hyperammonemia, a clinical pattern that differs strikingly from other urea cycle disorders. This review briefly highlights the current understanding of the etiology and pathophysiology of ARG1 deficiency derived from clinical case reports and therapeutic strategies stretching over several decades and reports on several exciting new developments regarding the pathophysiology of the disorder using ARG1 global and inducible knockout mouse models. Gene transfer studies in these mice are revealing potential therapeutic options that can be exploited in the future. However, caution is advised in extrapolating results since the lethal disease phenotype in mice is much more severe than in humans indicating that the mouse models may not precisely recapitulate human disease etiology. Finally, some of the functions and implications of ARG1 in non-urea cycle activities are considered. Lingering questions and future areas to be addressed relating to the clinical manifestations of ARG1 deficiency in liver and brain are also presented. Hopefully, this review will spark invigorated research efforts that lead to treatments with better clinical outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Arginase-1 and the urea cycle
The urea cycle disorders (UCDs) represent a group of inborn errors of hepatic metabolism that affect the detoxification of ammonia. Deficiency in any of the six principal enzymes associated with the urea cycle results in perturbation of ureagenesis, leading to incomplete removal of ammonia and eventual hyperammonemia of varying degrees. The liver is the main site of urea cycle activity where the proximal three enzymes are in the mitochondria [N-acetyl-glutamate synthase (NAGS), carbamoyl phosphate synthetase 1 (CPS1), and ornithine transcarbamylase (OTC)], while the distal three are cytosolic [argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL), and arginase] [1].
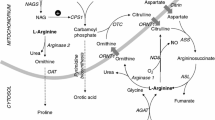
Arginase was discovered in mammalian liver tissue in 1904 [2]. Being the sixth and final enzyme of the cycle, arginase catalyzes the hydrolysis of arginine to ornithine and urea, where the latter is transported in the blood to the kidneys and excreted in the urine, while ornithine is recycled to continue the cycle for further rounds of urea production (Fig. 1). There are two major isoforms of arginase, which are encoded by separate genes in mammals: arginase-1 (ARG1) and arginase-2 (ARG2) that share approximately 60 % amino acid sequence homology [3]. They are similar in enzymatic properties but differ in tissue distribution, subcellular localization, and metabolic functions. ARG1, expressed in highest amounts in the liver cytosol, serves its primary function in the urea cycle as mentioned above [3] but is also found in lesser amounts in human erythrocytes and in the vasculature and immune cells (M2-like macrophages), to mention a few [4, 5]. ARG1 via its metabolic activity also yields ornithine, which serves as precursor to polyamines, proline, and other products. ARG2, on the other hand, is in the mitochondrial compartment of extrahepatic tissues such as kidney and prostate, with lower levels in brain, macrophages, gastrointestinal tract, and lactating mammary glands [6–9]. Besides regulating arginine homeostasis, ARG2 also plays pivotal roles in the biosynthesis of polyamines, proline, creatine, citrulline, γ-aminobutyric acid (GABA), glutamate, and nitric oxide [3, 8, 10].

Central role of arginase-1 to urea cycle function in liver hepatocytes. Note that the nitric oxide synthesis pathway primarily takes place in other tissues and cells of the body. NAGS N-acetyl-glutamate synthase, CPS1 carbamoyl phosphate synthetase
The first crystal structure of arginase to be solved derived from rat liver [11]. X-ray crystallographic analysis at 2.1 Å resolution revealed that Arg1 exists as a 105-kDa homotrimeric metalloprotein, which requires bivalent metal ions, in particular manganese (Mn2+) for maximal catalytic activity and structural stabilization. Each subunit contains a highly conserved binuclear Mn2+cluster with metal-coordinating histidine and aspartic acid residues at the active site [12–14] (see Fig. 2). According to a proposed mechanism of arginase-catalyzed hydrolysis, the metal-activated mechanism is facilitated when the binuclear manganese cluster activates nucleophilic attack of a metal-bridging hydroxide ion at the substrate guanidinium group of arginine to yield ornithine and urea [14].

ARG1 “mutations” superimposed on a crystal structure of native, unliganded human arginase at 1.90 Å resolution (PDB ID: 2PHA) [15]. ARG1 residues 5–318 are displayed in gray cartoon mode to represent secondary structural properties. Mn2+ is represented using cyan spheres. Residues associated with the most commonly occurring mutations are shown (R21, T134, D234, G235, and R308). Residues 1–4 and 319–322 were not resolved in this crystal structure
Arginase-1 deficiency: clinical characteristics
The first patients with ARG1 deficiency, two female siblings, were described in 1969. One presented at 22 months of age with epileptic seizures, abnormal gait at 2.5 years, and spastic diplegia at 3 years; the other had cerebral seizures at 3 months followed by periodic vomiting and hepatomegaly and later spasticity. Both had psychomotor retardation [16].
The clinical presentation of ARG1 deficiency is characterized by the development of spasticity predominantly in the lower limbs during early childhood [3, 17]. The clinical picture is strikingly uniform and has occasionally been mislabeled as cerebral palsy. Progressive loss of mental and motor skills, increasingly more severe spasticity, and pyramidal tract signs are the hallmarks of the disease [18]. The course and severity of spasticity distinguish ARG1 deficiency clinically from the other UCDs and from other disorders of amino acid metabolism [18].
The clinical manifestation of hyperammonemia in ARG1 deficiency is also different from the other UCDs. The initial presentation is usually not characterized by hyperammonemia. Rather, it is often only moderate, and neonatal presentations are rarely reported, although fatal hyperammonemic events [19, 20] and cases with early presentation [21–23] have been observed. Cyclic hyperammonemic episodes have been found related to the menstrual cycle [24, 25].
The development of progressive spastic paraplegia is the most obvious sign of the disease. Early symptoms of the disease include clumsiness, generalized developmental delays, failure to thrive, irritability, recurrent vomiting, feeding/protein aversion, and anorexia. Growth rate remains low leading to short stature. Ataxia, which is rare and usually only intermittent, is likely linked to hyperammonemia. Most of the patients show some degree of cognitive impairment and both loss of acquired skills and severe intellectual disability may occur [20, 26, 27].
Seizures occur in more than half of the patients and usually in the absence of hyperammonemia. They are mostly generalized tonic-clonic but can include simple focal epilepsy, complex focal epilepsy, generalized tonic, and generalized absence epilepsy and even generalized tonic-clonic status epilepticus [26]. The electroencephalograms are described as diffuse slowing, compatible with metabolic encephalopathy, and epileptic graphoelements [26], with no myopathy or neurogenic process in electromyography (EMG) and normal sensory and motor nerve conduction velocities [26].
Brain imaging may reveal cerebral atrophy, ranging from mild subcortical atrophy to severe cortical and subcortical atrophy, and less frequently cerebellar atrophy. Microcephaly observed in a few patients is likely a consequence of the cerebral atrophy. White matter changes with increased T2 signal have been observed in the periventricular and more peripheral white matter regions [26, 27].
Extraneurological symptoms of the disease are rare, affecting mostly the liver and the skeletal system. As in other UCDs, the liver can be involved with a spectrum ranging from mild hepatocellular injury with transient elevation of liver transaminases to mild dysfunction with coagulation abnormalities to acute liver failure [28, 29]. Histopathological and morphological findings include swollen hepatocytes, portal and sinusoidal fibrosis, macrovacuolar steatosis, increased cellular glycogen, and dilated endoplasmic reticulum [18, 26]. Intrahepatic cholestasis can lead to neonatal presentation with jaundice, hepatomegaly, and cirrhosis [30, 31]. Spinal deformities, such as scoliosis and lordosis, may occur as a consequence of the increasing spasticity [26].
Genetics of arginase-1 deficiency
The 11.1-kb arginase gene (ARG1)-encoding ARG1 sits on chromosome 6 (6q23) comprised of eight exons. There are at least 43 potentially disease-causing variations in ARG1 with the majority (23) being missense (18)/nonsense (5) mutations and small deletions (9) [see Table 1]. The mutations are spread out fairly uniformly across the eight exons as well as at several exon-intron boundary splice sites, and the disorder is inherited in an autosomal recessive Mendelian manner [21–23, 32–49]. The incidence of ARG1 deficiency has been reported to range somewhere between approximately 1:300,000–1:2,000,000 live births, with the most complete study combining databases in the USA/Europe indicating an incidence of 1:950,000 [50]. Formerly regarded as the rarest UCD, the recent estimates [50] would place ARG1 deficiency as the third rarest of the six main UCDs (after NAGS and CPS1 deficiencies). Various case studies indicate that ARG1 deficiency is pan-ethnic with subjects being reported of Arabic (Palestinean, Saudi), Iranian, Korean, Puerto Rican, Chinese, French Canadian, Italian, Portuguese (mainland and Madeira Islands), Brazilian, Pakistani, Hispanic, Japanese, Turkish, Ashkenazi Jewish, and Caucasian descent [21–23, 32–49]. Approximately half of reported subjects are compound heterozygotes with the other half homozygous with a moderate number of cases arising from consanguineous relations.
Attempts to correlate genotype and phenotype have been carried out only to a limited extent [36] due to the rarity of the disorder. Thus, the scientific literature is mainly populated by isolated case reports and small scale studies on a distinct ethnic population [21–23, 32–48]. Patients present with symptoms at different ages in early infancy, some with very mild symptoms, others much more severe and with progression to variable degrees of mental retardation and spastic diplegia as mentioned above. In general, however, those patients with nonsense mutations have severe disease. While only a fraction of ARG1 mutations have been systematically investigated biochemically in both patient erythrocyte enzyme assays and using in vitro overexpression systems, most have been predicted to modify enzyme function based on in silico methods. ARG1 mutations may alter structure/function and/or stability of the enzyme by compromising active site residues, by introducing packing defects or by causing incorrect translation due to frameshifts [51]. Thus, these various mutations can affect the binuclear manganese cluster and influence the metal-activated hydroxide mechanism by distorting the active site or bridging residues, created steric clashes, and buried hydrophilic groups as well as by influencing the regions necessary for oligomerization (see Fig. 2 for sites of some residues that are most often mutated).
Modeling of arginase-1 deficiency in animals and new insights learned about arginase-1 deficiency disease pathogenesis
The ARG1 gene was cloned in the 1980s, and the study of the genomic structure revealed a high degree of sequence homology between humans and rodents [52, 53]. Genetically manipulated mouse models have been employed to study the pathobiologic characteristics of ARG1 deficiency [54–62] (for summary, see Table 2). Thus, the first Arg1-deficient mouse model was generated by Iyer et al. [54] using standard gene knockout techniques to study the disease mechanisms. Exon 4 was replaced with a neomycin cassette, which would eliminate critical residues for enzymatic activity. This resulted in homozygous disruption of ARG1 expression with both mRNA and enzyme activities reduced to undetectable levels. The knockout (KO) mice were smaller at birth and continued to deviate in weight and other parameters from wildtype littermates until they died approximately 2 weeks postnatally, resulting in a much more severe phenotype than that observed in human patients. This model of arginase-deficient mice termed the “juvenile lethal model” [59] exhibited hyperargininemia, hypoornithinemia, and severe hyperammonemia [54]. The latter, which causes neurologic deficits, was thought to be the cause of death. Although a 2-fold upregulation in ARG2 activity was noted, it was still insufficient to compensate for lack of liver ARG1. A rescue strategy using intraperitoneal administration of ornithine was also unsuccessful [54].
Deignan et al. [55] later created Arg1/Arg2 gene double KO mice, resulting in a model completely devoid of all arginase activity. The double KO mice shared the same phenotype as the single Arg1 gene KO mice, with death invariably occurring by 14 days of age following severe hyperargininemia, hyperammonemia, and ornithine deficiency. There was no significant difference in plasma arginine and ornithine levels between the single and double KO mice. Taken together, their findings indicated that ARG2 deficiency has no effect on the phenotype of ARG1 deficiency [55]. It was later reported that several guanidino compounds were elevated in plasma and brain tissues collected from the mouse model [56], similar to that observed in hyperargininemic patients [60]. Clearly, ammonia is not solely responsible for having undue adverse effects on the central nervous system in ARG1 deficiency. These guanidino compounds are also neurotoxins and are equally likely to cause the developing neurological sequelae in the disorder. Previous studies in the juvenile lethal model using helper-dependent adenoviral vectors expressing ARG1 showed only transient rescue by extending lifespan from 14 to 28 days [61], while adeno-associated vector (AAV) expression of ARG1 has allowed for metabolic correction lasting longer than 8 months, albeit with some lingering defects [59, 62]. Unlike the human disorder, where survival into adulthood is common, mice of the juvenile lethal model that die in the perinatal period were not amenable to further studies on somatic growth and neurological development/impairment.
To overcome this limitation, knockout technology and inducible expression systems were employed to circumvent the neonatal lethality. Using a Cre/loxP-directed conditional targeting strategy, two new ARG1-deficient mouse models that allowed spatial and temporal control of Arg1 gene deletion were generated [57, 58]. Tamoxifen-mediated inducible Arg1 KO was performed in mice of different ages (ranging from the neonatal period to adulthood) to replicate a later-onset juvenile ARG1 deficiency phenotype. Despite using different Cre reporter strains (ROSA26 vs ubiquitin C), the deletion of Arg1 at various stages results in a phenotype similar to the original juvenile lethal global knockout model [57, 58]. However, different tamoxifen administration regimens attained different degrees of Cre-mediated recombination efficiency. Apparently, a regimen of five daily injections of 1 mg tamoxifen resulted in more substantial Arg1 gene disruption than did a single oral dose of 4 mg tamoxifen, as demonstrated by a consistent near complete knockout of Arg1 [57]. Thus, tamoxifen-induced excision of floxed exons 7 and 8 of Arg1 resulted in significant loss of ARG1 at both mRNA and protein expression levels, especially in liver. The mice exhibited several hallmark presentations of ARG1 deficiency, such as impaired hepatic arginase activity and profound hyperargininemia accompanied by hyperammonemia, prior to the humane euthanization end point. The symptoms presented in these KO mice were consistent with perturbation of the urea cycle. Coincidently, both research groups provided evidence that the phenotypic abnormalities in the KO mice were independent of age of the animal and were most likely attributable to the biochemical derangements of the disorder [57, 58]. Thus, in concordance with previous observations using ARG1 deficient mice [54], these inducible KO mice also exhibited significant alterations in plasma metabolic profiles, including proline, citrulline, alanine, glycine, serine, isoleucine, and guanidino compounds. Interestingly, amino acids such as alanine, glycine, proline, and serine, which are involved in the incorporation of ammonia nitrogen, are conversely reduced, hence suggesting an alternative ammonia-scavenging pathway [57].
In addition, progressive decline in animal body weight was detected in both inducible knockout models [57, 58]. Despite tamoxifen being known to cause alterations in gastric physiology [63], Sin et al. [57] showed that the substantial weight loss is unrelated to tamoxifen administration. According to the aminostatic hypothesis [64], it is possible that the abnormally high level of arginine triggers a satiety mechanism in the brain, resulting in a waning of appetite. Elevated levels of ammonia may also suppress food intake through the effect of insulin, a potent anorexigenic hormone [65]. Consequently, prolonged reduced food intake affects amino acid homeostasis, which may lead to undernutrition and eventually become life-threatening. However, the molecular mechanisms causing these weight differences remain to be elucidated.
There was some discrepancy between the two inducible models related to the expression of renal ARG2. As previously reported, a compensatory increase in ARG2 activity could be triggered to assuage symptoms of ARG1-deficient patients [19]. Although the exact mechanism is not clearly understood, it was hypothesized that the elevated expression of this second form of arginase could mitigate the severity of ARG1 deficiency via residual ureagenesis [19]. However, Sin et al. [57] found no evidence for renal ARG2 compensation, although Kasten et al. [58] reported a slight increment in renal expression, yet this failed to extend the lifespan of their KO mice. While ornithine was supplemented in an attempt to rescue the lethality of induced Arg1 deficiency, there was an absence of any phenotypic improvement despite showing elevated levels of ornithine in the blood [57, 66]. Other commonly used treatments for ARG1 deficiency, such as low-protein diet and administration of a nitrogen-scavenging drug, sodium phenylbutyrate, were also ineffective in alleviating the biochemical consequences in the induced KO mice [66]. These paradoxical observations indicate that compensatory responses in ARG1 deficiency are different between mice and humans.
Since there are so many uncertainties regarding the development of disease in ARG1 deficiency, it will be important to develop tissue-selective knockout mice. For instance, comparing liver-selective versus neuron-selective ARG1 knockout mice might aid in determining the steps of developing neurological symptoms; i.e., is it liver-derived circulating metabolites affecting the brain or metabolites derived within neurons that initiate neuropathogenesis?
Therapeutic strategies for arginase-1 deficiency
Reconstitution of enzyme function represents the only causal treatment but has not been successful in ARG1 deficiency. The attempt to replace the deficient enzyme by administration of packed red blood cells (which contain ARG1) led to a small immediate decrease of serum arginine but no significant clinical change [67, 68]. Erythrocyte exchange transfusion in addition to low-protein diet, an essential amino acid mixture, and sodium benzoate in one patient resulted in normalization of ammonia levels and serum arginine concentrations, but cerebrospinal fluid (CSF) concentrations of arginine remained unchanged [69]. Pegylated human recombinant ARG1, developed for treatment of liver cancer, has been tested in ARG1-deficient mice where it seems to normalize plasma and brain arginine levels but not in the liver and fails to rescue Arg1 KO mice from the lethal phenotype [70]. Liver transplantation in humans “cures” the disease in the liver but not in other organs including the brain, although it may normalize ammonia and arginine in blood [71, 72]. Extrahepatic ARG1 expression may be one explanation for incomplete phenotype reversal. An early attempt at “gene therapy” in three patients by intravenous injection of the Shope papilloma virus, which induces a viral-encoded arginase, was unsuccessful [73].
Management and treatment of ARG1, in general, is the same as for the other UCDs with the exception being that there is no supplementation with arginine or citrulline [74]. Diet plays the key role in the treatment of hyperargininemia since it has been shown that strict restriction of dietary protein in combination with the supplementation of an essential amino acid mixture that is free of arginine can ameliorate arginine levels in plasma and CSF. However, the response to dietary restriction is relatively poor and improvement of the clinical picture is unsatisfactory [18, 26, 75–77]. Reviewing the literature, Prasad et al. [20] found a clinical improvement in 50 %, stabilization in 25 %, and progression of the disease in 25 % of patients on treatment.
The use of nitrogen scavengers such as benzoate, phenylbutyrate, and phenylacetate, is an alternative pathway therapy for excretion of waste nitrogen via formation and excretion of hippuric acid and phenylacetylglutamine and can be used to lower plasma ammonia levels in ARG1 deficiency [3, 78, 79]. Since the removal of nitrogen via alternative pathways lowers the flux through the urea cycle and this cycle is the only synthetic pathway for arginine, nitrogen scavengers can also be used to lower the formation of arginine in ARG1 deficiency.
Treatment with the amino acid ornithine may help to replenish hepatocellular ornithine to prevent hyperammonemia [23], and it may also inhibit the formation of neurotoxic guanidino compounds through inhibition of the enzyme arginine:glycine amidinotransferase [32]. Lysine supplementation has been trialed to augment argininuria but also in the hope that lysine might compete with arginine for uptake in the brain, thus lowering brain arginine levels [67, 80, 81]. Symptomatic treatments to alleviate the consequences of spasticity progression, despite the aforementioned treatments, include injections with botulinum toxin and orthopedic surgery (i.e., tendon release procedures).
No gene therapy trials for delivery of an arginase repair construct have been attempted yet in humans. However, based on some of the promising results in the juvenile lethal and inducible ARG1-deficient mouse models using AAV delivery [59, 62, 66], it is likely only a matter of time before human trials will be initiated.
Non-urea cycle functions of arginase-1
ARG1 not only plays a vital role in urea cycle metabolism but is also known to affect a variety of other systems in ways that are not yet fully understood. Related to the cardiovascular system, there has been intense interest in L-arginine metabolic pathways since this amino acid is the substrate required for both the synthesis of vasorelaxant nitric oxide by endothelial nitric oxide synthase (eNOS) as well as for being the key substrate of ARG1 [82]. L-Arginine is found in endothelial cells at levels far exceeding the Km for eNOS, indicating that substrate is not a limiting factor in vivo. Yet, infusion studies of L-arginine often enhance vasodilatation in an eNOS-dependent manner, a phenomenon referred to as the “arginine paradox” [83]. High arginase activity is associated with cardiovascular dysfunction potentially by limiting available substrate for eNOS and by excessive ornithine that can lead to vascular structural problems [84]. While the relationship of arginase with NOS activity is complex, inhibition of arginase has been shown in small-scale human studies to improve cardiovascular health in patients with coronary artery disease, type 2 diabetes, heart failure, hypertension, or following resuscitation after cardiac arrest [84]. It should be mentioned that some of the cardiovascular actions of arginase may be mediated by ARG2 and not by ARG1.
ARG1 is also known to play a large role in the immune system and cancer through the ability of the enzyme to alter macrophage and myeloid-derived suppressor cell (MDSC) states [85]. Macrophages are generally divided into two groups—M1 macrophages that are responsible for inflammation and immunity and M2 macrophages that promote resolution of inflammation, wound healing, and aid in tumor growth, with ARG1 expression being a hallmark marker of M2 macrophage expression. Ron receptor kinase activation induces ARG1 activity [86], leading to attenuation of the M1 phenotype. An increase in tumor-associated MDSCs is a key feature of the malignancy-mediated inflammatory response and a factor leading to T cell suppression in cancer [87]. ARG1-expressing MDSCs can deplete arginine in the tumor microenvironment leading to accumulation of ornithine for polyamine synthesis in cancer cells. Thus, via its ability to deplete arginine, ARG1-expressing cells can regulate the production of nitric oxide and modulate T lymphocyte function [88].
ARG1 is also implicated in airway hyperresponsiveness in asthma and airway inflammation [89]. Chronic asthma patients have a 4.4-fold increase in ARG1 expression compared to controls, while murine models of acute airway inflammation show an 11-fold increase in expression of the enzyme [90].Very high doses of L-arginine are beneficial in murine airway hyperresponsiveness models by reducing levels of TH2 cytokines, eotaxin, TGF-β1, and ovalbumin-specific IgE [91]. Additionally, inhibition of ARG1 expression through the use of a shRNA decreases IL13-induced airway hyperresponsiveness in mice [92].
Arginase plays a key role in maintaining levels of ornithine for use in polyamine synthesis [84]. Polyamines such as putrescine, spermidine, and spermine are derived from ornithine via ornithine decarboxylase (ODC). ARG1 KO mice have increased expression of vital polyamine enzymes ornithine aminotransferase (OAT) and spermidine/spermine-N1-acetyltransferase (SSAT) in the liver; however, the levels of polyamines were highly variable and few significant trends were seen in the KO mouse tissues [93]. Conversely, overexpression of arginase in mouse macrophages leads to significant increases in putrescine and spermidine when cells are stimulated with either lipopolysaccharide or 8-Bromo-cAMP [94].
Concluding thoughts and future outlooks
Several lingering questions remain relating to the unique clinical manifestations of ARG1 deficiency among urea cycle disorders. In particular, is it excess arginine contributing to the main neurological features of the disease, or is it other toxic guanidino metabolites, diminished nitric oxide production, altered polyamine biosynthesis, or a combination of all these metabolic derangements in various regions of the brain and other tissues? The development of ARG1-deficient mouse models has revealed hyperargininemia in concert with diminished amounts of several other amino acids that affect feeding, leading to a wasting phenotype [57, 66]. It remains an enigma as to why the disorder is so much more severe in mice than in humans. Differences in extrahepatic ARG1 (and ARG2) expression patterns between humans and rodents are likely to explain these differences. Going forward, strategies that can rescue the severe phenotype of ARG1 KO mice should offer promise for similar therapeutic approaches in humans, although this remains to be seen. How much expression of liver ARG1 is necessary to restore adequate urea cycle function in ARG1 deficiency? Based on AAV delivery experiments in mice [66], it is likely that at least 15 % of normal levels are required, but this may not be enough to rescue the neurological sequelae. Adequate ARG1 expression may need to be present in precise metabolic zones of liver, in addition to proper expression in erythrocytes, immune cells [95], and various structures in the brain to adequately control pathophysiology.
Rapid advances have been achieved in modeling diseases in vitro using appropriately differentiated induced pluripotent stem cells (iPSCs) from patients, combined with gene-editing tools to create “repaired” cells that are genetically identical to the patient cells except for the gene mutation site [96, 97]. Since ARG1 deficiency is so rare and obtaining adequate, unlimited supply of tissue (e.g., liver, blood, brain) from these patients is impossible, adopting these methodologies to study ARG1 deficiency is paramount. We have developed iPSC lines from three separate patients and have initiated the process to gene-edit the cells (unpublished observations). Thus, comparisons can be carried out at the cellular, transcriptomic, proteomic, and metabolomics levels of gene-edited versus parental-mutated iPSCs that have been differentiated to hepatocyte-like cells (and/or other cell types expressing ARG1, e.g., neuronal cells). This will enable insights into biochemical regulatory pathways, besides the urea cycle, that are disrupted/augmented, as well as detection of novel metabolites in ARG1 deficiency and may also lead to cellular phenotypes that would predict pathogenesis. Moreover, strategies to introduce gene-edited hepatocyte-like cells in a patient-specific manner may eventually be feasible, if engraftment strategies can be ameliorated. Hopefully, these types of studies will lead to new insights into this disorder and lead to new therapeutic options.
References
Jackson MJ, Beaudet AL, O’Brien WE (1986) Mammalian urea cycle enzymes. Annu Rev Genet 20:431–464
Kossel A, Dakin HD (1904) Über die Arginase. Z Physiol Chem 41:321–331
Iyer RK, Jenkinson CP, Vockley JG, Kern RM, Grody WW, Cederbaum SD (1998) The human arginases and arginase deficiency. J Inherit Metab Dis 21:86–100
Waddington SN, Mosley K, Cook HT, Tam FWK, Cattell V (1998) Arginase AI is upregulated in acute immune complex-induced inflammation. Biochem Biophys Res Commun 247:84–87
Zhang C, Hein TW, Wang W, Chang C-I, Kuo L (2001) Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J 15:1264–1266
Morris SM Jr, Bhamidipati D, Kepka-Lenhart D (1997) Human type ll arginase: sequence analysis and tissue-specific expression. Gene 193:157–161
Yip MCM, Knox WE (1972) Function of arginase in lactating mammary gland. Biochem J 127:893–899
Jenkinson CP, Grody WW, Cederbaum SD (1996) Comparative properties of arginases. Comp Biochem Physiol 114B:107–132
Vockley JG, Jenkinson CP, Shukla H, Kern RM, Grody WW, Cederbaum SD (1996) Cloning and characterization of the human type II arginase gene. Genomics 38:118–123
Li H, Meininge CJ, Hawker JR Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris SM Jr, Wu G (2001) Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 280:E75–E82
Kanyo ZF, Scolnick LR, Ash DE, Christianson DW (1996) Structure of a unique binuclear manganese cluster in arginase. Nature 383:554–557
Reczkowski RS, Ash DE (1992) EPR evidence for binuclear manganese (II) centers in rat liver arginase. J Am Chem Soc 114:10992–10994
Carvajal N, Torres C, Uribe E, Salas M (1995) Interaction of arginase with metal ions: studies of the enzyme from human liver and comparison with other arginases. Comp Biochem Physiol B Biochem Mol Biol 112:153–159
Cama E, Emig FA, Ash DE, Christianson DW (2003) Structural and functional importance of first-shell metal ligands in the binuclear manganese cluster of arginase I. Biochemistry 42:7748–7758
Di Costanzo L, Pique ME, Christianson DQ (2007) Crystal structure of human arginase 1 complexed with thiosemicarbazide reveals an unusual thiocarbonyl μ-sulfide ligand in the binuclear manganese cluster. J Am Chem Soc 129:6388–6389
Terheggen HG, Lowenthal A, van Sande M, Colombo JP (1969) Argininaemia with arginase deficiency. Lancet 2:748–749
Cederbaum SD, Shaw KN, Valente M (1977) Hyperargininemia. J Pediatr 90:569–573
Cederbaum SD, Shaw KN, Spector EB, Verity MA, Snodgrass PJ, Sugarman GI (1979) Hyperargininemia with arginase deficiency. Pediatr Res 13:827–833
Grody WW, Kern RM, Klein D, Dodson AE, Wissman PB, Barsky SH, Cederbaum SD (1993) Arginase deficiency manifesting delayed clinical sequelae and induction of a kidney arginase isozyme. Hum Genet 91:1–5
Prasad AN, Breen JC, Ampola MG, Rosman NP (1997) Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. J Child Neurol 12:301–309
Scholl-Bürgi S, Sigl SB, Häberle J, Haberlandt E, Rostásy K, Ertl C, Eichinger-Öttl U, Heinz-Erian P, Karall D (2008) Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiency. J Inherit Metab Dis 31(Suppl 2):S323–S328
Schiff M, Benoist JF, Cardoso ML, Elmaleh-Bergès M, Forey P, Santiago J, Ogier de Baulny H (2009) Early-onset hyperargininaemia: a severe disorder? J Inherit Metab Dis 32(Suppl 1):S175–S178
Jain-Ghai S, Nagamani SC, Blaser S, Siriwardena K, Feigenbaum A (2011) Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported? Mol Genet Metab 104(1–2):107–111, Erratum in: (2012) Mol Genet Metab105:159
Grody WW, Chang RJ, Panagiotis NM, Matz D, Cederbaum SD (1994) Menstrual cycle and gonadal steroid effects on symptomatic hyperammonaemia of urea-cycle-based and idiopathic aetiologies. J Inherit Metab Dis 17:566–574
Boles RG, Stone ML (2006) A patient with arginase deficiency and episodic hyperammonemia successfully treated with menses cessation. Mol Genet Metab 89:390–391
De Deyn PP, Marescau B, Qureshi IA et al (1997) Hyperargininemia: a treatable inborn error of metabolism? In: De Deyn PP, Marescau B, Qureshi IA, Mori A (eds) Guanidino compounds in biology & medicine II. John Libbey & Company Ltd., London, pp 53–69
Gungor S, Akinci A, Firat AK, Tabel Y, Alkan A (2008) Neuroimaging findings in hyperargininemia. J Neuroimaging 18:457–462
Scaglia F, Lee B (2006) Clinical, biochemical, and molecular spectrum of hyperargininemia due to arginase I deficiency. Am J Med Genet C: Semin Med Genet 142:113–120
Gallagher RC, Lam C, Wong D, Cederbaum S, Sokol RJ (2014) Significant hepatic involvement in patients with ornithine transcarbamylase deficiency. J Pediatr 164:720–725
Braga AC, Vilarinho L, Ferreira E, Rocha H (1997) Hyperargininemia presenting as persistent neonatal jaundice and hepatic cirrhosis. J Pediatr Gastroenterol Nutr 24:218–221
Gomes ME, Santos SE, Vilarinho S, Saudubray JM, Vilarinho L (2010) Neonatal cholestasis: an uncommon presentation of hyperargininemia. J Inherit Metab Dis 33(Suppl 3):S503–S506
Amayreh W, Meyer U, Das AM (2014) Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev Med Child Neurol 56:1021–1024
Lee BH, Jin HY, Kim GH, Choi JH, Yoo HW (2011) Argininemia presenting with progressive spastic diplegia. Pediatr Neurol 44:218–220
Uchino T, Snyderman SE, Lambert M, Qureshi IA, Shapira SK, Sansaricq C, Smit LM, Jakobs C, Matsuda I (1995) Molecular basis of phenotypic variation in patients with argininemia. Hum Genet 96:255–260
Uchino T, Haraguchi Y, Aparicio JM, Mizutani N, Higashikawa M, Naitoh H, Mori M, Matsuda I (1992) Three novel mutations in the liver-type arginase gene in three unrelated Japanese patients with argininemia. Am J Hum Genet 51:1406–1412
Carvalho DR, Brand GD, Brum JM, Takata RI, Speck-Martins CE, Pratesi R (2012) Analysis of novel ARG1 mutations causing hyperargininemia and correlation with arginase I activity in erythrocytes. Gene 509:124–130
Wu TF, Liu YP, Li XY, Wang Q, Ding Y, Ma YY, Song JQ, Yang YL (2013) Five novel mutations in ARG1 gene in Chinese patients of argininemia. Pediatr Neuro l49:119–123
Korman SH, Gutman A, Stemmer E, Kay BS, Ben-Neriah Z, Zeigler M (2004) Prenatal diagnosis for arginase deficiency by second-trimester fetal erythrocyte arginase assay and first-trimester ARG1 mutation analysis. Prenat Diagn 24:857–860
Cardoso ML, Martins E, Vasconcelos R, Vilarinho L, Rocha J (1999) Identification of a novel R21X mutation in the liver-type arginase gene (ARG1) in four Portuguese patients with argininemia. Hum Mutat 14:355–366
Segawa Y, Matsufuji M, Itokazu N, Utsunomiya H, Watanabe Y, Yoshino M, Takashima S (2011) A long-term survival case of arginase deficiency with severe multicystic white matter and compound mutations. Brain Dev 33:45–48
Haraguchi Y, Aparicio JM, Takiguchi M, AkaboshiI YM, Mori M, Matsuda I (1990) Molecular basis of argininemia. Identification of two discrete frame-shift deletions in the liver-type arginase gene. J Clin Invest 86:347–350
Hertecant JL, Al-Gazali LI, Karuvantevida NS, Ali BR (2009) A novel mutation in ARG1 gene is responsible for arginase deficiency in an Asian family. Saudi Med J 30:1601–1603
Edwards RL, Moseley K, Watanabe Y, Wong LJ, Ottina J, Yano S (2009) Long-term neurodevelopmental effects of early detection and treatment in a 6-year-old patient with argininaemia diagnosed by newborn screening. J Inherit Metab Dis 32(Suppl 1):S197–S200
Cohen YH, Bargal R, Zeigler M, Markus-Eidlitz T, Zuri V, Zeharia A (2012) Hyperargininemia: a family with a novel mutation in an unexpected site. JIMD Rep 5:83–88
Vockley JG, Tabor DE, Kern RM, Goodman BK, Wissmann PB, Kang DS, Grody WW, Cederbaum SD (1994) Identification of mutations (D128G, H141L) in the liver arginase gene of patients with hyperargininemia. Hum Mutat 4:150–154
Vockley JG, Goodman BK, Tabor DE, Kern RM, Jenkinson CP, Grody WW, Cederbaum SD (1996) Loss of function mutations in conserved regions of the human arginase I gene. Biochem Mol Med 59:44–51
Tsang JP, Poon WL, Luk HM, Fung CW, Ching CK, Mak CM, Lam CW, Siu TS, Tam S, Wong VC (2012) Arginase deficiency with new phenotype and a novel mutation: contemporary summary. Pediatr Neurol 47:263–269
Häberle J, Koch HG (2004) Genetic approach to prenatal diagnosis in urea cycle defects. Prenat Diagn 24:378–383
Wu T, Li X, Ding Y, Liu Y, Song J, Wang Q, Li M, Qin Y, Yang Y, Zhonghua (2015) Seven patients of argininemia with spastic tetraplegia as the first and major symptom and prenatal diagnosis of two fetuses with high risk. Er Ke Za Zhi 53:425–430
Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, Kirmse B, European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD); Members of the Urea Cycle Disorders Consortium (UCDC) (2013) The incidence of urea cycle disorders. Mol Genet Metab 110:179–180
Ash DE, Scolnick LR, Kanyo ZF, Vockley JG, Cederbaum SD, Christianson DW (1998) Molecular basis of hyperargininemia: structure function consequences of mutations in human liver arginase. Mol Genet Metab 64:243–249
Dizikes GJ, Spector EB, Cederbaum SD (1986) Cloning of rat liver arginase cDNA and elucidation of regulation of arginase gene expression in H4 rat hepatoma cells. Somat Cell Mol Genet 12:375–384
Takiguchi M, Haraguchi Y, Mori M (1988) Human liver-type arginase gene: structure of the gene and analysis of the promoter region. Nucleic Acids Res 16:8789–8802
Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O’Brien WE, Yu H, Grody WW, Cederbaum SD (2002) Mouse model for human arginase deficiency. Mol Cell Biol 22:4491–4498
Deignan JL, Livesay JC, Yoo PK, Goodman SI, O’Brien WE, Iyer RK, Cederbaum SD, Grody WW (2006) Ornithine deficiency in the arginase double knockout mouse. Mol Genet Metab 89:87–96
Deignan JL, Marescau B, Livesay JC, Iyer RK, De Deyn PP, Cederbaum SD, Grody WW (2008) Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Mol Genet Metab 93:172–178
Sin YY, Ballantyne LL, Mukherjee K, St Amand T, Kyriakopoulou L, Schulze A, Funk CD (2013) Inducible arginase 1 deficiency in mice leads to hyperargininemia and altered amino acid metabolism. PLoS One 8:e80001
Kasten J, Hu C, Bhargava R, Park H, Tai D, Byrne JA, Marescau B, De Deyn PP, Schlichting L, Grody WW et al (2013) Lethal phenotype in conditional late-onset arginase 1 deficiency in the mouse. Mol Genet Metab 110:222–230
Lee EK, Hu C, Bhargava R, Rozengurt N, Stout D, Grody WW, Cederbaum SD, Lipshutz GS (2012) Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol Ther 20:1844–1851
Deignan JL, De Deyn PP, Cederbaum SD, Fuchshuber A, Roth B, Gsell W, Marescau B (2010) Guanidino compound levels in blood, cerebrospinal fluid, and post-mortem brain material of patients with argininemia. Mol Genet Metab 100(Suppl 1):S31–S36
Gau CL, Rosenblatt RA, Cerullo V, Lay FD, Dow AC, Livesay J, Brunetti-Pierri N, Lee B, Cederbaum SD, Grody WW et al (2009) Short-term correction of arginase deficiency in a neonatal murine model with a helper-dependent adenoviral vector. Mol Ther 17:1155–1163
Lee EK, Hu C, Bhargava R, Ponnusamy R, Park H, Novicoff S, Rozengurt N, Marescau B, De Deyn P, Stout D et al (2013) AAV-based gene therapy prevents neuropathology and results in normal cognitive development in the hyperargininemic mouse. Gene Ther 20:785–796
Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, Mills JC (2012) Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 142:21–24
Mellinkoff SM, Frankland M, Boyle D, Greip M (1956) Relationship between serum amino acid concentration and fluctuations in appetite. J Appl Physiol 8:535–538
Feldman JM, Lebovitz HE (1971) Ammonium ion, a modulator of insulin secretion. Am J Physiol 221:1027–1032
Ballantyne LL, Sin YY, St Amand T, Si J, Goossens S, Haenebalcke L, Haigh JJ, Kyriakopoulou L, Schulze A, Funk CD (2015) Strategies to rescue the consequences of inducible arginase-1 deficiency in mice. PLoS One 10:e0125967
Michels VV, Beaudet AL (1978) Arginase deficiency in multiple tissues in argininemia. Clin Genet 13:61–67
Sakiyama T, Nakabayashi H, Shimizu H, Kondo W, Kodama S, Kitagawa T (1984) A successful trial of enzyme replacement therapy in a case of argininemia. Tohoku J Exp Med 142:239–248
Mizutani N, Hayakawa C, Maehara M, Watanabe K (1987) Enzyme replacement therapy in a patient with hyperargininemia. Tohoku J Exp Med 151:301–307
Burrage LC, Sun Q, Elsea SH, Jiang MM, Nagamani SC, Frankel AE, Stone E, Alters SE, Johnson DE, Rowlinson SW et al. (2015) Human recombinant arginase enzyme reduces plasma arginine in mouse models of arginase deficiency. Hum Mol Genet. doi: 10.1093/hmg/ddv352.
Whitington PF, Alonso EM, Boyle JT, Molleston JP, Rosenthal P, Emond JC, Millie JM (1998) Liver transplantation for the treatment of urea cycle disorders. J Inherit Metab Dis 21(Suppl 1):112–118
Mace H, Srinivas C, Selzner M, Minkovich L (2014) Anesthetic management of a patient with arginase deficiency undergoing liver transplantation. A A Case Rep 3:85–87
Terheggen HG, Lowenthal A, Lavinha F, Colombo JP, Rogers S (1975) Unsuccessful trial of gene replacement in arginase deficiency. Z Kinderheilkd 119:1–3
Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R et al (2012) Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 7:32
Terheggen HG, Lavinha F, Colombo JP, Van SM, Lowenthal A (1972) Familial hyperargininemia. J Genet Hum 20:69–84
Cederbaum SD, Moedjono SJ, Shaw KN, Carter M, Naylor E, Walzer M (1982) Treatment of hyperargininaemia due to arginase deficiency with a chemically defined diet. J Inherit Metab Dis 5:95–99
Snyderman SE, Sansaricq C, Chen WJ, Norton PM, Phansalkar SV (1977) Argininemia. J Pediatr 90:563–568
Qureshi IA, Letarte J, Ouellet R, Lelievre M, Laberge C (1981) Ammonia metabolism in a family affected by hyperargininemia. Diabete Metab 7:5–11
Batshaw ML, MacArthur RB, Tuchman M (2001) Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr 138:S46–S54
Pardridge WM (1977) Lysine supplementation in hyperargininemia. J Pediatr 91:1032–1033
Kang SS, Wong PW, Melyn MA (1983) Hyperargininemia: effect of ornithine and lysine supplementation. J Pediatr 103:763–765
Wijnands KA, Hoeksema MA, Meesters DM, van den Akker NM, Molin DG, Briedé JJ, Ghosh M, Köhler SE, van Zandvoort MA, de Winther MP et al (2014) Arginase-1 deficiency regulates arginine concentrations and NOS2-mediated NO production during endotoxemia. PLoS One 9(1):e86135
Bode-Böger SM, Scalera F, Ignarro LJ (2007) The L-arginine paradox: importance of the L-arginine/asymmetrical dimethylarginine ratio. Pharmacol Ther 114:295–306
Caldwell RB, Toque HA, Narayanan SP, Caldwell RW (2015) Arginase: an old enzyme with new tricks. Trends Pharmacol Sci 36:395–405
Popovic PJ, Zeh HJ 3rd, Ochoa JB (2007) Arginine and immunity. J Nutr 137:1681S–1686S
Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM Jr, Hankey PA (2011) Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol 187:2181–2192
Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, Ochoa AC, Fletcher M, Velasco C, Wilk A et al (2014) Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer 134:2853–2864
Raber P, Ochoa AC, Rodríguez PC (2012) Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest 41:614–634
Maarsingh H, Zaagsma J, Meurs H (2009) Arginase: a key enzyme in the pathophysiology of allergic asthma opening novel therapeutic perspectives. Br J Pharmacol 158:652–664
North ML, Khanna N, Marsden PA, Grasemann H, Scott JA (2009) Functionally important role for arginase 1 in the airway hyperresponsiveness of asthma. Am J Physiol Lung Cell Mol Physiol 296:L911–L920
Mabalirajan U, Ahmad T, Leishangthem GD, Joseph DA, Dinda AK, Agrawal A, Ghosh B (2010) Beneficial effects of high dose of L-arginine on airway hyperresponsiveness and airway inflammation in a murine model of asthma. J Allergy Clin Immunol 125:626–635
Yang M, Rangasamy D, Matthaei KI, Frew AJ, Zimmmermann N, Mahalingam S, Webb DC, Tremethick DJ, Thompson PJ, Hogan SP et al (2006) Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. J Immunol 177:5595–5603
Deignan JL, Livesay JC, Shantz LM, Pegg AE, O’Brien WE, Iyer RK, Cederbaum SD, Grody WW (2007) Polyamine homeostasis in arginase knockout mice. Am J Physiol Cell Physiol 293:C1296–C1301
Kepka-Lenhart D, Mistry SK, Wu G, Morris SM Jr (2000) Arginase I: a limiting factor for nitric oxide and polyamine synthesis by activated macrophages? Am J Physiol Regul Integr Comp Physiol 279:R2237–R2242
Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM, Luckner C, Doschko G, Soler G, Eichmann K et al (2005) Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 105:2549–2556
Li M, Suzuki K, Kim NY, Liu GH, Izpisua Belmonte JC (2014) A cut above the rest: targeted genome editing technologies in human pluripotent stem cells. J Biol Chem 289:4594–4599
Cox DB, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21:121–131
Acknowledgments
CDF is a Tier 1 Canada Research Chair holder in Molecular, Cellular, and Physiological Medicine and kindly acknowledges the CRC program support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
A.S. is a paid consultant of Hyperion Therapeutics Inc. (Brisbane, CA, USA) and Medunik Canada (Blainville, QC, Canada). The other authors have no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Sin, Y.Y., Baron, G., Schulze, A. et al. Arginase-1 deficiency. J Mol Med 93, 1287–1296 (2015). https://doi.org/10.1007/s00109-015-1354-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-015-1354-3