
Abstract
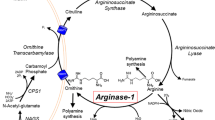
Argininemia is an autosomal recessive disorder caused by a deficiency in the liver-type arginase enzyme. Clinical manifestations include progressive spastic diplegia and mental retardation. While the quality of life can severely deteriorate in most such patients, some do show remarkable improvement in neurological symptoms while on controlled diets. We examined the thesis that differences in clinical responses to dietary treatment are based on molecular heterogeneity in mutant arginase alleles. Genomic DNAs from 11 patients with argininemia were examined using the polymerase chain reaction, cloning, and sequencing. Nine mutations representing 21/22 mutant alleles were identified in 11 patients with argininemia, and four of these mutations were expressed in vitro to determine the severity of enzymatic defects. We found that these mutations accounted for 64% of the mutant alleles in our patients. Based on findings in vitro expression tests, the mutations can be considered either severe or moderate. Patients with at least one moderate mutant allele responded well to dietary treatment; concentrations of plasma arginine were controlled within 300 μM. In contrast, patients with two severely mutated alleles did not respond to dietary treatment and plasma arginine was over 400 μM. Argininemia is heterogeneous at the molecular level. The degree of clinical improvement during dietary treatment is reflected in the concentration of arginine in plasma, as a measure of metabolic control. Plasma arginine levels during treatment is reflected in the concentration of arginine in plasma, as a measure of metabolic control. Plasma arginine levels during treatment correlated with types of molecular defects in the arginase genes.
Article PDF
Similar content being viewed by others


Avoid common mistakes on your manuscript.
References
Brockstedt M, Smit LME, Grauw AJC, Klei-van Moorssel JM, Jakobs C (1990) A new case of hyperargininaemia: neurological and biochemical findings prior to and during dietary treatment. Eur J Pediatr 149:341–343
Brusilow SW, Horwich AL (1989) Urea cycle enzymes. In: Scriver CR, Sly WS, Beaudet AL, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill. New York, pp 629–663
Haraguchi Y, Takiguchi M, Amaya Y, Kawamoto S, Matsuda I, Mori M (1987) Molecular cloning and nucleotide sequence of cDNA for human liver arginase. Proc Natl Acad Sci USA 84:412–415
Haraguchi Y, Aparicio JM, Takiguchi M, Akaboshi I, Yoshino M, Mori M, Matsuda I (1990) Molecular basis of argininemia: identification of two discrete frame-shift deletions in the livertype arginase gene. J Clin Invest 86:347–350
Ikemoto M, Tabata M, Miyake T, Kono T, Mori M, Totani M, Murachi T (1990) Expresssion of human liver arginase in Escherichia coli: purification and properties of the product. Biochem J 270:697–703
Lambert MA, Desjardins M, Desjardins M, Laberge M, Dhondt JL, Dallaire L, De Deyn PP, et al (1991) Hyperargininemia: inellectual and motor improvement related to changes in biochemical data. J Pediatr 118:420–424
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Marescau B, De Deyn PP, Lowenthal A, Qureshi IA, Antonozzi I, Bachmann C, Cederbaum SD, et al (1990) Guanidino compound analysis as a complementary diagnostic parameter for hyperargininemia: follow-up of guanidino compound levels during therapy. Pediatr Res 27:297–303
Matsuura T, Hoshide R, Setoyama C, Komaki S, Kiwaki K, Endo F, Nishikawa S, et al (1994) Expression of four mutant human ornithine transcarbamylase genes in cultured Cos 1 cells relates to clinical phenotypes. Hum Genet 93:129–134
Okano Y, Eisensmith RC, Guttler F, Lichter-Konecki U, Konecki DS, Trefz FK, Dasovigh M, et al (1991) Molecular basis of phenotypic heterogeneity in phenylketonuria. N Engl J Med 324:1232–1238
Saiki RKF, Gelfand DH, Stoffel S, Sharf SJ, Higuchi R, Horn GT, Mullis KB, et al (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487–491
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Scheuerle AE, McVie R, Beaudet AL, Shapira SK (1993) Arginase deficiency presenting as cerebral palsy. Pediatrics 91:995–996
Schimke RT (1961) Adaptive characteristics of urea cycle enzymes in the rat. J Biol Chem 237:459–468
Shapiro MB, Sneapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15:7155–7174
Snyderman SE, Sansasricq C, Chen WJ, Norton PM Phansalkar SV (1977) Argininemia. J Pediatr 90:563–568
Snyderman SE, Sansaricq C, Norton PM, Goldstein F (1979) Argininemia treated from birth. J Pediatr 92:61–63
Sparkes RS, Dizikes GJ, Klisak I, Grody WW, Mohandas T, Heinzmann C, Zollman, et al (1986) The gene for human liver arginase (ARG1) is assigned to chromosome band 6q23. Am J Hum Genet 39:186–193
Takiguchi M, Haraguchi Y, Mori M (1988) Human liver-type arginase gene: structure of the gene and analysis of the promoter region. Nucleic Acids Res 16:8789–8802
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:350–4354
Uchino T, Haraguchi Y, Aparicio JM, Mizutani N, Higashikawa M, Naitoh H, Mori M, et al (1992) Three novel mutations in the liver-type arginase gene in three unrelated Japanes patients with argininemia. Am J Hum Genet 51:1406–1412
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Uchino, T., Snyderman, S.E., Lambert, M. et al. Molecular basis of phenotypic variation in patients with argininemia. Hum Genet 96, 255–260 (1995). https://doi.org/10.1007/BF00210403
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF00210403