
Abstract
Design and anti-HIV activity of a series of arylsulphonamide derivatives as possible non-nucleoside reverse transcriptase inhibitors have been discussed. Compounds designed on the basis of Lipinski’s rule of five and having H-bond donor and acceptor sites were synthesized and screened in vitro to assess their human immunodeficiency virus type 1 reverse transcriptase inhibitory activity using TZM-bl cells. In silico studies using Discovery Studio 3.0 software showed that these molecules formed H-bonds and exhibited π–π, π–+ interactions, with amino acids in the non-nucleoside inhibitor binding pocket, and formed more stable complexes (total interaction energy in the range of (−) 47.85–(−) 77.01 kcal/mol) with human immunodeficiency virus type 1 reverse transcriptase in comparison to nevirapine and etravirine, (−) 45.79 and (−) 61.43 kcal/mol, respectively, and thus, lower EC50 values were predicted. The molecule, 4-(4-chloro-benzenesulphonyl amino)-N-(1H-indazole-5-yl)-benzamide showed significant inhibition of human immunodeficiency virus type 1 growth under in vitro conditions with EC50 value in the range of 4.89 × 10−5 μm. However, its selectivity index value was 2.45 only, which was much lower than nevirapine and etravirine. The reverse transcriptase analysis, based on luciferase reporter assay, of this compound proved its nature as non-nucleoside reverse transcriptase inhibitors. Though the molecules showed low inhibitory action against human immunodeficiency virus type 1 under in vitro conditions, the idea to develop non-nucleoside reverse transcriptase inhibitors was vindicated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) is one of the most attractive targets in the chemotherapy of AIDS patients and HIV-1 RT inhibitors, thus, play a vital role in HIV/AIDS effective therapy in vogue—the highly active antiretroviral therapy (HAART). It is noteworthy that HIV-1 RT is essential for viral replication as it is responsible for conversion of its genomic ssRNA into dsDNA—the provirus, which initiates the viral replication process (Kumari and Singh, 2013). HIV-1 RT has a flexible structure and contains two known sites where drugs can bind: first is the active binding site, characterized by its catalytic triad, Asp110, Asp185, Asp186, where deoxynucleoside triphosphates bind in the normal procedure, and the second is an allosteric site or non-nucleoside inhibitor binding pocket (NNIBP), characterized by hydrophobic amino acid residues, and located about 10 Å away from the catalytic or active site (Jonckheere et al., 2000; Singh et al., 2010).
The NNIBP is hydrophobic in nature and contains primarily amino acid residues from the loop β5 (Pro95, Leu100, Lys101, and Lys103), β6 (Val106 and Val108), the β9–β10 hairpin (Val179, Tyr181, Tyr188, and Gly190), and the β12–β13 hairpin (Phe227, Trp229, Leu234, His235, and Pro236) of the p66 palm sub domain and the β15 (Tyr318) of the p66 thumb sub domain, as well as the β7–β8 connecting loop (Gly138) of the p51 fingers sub domain. Most of the amino acid residues that form the binding pocket are hydrophobic (Pro59, Leu100, Val106, Val179, Leu234, and Pro236) and five of them are aromatic residues (Tyr181, Tyr188, Phe227, Trp229, and Tyr232). Some hydrophilic residues, like Lys101, Lys103, Ser105, Asp132, and Glu224 are also present. The binding of non-nucleoside reverse transcriptase inhibitors (NNRTIs) results in a concomitant shift of the β4–β7–β8 sheet and the three catalytic aspartic acid residues cause conformational changes in HIV-1 RT. These changes form the basis of inhibitory action of NNRTIs (Tronchet and Seman, 2003; Zhou et al., 2006). The NNRTIs include a variety of conformationally restrained two-ring and three-ring structures with varying degree of flexibility and some common underlying chemistry and interact at allosteric site on HIV-1 RT. NNRTIs are specific only for HIV-1 RT because of the presence of a flexible hydrophobic pocket in which NNRTIs can fit (Martins et al., 2008; Singh et al., 2015).
Most of the NNRTIs adopt a “butterfly” conformation with a central lipophilic domain (body) flanked by two hydrophobic (normally a benzene ring and an extended π system) moieties (wings). The hydrophobic wings interfere efficiently with functional aromatic amino acid residues within the binding pocket (Tyr181, Tyr188, Trp229, and Try318). Apart from stacking interactions, electrostatic interactions (with Lys101, Lys103, and Gly138), van der Waals (vdW) interactions (with Leu100, Val106, Val179, Tyr181, Gly190, Trp229, Leu234, and Trp318), and hydrogen bonding between NNRTIs and the main chain peptide bonds contribute to the interaction and binding efficacies of the drugs with the NNRTI-specific pocket. The hydrophobic interactions of the side chains of the residues, Tyr181, Tyr188, and Trp229, with the hydrophobic moieties of the NNRTIs appear to be important for inhibitor binding. Since most of the NNRTIs contain polar groups, they have the potential to form hydrogen bonds with surrounding amino acid residues either directly or via water bridges (Kumari and Singh, 2013; Zhan et al., 2013).
The recently developed second generation NNRTIs shows a “U” (or “horse-shoe”) conformation, which gives an increased plasticity to these derivatives and NNIBP and this appears to be a deciding factor for potency against wild type (WT) and mutant HIV-1 RTs. The latest approved NNRTIs, etravirine and rilpivirine and another compound, dapivirine, under clinical investigation in phase I/II clinical trials are success stories of such an approach (Wang et al., 2014).
The overall structure of HIV-1 RT has segmental flexibility that also varies according to the nature of the bound NNRTI. The NNIBP is elastic and its conformation depends on the size, specific chemical structure, and binding mode of the NNRTI. Limits to the pocket’s flexibility are not fully understood and this makes accurate predictions of the structure of RT-NNRTI complexes very challenging with the available molecular modeling techniques. Molecular modeling analyses of the RT-NNRTI complexes suggest that potent inhibitors tend to bind in the NNIBP in low energy conformations. The analyses suggest that inhibitors with selected conformational degrees of freedom, such as torsional flexibility around strategically located chemical bonds, can compensate for the effects of drug-resistance mutations. Using inhibitor flexibility in designing drugs to overcome the effects of resistant mutations has broader implications for diseases where drug resistance is a primary concern (Zhan et al., 2016).
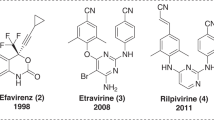
Unique antiviral activity, high specificity and low toxicity of NNRTIs make them attractive ingredient of HAART. Currently, there are five FDA approved drugs in this class (Fig. 1), namely nevirapine, efavirenz, delaviridine, etravirine, and rilpivirine, which effectively inhibit WT HIV, but are less effective against clinically important RT mutant viruses having mutation at Tyr188, Tyr181, and Lys103 amino acid residues. Therefore, there is an urgent need to develop novel, highly potent NNRTIs with broad spectrum of antiviral activity. We, therefore, focused our attention on the design and synthesis of some possible NNRTIs. The synthetic pathway was planned in a way to accommodate well documented pharmacophoric moieties of interest, such as, amide, aryl and heteroaryl moieties that proved to contribute to anti-HIV (Kumari and Singh, 2012; Kumari et al., 2011; Singh and Singh, 2011; Esposito et al., 2012).

a First generation NNRTIs and b second generation NNRTIs are FDA approved NNRTIs and c Designed prototype
Experimental section
Design of NNRTIs
We have analyzed some physically significant descriptors and pharmaceutically relevant properties of compounds, like molecular weight, H-bond donors, H-bond acceptors, log P (octanol/water), on the basis of Lipinski’s rule of five, using Molinspiration and ChemDraw softwares.
Chemistry
Chemicals were obtained from E. Merck India Ltd., India and Sigma-Aldrich Chemical Company, USA. Melting points (m.p.) determined on electro thermal apparatus are uncorrected. Silica gel for TLC was obtained from E. Merck India Ltd. and spots were identified by iodine and UV lamp exposure. UV measurements were carried out on Lambda 25 spectrophotometer. 1H nuclear magnetic resonance (NMR) spectra and 13CMR were recorded on DRX 400 MHz instrument using CDCl3 as solvent and tetra methyl silane as an internal standard. Mass spectra were obtained using a Thermofinnigan TRACE-DSQ Electro Spray Ionization mass spectrometer. Elemental analyses were carried out on a Perkin-Elmer 240-C analyzer. All solvents were dried and distilled prior to use.
4-(4-Chloro-benzenesulphonylamino)-benzoic acid (2)
To an aqueous solution of 4-amino benzoic acid 1 (3.0 g, 21.8 mmol) maintained at pH 9 with aqueous sodium bicarbonate solution, 4-chloro benzenesulphonyl chloride (4.617 g, 21.8 mmol) was added. Contents were stirred at room temperature until the complete consumption of the sulphonyl chloride (as indicated by TLC). The medium of the reaction mixture was changed to pH 1 using hydrochloric acid (1 M) and the precipitate obtained was filtered, washed with water and dried. The resulting solid was recrystallized from ethanol to get white solid (96 %); m.p. 105 °C, R f 0.6 (DCM/MeOH, 9.0:1.0); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 7.92–7.86 (m, 2H, Ar–H), 7.72–7.66 (m, 2H, Ar–H), 7.64–7.58 (m, 2H, Ar–H), 7.01–6.94 (m, 2H, Ar–H), 5.76 (s, 1H, −NH).
4-(4-Chloro-benzenesulphonylamino)-benzoyl chloride (3)
To compound 2 (1 mmol) dissolved in dry acetonitrile and a small amount of pyridine, oxalyl chloride (1.2 mmol) was added drop wise in cold and anhydrous conditions. Reaction mixture was stirred at 25 °C for 1 h and the solvent removed in vacuo. The residual oil was dissolved in dry pyridine and used directly in the next step without further purification.
General procedure for synthesis of title compounds
To a stirred solution of 3 (1 mmol) in dry pyridine (3 mL) maintained in an ice bath, added drop wise amine derivative (1 mmol) dissolved in dry acetonitrile (4 mL) and pyridine (1 mL), and stirred the reaction mixture at room temperature. After completion of the reaction, the solvent was evaporated under pressure and the residual mass dissolved in ethyl acetate (30 mL). This organic fraction was washed consecutively with 5 % NaHCO3 solution (20 mL), NaCl (20 mL), H2O (20 mL), dried on anhydrous Na2SO4, filtered and reduced under vacuum (40–50 °C). The title compound was purified by recrystallization from ethanol: water.
4-(4-Chloro-benzenesulphonylamino)-N-pyridin-2-yl-benzamide (4)
White solid (60 %); m.p. 130 °C; R f 0.48 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 8.96 (s, 1H, –CONH–), 8.87 (dd, J = 8.1, 1.0 Hz, 1H, C3–H pyridine), 7.90 (td, J = 8.0, 1.2 Hz, 1H, C4–H pyridine), 7.33 (ddd, J = 7.9, 4.9, 0.9 Hz, 1H, C5-H pyridine), 8.69 (dd, J = 5.0, 1.3 Hz, 1H, C6-H pyridine), 7.72–7.66 (m, 2H, Ar–H), 7.64–7.58 (m, 2H, Ar–H), 7.54–7.48 (m, 2H, Ar–H), 7.09–7.03 (m, 2H, Ar–H), 6.17 (s, 1H, –NH); 13C NMR (CDCl3, 100 MHz): δ = 167.17 (C-1), 151.60 (C-10), 148.28 (C-14), 140.99 (C-23), 139.32 (C-12, C-20), 138.32 (C-21, C-25), 137.24 (C-5), 130.15 (C-22, C-24), 128.44 (C-3, C-7), 122.30 (C-4, C-6), 121.95 (C-2), 117.96 (C-13), 114.40 (C-11). UV (EtOH): λmax 213 nm; MS (EI+, 70 eV): m/z (%): 387 (M+); anal. calcd. for C18H14ClN3O3,S: C 55.74, H 3.64, Cl 9.14, N 10.83, O 12.381, S 8.27; found; C 56, H 3, Cl 9, N 10.1, S 8.
N-(5-Bromo-pyridin-2-yl)-4-(4-chloro benzenesulphonylamino)-benzamide (5)
Light brown solid (83 %); m.p. 147 °C; R f 0.48 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 9.10 (s, 1H, –CONH–), 7.36 (d, J = 8.1 Hz, 1H, C3-H pyridine), 7.76 (dd, J = 8.0, 1.2 Hz, 1H, C4-H pyridine), 8.49 (d, J = 1.3 Hz, 1H, C6-H pyridine), 7.72–7.66 (m, 2H, Ar–H), 7.64–7.58 (m, 2H, Ar–H), 7.52–7.45 (m, 2H, Ar–H), 7.09–7.03 (m, 2H, Ar–H), 6.16 (s, 1H, –NH); 13C NMR (CDCl3, 100 MHz): δ = 167.17 (C-1), 150.96 (C-21), 149.34 (C-21), 140.99 (C-17), 140.38 (C-23), 139.32 (C-14), 137.24 (C-5), 133.95 (C-15, C-19), 130.15 (C-16, C-18), 128.44 (C-3, C-7), 122.30 (C-4, C-6), 121.95 (C-2), 118.47 (C-22), 114.61(C-24); UV (EtOH): λmax 345 nm; MS (EI+, 70 eV): m/z (%): 447 (M+); anal. calc. for C18H14BrClN3O3,S: C 46.32, H 2.81, Br 17.12, Cl 7.60, N 9.00, S 6.87; found; C 46, H 2.75, Cl 7.5, Br 17, N 8.9, S 6.5.
4-(4-Chloro-benzenesulphonylamino)-N-(5-nitro-pyridin-2-yl)-benzamide (6)
Light yellow solid (56 %); m.p. 138–139 °C; R f 0.45 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 7.64 (d, J = 8.1 Hz, 1H, C3-H pyridine), 8.49 (dd, J = 8.0, 1.2 Hz, 1H, C4-H pyridine), 9.40 (d, J = 1.3 Hz, 1H, C6-H pyridine), 9.31 (s, 1H, –CONH), 7.72–7.66 (m, 2H, Ar–H), 7.52–7.45 (m, 2H, Ar–H), 7.09–7.03 (m, 2H, Ar–H), 6.18 (s, 1H, –NH); 13C NMR (CDCl3, 100 MHz): δ = 167.17 (C-1), 153.48 (C-10), 149.59 (C-14), 140.99 (C-26), 139.32 (C-23), 137.24 (C-5), 133.95 (C-24, C-28), 133.34 (C-12), 133.13 (C-13), 130.15 (C-25, C-27), 128.44 (C-3, C-7), 122.30 (C-4, C-6), 121.94 (C-2), 112 (C-11); UV (EtOH): λmax 382 nm; MS (EI+, 70 eV): m/z (%): 433 (M+); anal. calcd. for C18H14ClN4O5,S: C 49.83, H 3.25, Cl 8.17, N 12.91, S 7.39; found; C 50, H 3.00, Cl 8.18, N 12.8, S 7.35.
4-(4-Chloro-benzenesulphonylamino)-N-(6-ethoxy-benzothiazol-2-yl)-benzamide (7)
Light brown solid (76 %); m.p. 257 °C; R f 0.48 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 7.72–7.57 (m, 2H, Ar–H), 7.54–7.47 (m, 2H, Ar–H), 7.09–7.03 (m, 3H, Ar–H), 6.07 (s, 1H, –NH), 4.11 (q, J = 5.9 Hz, 2H, –CH2), 1.34 (d, J = 11.8 Hz, 3H, –CH3); 13C NMR (CDCl3, 100 MHz): δ = 167.84 (C-1), 156.28 (C-11), 153.50 (C-17), 150.11(C-13), 140.99 (C-29), 139.32 (C-26), 137.24 (C-5), 133.95 (C-27, C-31), 132.83 (C-14), 130.15 (C-28, C-30), 128.44 (C-3, C-7), 128.19 (C-2), 122.30 (C-4, C-6), 118.42 (C-15), 113.35 (C-16), 106.93 (C-18), 63.77 (C-20), 14.71 (C-21); UV (EtOH): λmax 208 nm, 255 nm; MS (EI, 70 eV): m/z (%): 487.04(M+), 489(41 %); anal. calcd. for: C22H18ClN3O4,S2: C 54.15, H 3.72, Cl 7.27, N 8.61; found; C 54, H 3.8, Cl 7.00, N 8.5, S 13.02.
3-(4-Chloro-benzenesulphonylamino)-N-(6-chloro-benzothiazol-2-yl)-benzamide (8)
Brown crystals (66 %); m.p. 265 °C; R f = 0.47 (DCM/MeOH, 9.9:0.1); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 8.10 (d, J = 1.5 Hz, 1H, Ar–H), 7.86 (d, J = 7.5 Hz, 1H, Ar–H), 7.72–7.66 (m, 2H, Ar–H), 7.64–7.58 (m, 2H, Ar–H), 7.54 (dd, J = 7.5, 1.5 Hz, 1H, Ar–H), 7.48 (dt, J = 7.4, 2.1 Hz, 1H, Ar–H), 7.38–7.27 (m, 2H, Ar–H), 7.14 (t, J = 2.0 Hz, 1H, Ar–H), 6.01 (s, 1H, –NHSO2); 13C NMR (CDCl3, 100 MHz): δ = 169.01 (C-1), 156.28 (C-11), 151.65 (C-13), 140.99 (C-27), 140.36 (C-4), 139.32 (C-24), 133.95 (C-2), 132.69 (C-25, C-29), 130.15 (C-14), 128.47 (C-26, C-28), 125.83 (C-6), 125.30 (C-7), 124.65 (C-16), 120.21 (C-17), 120.18 (C-5, C-18), 120.06 (C-3), 118.46 (C-15); UV (EtOH): λmax 210 nm; MS (EI, 70 eV): m/z (%): 476.94(M+); anal. calcd. for: C20H13Cl2N3O3,S2: C 50.21, H 2.74, Cl 14.82, N 8.78; found; C 50, H 2.5, Cl 14.70, N 8.6, S 13.02.
N-(6-Bromo-benzothiazol-2-yl)3-(4-Chloro-benzenesulphonylamino)-benzamide (9)
Dark brown solid (69 %); m.p. 279 °C; R f = 0.49 (DCM/MeOH, 9.9:0.1); 1H NMR (400 MHz, Chloroform-d): δ (ppm): δ = 7.53–7.81 ppm (m, 4H), 4.1 (s, 1H, Ar–NH), 6.5–7.4 (m, 4H, Ar), 7.86 (s, 1H, NH), 7.7–8.2 (m, 3H, Ar); 13C NMR (CDCl3, 100 MHz): δ = 169.01 (C-1), 156.28 (C-22), 152.88 (C-24), 140.99 (C-17), 140.36 (C-4), 139.32 (C-14), 133.95 (C-2, C-15, C-19), 132.26 (C-25), 130.15 (C-16, C-18), 128.47 (C-6), 128.10 (C-27), 125.83 (C-7), 123.09 (C-29), 120.19 (C-3, C-5), 118.86 (C-26), 112.03 (C-28); UV (EtOH) λmax 216 nm; MS (EI, 70 eV): m/z (%): 522.92(M+); anal. calcd. for: C20H13BrN3O3,S2: C 45.95, H 2.51, Br 15.28, Cl 6.78, N 8.04 S 12.27; found; C 45, H 2.3, Br 15.21,Cl 6.73, N 8.0, S 12.02.
4-(4-Chloro-benzenesulphonylamino)-N-(1H-indazole-5-yl)-benzamide (10)
Dark brown (70 %); m.p. 238 °C; R f = 0.47 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d) δ 9.19 (s, 1H, –CONH–), 9.04 (s, 1H, –NH, indazole), 8.32 (d, J = 1.5 Hz, 1H, C-3 indazole), 7.83 (dd, J = 7.4, 1.6 Hz, 1H, C-6 indazole), 7.72–7.66 (m, 2H, Ar–H), 7.64–7.53 (m, 6H, Ar–H), 7.09–7.03 (m, 2H, Ar–H), 6.17 (s, 1H); 13C NMR (CDCl3, 100 MHz): δ = 165.98 (C-1), 140.99 (C-26), 139.32 (C-23), 137.24 (C-5), 135.40 (C-18), 133.95(C-24, C-28) , 132.08 (C-12), 130.15 (C-15, C-25, C-28), 128.44 (C-3, C-7), 122.30 (C-16, C-13), 121.39 (C-4, C-6), 115.21 (C-2), 110.31 (C-14, C-17); UV (EtOH): λmax 245 nm; MS (EI, 70 eV): m/z (%): 426 (M+); anal. calcd. for: C20H15ClN4O3,S: C 56.27, H 3.54, Cl 8.31, N 13.12, S 7.51; found; C 55.5, H 3.55, Cl 8.28, N 13.8, S 7.33.
N-[(4-Benzoimidazole-1-carbonyl)-phenyl]-4-chloro-benzenesulphonamide (11)
Creamish white crystals (74 %); m.p. 188 °C; R f 0.46 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d) δ 8.74 (s, 1H, C-2 benzimidazole), 7.72–7.65 (m, 4H, benzimidazole), 7.64–7.58 (m, 2H, Ar–H), 7.57–7.51 (m, 2H, Ar–H), 7.15–7.08 (m, 2H), 6.21 (s, 1H, –NHSO2); 13C NMR (CDCl3, 100 MHz): δ = 166.41 (C-18), 142.62 (C-20), 140.99 (C-4), 139.32 (C-1), 137.31 (C-23), 136 (C-22, C-12), 133.95(C-2, C-6), 131.43 (C-15), 130.15 (C-3, C-5), 128.40 (C-14, C-16), 124.30 (C-27), 121.75 (C-26), 119.95 (C-1), 114.82 (C-13,C-17); UV (EtOH): λmax 235 nm; MS (EI, 70 eV): m/z (%): 411 (M+), 413 (37 %); anal. calcd. for: C20H14ClN3O3,S: C 58.32, H 3.43, Cl 8.61, N 10.20, S 7.79; found; C 58.0, H 3.44, Cl 8.5, N 10.8, S 7.6.
4-Chloro N-[4-(4-nitro-benzoimidazole-1-carbonyl)-phenyl]-benzenesulphonamid (12)
Cream solid (68 %); m.p. 196 °C; R f 0.47 (DCM/MeOH, 9.5:0.5); 1H NMR (400 MHz, Chloroform-d) δ 8.79 (s, 1H, C-2 benzimidazole), 8.59 (dd, J = 7.5, 1.5 Hz, 1H, C-7 benzimidazole), 7.94 (dd, J = 7.5, 1.5 Hz, 1H, C-5 benzimidazole), 7.52 (t, J = 7.5 Hz, 1H, C-6 benzimidazole), 7.72–7.66 (m, 2H, Ar–H), 7.64–7.57 (m, 4H, Ar–H), 6.97–6.91 (m, 2H, Ar–H), 5.97 (s, 1H, –NHSO2); 13C NMR (CDCl3, 100 MHz): δ = 166.41 (C-17), 142.32 (C-24), 141.49 (C-19), 140.99 (C-4), 139.32 (C-1), 137.35 (C-22), 136.40 (C-11), 136.15 (C-2, C-6), 133.95 (C-14), 131.43 (C-3, C-5), 130.15 (C-13, C-15), 128.40 (C-26), 126.72 (C-25), 123.45 (C-12), 121.75 (C-16), 116.03 (C-27); UV (EtOH): λmax 247 nm; MS (EI, 70 eV): m/z (%): 456 (M+), 458 (37 %); anal. calcd. for: C20H13ClN4O5,S: C 52.57, H 2.87, Cl 7.76, N 12.26, S 7.02; found; C 52.5, H 2.55, Cl 7.28, N 12.8, S 7.3.
4-(4-Chloro-benzenesulphonylamino)-N-napthalen-2-yl-benzamide (13)
Brown solid (70 %); m.p. 246 °C; R f 0.35 (DCM/MeOH, 9.9:0.1); 1H NMR (400 MHz, Chloroform-d): δ (ppm): 9.28 (s, 1H, –CONH–), 7.74–7.54 (m, 7H, Ar–H), 7.40–7.31 (m, 2H, Ar–H), 7.09–7.03 (m, 2H, Ar–H), 6.19 (s, 1H, –NH); 13C NMR (CDCl3, 100 MHz): δ = 165.98 (C-1), 140.99 (C-27), 139.32 (C-24), 137.24 (C-5), 134.69 (C-19), 133.95 (C-25, C-29), 133.61 (C-11), 130.15 (C-26, C-28), 129.44 (C-13), 129.20 (C-14), 128.44 (C-3, C-7), 128.07 (C-15), 126.45 (C-18), 126.12 (C-16, C-17), 122.30 (C-4, C-6), 121.39 (C-2), 121.05 (C-12); UV (EtOH): λmax 250 nm; MS (EI, 70 eV): m/z (%): 436 (M+); anal. calcd. for: C23H17ClN2O3S: C 63.23, H 3.92, Cl 8.11, N 6.41; found; C 63.20, H 3.85, Cl 8, N 6.5, S 7.21.
Molecular modeling analysis
The structure retrieved from the Protein Data Bank (www.rcsb.org) with entry code (PDB: ID 3MEC), has been utilized for modeling. The protein was prepared, docked, scored, and the molecular dynamics (MD) simulation carried out using standard procedures. All computational studies were done using Discovery Studio 3.0 (DS 3.0, Accelrys Ltd., UK).
Receptor set up
The target protein (PDB: ID 3MEC) was taken, ligand extracted, missing hydrogens added, and their positions optimized using the all-atom CHARMm forcefield and the Adopted Basis set Newton–Raphson (ABNR) method available in DS 3.0 protocol until the root mean square (r.m.s.) gradient was less than 0.05 kcal/mol/Å (Brooks et al., 1983). The minimized protein was defined as the “receptor” using the binding site module of DS 3.0. The binding site was defined from the volume of ligand method, which was modified to accommodate all important interacting residues in the active site of HIV-1 RT. The Input Site Sphere was defined over the binding site, with a radius of 5 Å from the center of the binding site. The protein thus characterized was taken as the target receptor for the docking procedure. Minimization was performed to relax these newly added hydrogen atoms by fixing all other non-hydrogen atoms. The minimized structure of the protein was used in docking simulations.
Ligand set up
In order to propose new inhibitors, build-and-edit module of DS 3.0 was used. A series of desired arylsulphonamides, Scheme 1, having substitution at C1 positions of para amino benzoic acid nucleus through amide linkage with different heterocyclic amines was built, all-atom CHARMm forcefield was assigned and then minimized using the ABNR method. A conformational search of inhibitors was carried out using a simulated annealing MD approach. The ligand was heated to a temperature of 700 K and then annealed to 200 K. Thirty such cycles were carried out. The conformation obtained at the end of each cycle was further subjected to local energy minimization, using Smart Minimizer algorithm with 2000 steps and r.m.s. gradient of 0.001 kcal/mol/Å with the help of ABNR method as described above. The 30 energy-minimized structures were then superimposed and the lowest energy conformation occurring in the major cluster was taken to be the most probable conformation.

Reagents and conditions: a water, sodium bicarbonate (1 mol), room temperature, 4 h, HCl (1 mol); b CH3CN and pyridine (3:1), oxalyl chloride, room temperature, 1 h; c appropriate amine, pyridine, triethyl amine, overnight stirring
Docking and scoring
Molecular docking is a computational method used to predict the binding of the ligand to the receptor binding site by varying position and conformation of the ligand keeping the receptor rigid. Binding mode of inhibitors can be explored by using the LigandFit docking protocol (Venkatachalam et al., 2003). LigandFit algorithm performed a shape comparison filter with a Monte Carlo conformational search (bond lengths and bond angles fixed and only the rotatable bonds were allowed to rotate freely) to generate docked poses parallel to the shape of the binding site. Dreiding forcefield and grid-based calculated interaction energy, were used to refine these poses by rigid body minimization (Hamdouchi et al., 2005). The receptor protein was fixed during docking. The docked poses were further minimized by using all-atom CHARMm forcefield and smart minimizer algorithm until the r.m.s gradient for potential energy was less than 0.001 kcal mol−1 Å−1 and evaluated with a set of scoring functions. The description of ligand scoring (-PLP1, -PLP2, -PMF, Lig_Internal_Energy, Binding Energy, and Dock Score) have already been discussed in “Result and discussion” section. The atoms of ligand and the side chains of the residues of the receptor within 5 Å from the center of the binding site were kept flexible during minimization. The LUDI III score was used to score the refined poses. The ligand pose, which corresponded to the highest LUDI III score was taken as the best docked pose. Furthermore, determination of binding energy to assess the binding affinity of ligands for receptor was calculated by employing highest stable ligand-receptor complex through the protocol “Calculate Binding Energies” within DS 3.0 using the default settings (Bohm, 1994a, b, 1998; Wang et al., 2003; Prathipati and Saxena, 2006).
Validation of the docking methodology
Docking was first tested on known inhibitor—etravirine, which was docked in the allosteric binding site of HIV-1 RT, after extracting the ligand from the crystal structure and then the molecules under study were docked with HIV-1 RT (Table 2). Results were in good agreement with the observed values of etravirine. The root mean square deviation (RMSD) of the best docked poses of the molecules reported herein was less than 2 Å, which was supposed to be an appropriate condition for developing NNRTIs. Two structures overlapped very well with positional RMSD of 1.2. The docked pose having the highest LUDI III score gave the least RMSD with respect to the crystal conformation for both the ligands. The analysis of the results showed a sound validation of the docking and scoring methodology used in this study and the ligand as a whole moved into a more stable position with a much lower docked energy.
MD simulation
We performed MD simulation of the bound and free state for compound 10 using the DS 3.0 software in CHARMm force field to study the relationship between biological activity and energetic properties. First, the compound was solvated in an orthorhombic solvent model with explicit spherical boundary with harmonic restraint solvation condition, which was used as input for standard dynamics cascade protocol.
The Standard Dynamics Cascade includes two stages of energy minimization followed by three stages of dynamics, including heating, equilibration, and production. The structures were firstly energy relaxed with 1000 steps of steepest-descent energy minimization followed by another 1000 steps of conjugate gradient energy minimization. The RMS gradients 0.1 and 0.001 kcal/mol/Å were used for the steepest descent (SD) and conjugate gradient algorithms, respectively. An initial minimization stage, typically using the robust SD algorithm to resolve any initial poor contacts within the system without creating large distortions in the overall structure and a second minimization stage, typically using the ABNR method was adopted. A MD simulation heating stage is then employed to add thermal energy to the system to reach a target temperature. The heating dynamics started from 50 K and gradually increased to reach the target temperature of 300 K in 50,000 steps with a time step of 1 fs. Then the structures were forced to a 1 ns equilibration MD run followed by a 5 ns production MD run. The purpose of the equilibration stage is to ensure that the energy in the system is distributed appropriately among all degrees of freedom. This allows the system to achieve thermal equilibration at the target temperature. A NPT stimulation condition (using a leap-frog Verlet integration algorithm) with 2 fs time step and 310 K temperature was used in the MD run. The total energy of the stimulated systems turned out to be stable after 5 ns MD stimulation. The results of the production stage are stored in the simulation trajectory, from which structural and energetics properties can be calculated and subsequently analyzed (Carlsson et al., 2008).
MD with desmond
The bound and free state of compound 10 was prepared similarly for MD using the Desmond 3.1 MD package. To maintain an electrical neutrality of unit cell, sodium and chloride ions were added to reach a final concentration of 0.15 M Na+ and the system was solvated with TIP3P water molecules after reorientation to minimize the volume in an orthorhombic box.
The Amber force fields were applied to these periodic systems. The default Desmond minimization and equilibration procedure was followed, except for the maximum number of steps for SD and total minimization were increased to 1000 and 5000 steps, respectively. Simulations were kept at constant pressure (1 atom) and temperature (335 K) maintained with a Berendsen barostat and thermostat, respectively. SHAKE was applied to all systems allowing a 2-fs time-step. Long-range interactions were treated with the Particle Mesh Ewald method for periodic boundaries using a nonbonded cutoff of 8.0 Å and the nonbonded list was updated frequently using the default settings (Miller et al., 2014).
Biological evaluation
Anti-HIV screening using TZM-bl cells
TZM-bl cells (4 × 104/well) were seeded in 24-well plate and cultured overnight at 37 °C in humidified atmosphere of carbondioxide (5 %). Different vials containing HIV-1 NL4.3 at a concentration equivalent to 0.05 multiplicity of infection with respect to seeded TZM-bl cells were treated with various compounds along with appropriate vehicle controls for 1 h at 37 °C. Subsequently, pre-treated virus was added to TZM-bl cells growing in 24-well plate and incubated for 4 h at 37 °C in humidified atmosphere of carbon dioxide (5 %). After incubation, the culture supernatant was removed and the cells were washed with cold 50 mm PBS followed by addition of fresh culture medium with or without compounds/formulations. Cells were further cultured for 48 h in humidified atmosphere of carbondioxide (5 %). Varying concentrations of known anti-HIV drug, such as AZT, was used as positive reference control whereas negative control comprised of untreated cells. After incubation, the cells were washed twice with PBS and 1X Promega cell culture lysis buffer (Promega Corporation, USA) and left for 30 min at 4 °C and lysates were centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant was checked for the luciferase activity using the appropriate substrate (Promega Corporation, USA). The result expressed as percentage inhibition was calculated by taking the read out (luciferase activity) in experimental group (i.e., in the presence of compounds) divided by the read out in infected cells in the absence of test compound multiplied by hundred. Percent inhibition was calculated by subtracting the above value from hundred.
MTT cell cytotoxicity assay
The cytotoxicity of compounds on TZM-bl cells was assayed by MTT assay (Mosmann, 1983). The MTT assay is based on reduction of the yellow colored 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) by mitochondrial dehydrogenases of metabolically active cells (live cells) to a blue colored formazan, which can be measured spectroscopically. Briefly, 3 × 103 TZM-bl cells were seeded in 96 well cell culture plates (Greiner Bio-One, GmbH, Frickenhausen, Germany) and incubated at 37 °C in a humidified atmosphere of carbon dioxide (5 %). After 24 h, cells were treated with varying concentrations of the compounds ranging from 10–400 μg/mL, for 48 h. Negative control included cells treated with solvent/medium. After incubation, cell viability was assessed by adding 20 μL MTT (5 mg/mL in 50 mM PBS) per well and incubated at 37 °C for 3 h followed by addition of MTT solvent (100 μL/well; 20 % SDS and 50 % dimethyl formamide in 50 mm PBS). The absorbance (OD) was read at 570 nm with reference filter at 690 nm (690 nm reading was used as blank). Experiments were performed in duplicate and the percent viability was calculated from the equation:
HIV-reverse transcriptase (RT) inhibitory activity
The ability of the compounds to inhibit HIV-RT activity was analyzed using the commercially available ELISA kit (Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany) according to the instructions of the manufacturer. In brief, the reaction mixture containing template/primer complex, 2′- deoxynucleoside-5′-triphosphates (dNTPs) and RT enzyme in the lysis buffer were incubated for 1 h at 37 °C and subsequently, the reaction mixture was transferred to streptavidine-coated microtitre plate (MTP). The biotin labeled dNTPs that are incorporated in the template due to activity of RT bind to streptavidine. The unbound dNTPs were washed using wash buffer and anti-digoxigenin-peroxidase (DIG-POD) was added onto MTP. The DIG-labeled dNTPs incorporated in the template was bound to anti-DIG-POD antibody. The unbound anti-DIG-POD was washed and the peroxide substrate (ABST) was added to the MTP. A colored reaction product was produced during the cleavage of the substrate catalyzed by a peroxide enzyme. The OD of the sample was determined as OD at 405 nm using microtiter plate ELISA reader. The resulting color intensity is directly proportional to the actual RT activity. The percentage inhibitory activity of RT inhibitors was calculated by comparing to a sample that did not contain an inhibitor.
The percentage inhibition was calculated by formula as given:
Results and discussion
Designing of NNRTIs
We have examined 55 arylsulphonamide derivatives to develop some potent and selective inhibitors of HIV-1 RT by using in silico structure-based approach. On the basis of preliminary in silico investigations, we selected ten promising molecules for further studies. All compounds have amide and sulphonamide linkages. The presence of C=O dipole, N–C dipole, S=O dipoles, and S–N dipole allowed these molecules to function as H-bond acceptors as well as H-bond donors. Since these molecules have both H-donor and H-acceptor sites, they can ionize at an appropriate pH to further enhance their solubility. Thus, these molecules have potential to internalize better in cells. Some physically significant descriptors and pharmaceutically relevant properties of designed arylsulphonamides, like molecular weight, H-bond donors, H-bond acceptors, logP (octanol/water) have been analyzed on the basis of Lipinski’s rule of five and the results are summarized in Table 1. Molecules showing more than one type of violations are supposed to have problems with bioavailability, hence rejected. The intermediate TPSA values of these compounds in comparison with nevirapine and etravirine were expected to help in better cellular internalization.
Chemistry
Arylsulphonamide derivatives have been synthesized via 4-(4chloro benzenesulphonylamino)-benzoyl chloride starting from p-amino benzoic acid as shown in Scheme 1. For synthesis of 4-(4-chloro-benzenesulphonylamino)-benzoic acid, green protocols were applied. To an aqueous solution of 4-amino benzoic acid 1, maintained at pH 9 with aqueous sodium bicarbonate solution, 4-chloro benzenesulphonyl chloride was added drop wise under cold conditions. In this step, we selected water as solvent instead of organic solvents because the reaction of aminobenzoic acids and sulphonyl chloride in organic solvents in the presence of organic amine bases gave poor yield in our earlier experiment, and this was also corroborated by cited literature (Deng and Mani, 2006) . Contents were stirred at room temperature and on completion of the reaction, the medium of the reaction mixture was adjusted to pH 2 using hydrochloric acid (1 M) and the precipitate obtained was filtered, washed with water and dried. The resulting solid was recrystallized from ethanol.
To a solution of compound 2 in dry acetonitrile containing pyridine as a catalyst, was added oxalyl chloride drop wise in cold and anhydrous conditions. The reaction mixture was stirred for 1 h, extra oxalyl chloride removed under pressure and the resultant solution was added drop wise to a acetonitrile:pyridine (1:1) solution of different heterocyclic amines in cold and anhydrous condition to get the desired compounds, (Scheme 1), which were purified by recrystallization from ethanol:water in good yields (Rai and Singh, 2011, Parang et al., 1998).
Molecular modeling analysis
Recently, molecular docking has emerged as a powerful tool in drug designing, predicting the best mode by which a given compound fits well into a binding site of a macromolecular target (Chen et al., 2006). We have docked the molecules within the NNIBP of HIV-1RT and docking results have been discussed using the parameters, like hydrogen bond, nonbonded “π–π” and “π–+” interactions as they help stabilize and strengthen the receptor-ligand complex. The “π–π” and “π–+” interactions are non-covalent interactions, which have pivotal role in protein-ligand recognition and are termed as major forces that stabilize the association. According to Gallivan and Dougherty, “π–+” interaction energies are considered of the same order of magnitude as hydrogen bonds or salt bridges and play an important role in molecular recognition and interaction with ligands. The distance for “π–+” interactions in the present studies was observed well below the limit of 6 Å as suggested by Gallivan and Dougherty (1999) for optimum receptor-ligand recognition.
Structure-activity relationship (SAR)
Docking of compounds 4–13 using DS 3.0 into the active site of target protein (PDB: ID 3MEC) generated a number of possible binding conformations with the corresponding Ludi_3 values. The cluster analysis revealed a predominant orientation of the ligands within the binding pocket of active site and the conformation with the highest Ludi_3 value for each molecule was chosen for further analysis. Considering the magnitude of interacting forces for receptor-ligand interaction, the entire set of molecules was rigorously docked into the allosteric site of HIV-1 RT, using the same protocol, and analyzed through the above-mentioned parameters, and the results are presented in Tables 2 and 3.
Visual inspection of the minimized complexes of inhibitors with HIV-1 RT showed the optimal electronic and hydrophobic interactions between the enzyme and the ligands. These derivatives, like other second generation non-nucleoside inhibitors, adopted a common “Horse-shoe” conformation. Nevertheless, there are important differences in their conformations and specific positioning within the NNIBP. Compounds 6 and 10 adopted a “Horse-shoe” conformation (Fig. 2), whereas compounds 4, 5, 7, 8, 9, 11, 12, and 13 showed a “Sea-horse” conformation (Figs. 3, 4), which closely resembled “Horse-shoe” conformation.

Interaction of compound 4, 5, 7, and 8 with HIV-RT showing “Sea-Horse” conformation

Interaction of compound 9, 11, 12, and 13 with HIV-RT showing “Sea-Horse” conformation

Interaction of compound 6 and 10 with HIV-RT showing “Horse-shoe” conformation
To explore the SAR profile, the role of substituents at 5-position of pyridine ring of compounds 4, 5, 6 was analyzed. Three types of derivatives, bearing a hydrogen (4), bromine (5), and nitro group (6) were studied. The compound 4 bearing a hydrogen at 5-position formed four H-bonds: the first H-bond between the –SO– group of sulphonamide moiety and Lys103 with a distance of 2.7 Å, the second H-bond between –NH of the sulphonamide moiety with Tyr318 and other H-bonds formed between benzamide moiety and Gly190 and Tyr188 present in the NNIBP with interatomic distance of 3.0 Å each. The compound 5, having Br atom in place of hydrogen, formed no H-bonds but showed one π interaction with Tyr181 with an interatomic distance of 5.9 Å. The nitro group introduced at 5-position, in compound 6, formed one H-bond with Lys101 and showed π-cation interaction with Trp229 with interatomic distance of 5.9 Å. The compound 4 showed highest stabilization energy, i.e., (−) 77.01 kcal/mol whereas compounds 5 and 6 showed (−) 58.35 and (−) 47.85 kcal/mol, respectively. Thus, it may be assumed that at 5-position on pyridine ring smaller groups are preferred for better orientation of the molecules in the hydrophobic pocket.
In compounds 7, 8, and 9 pyridine ring is replaced by benzothiazole group where compound 7 has an ethoxy, compound 8 a chloro, and compound 9 a bromo group at 4-position. All three compounds have shown similar orientation within NNIBP. However, heterocyclic core of the molecules despite being flanked by the aromatic amino acid residues did not show π interaction. Sulphonamide linkage formed one H-bond with Lys103 in all three compounds. The compound 7 showed highest stabilization energy, i.e., (−) 76.07 kcal/mol whereas compounds 8 and 9 showed (−) 66.50 and (−) 72.37 kcal/mol, respectively. So it may be assumed that a larger group in benzothiazole ring better stabilizes the protein-ligand complex.
So, it was clear from the SAR analysis that more stable protein-ligand complexes were formed with compounds 4 and 7, and this behavior can be explained on the basis of different orientations adopted by the molecules as a whole inside the NNIBP and this fact also indicated about the degree of flexibility of NNIBP.
Compound 10 contains indazole group as heterocyclic amine part, which in the protein-ligand complex is surrounded by the β5–β6 loop (Pro95, Leu100, Lys101, and Lys103). The indazole ring showed π-cation interaction with Lys101 and N atom of indazole nucleus formed one hydrogen bond with Gly99 (3 Å). Central benzene ring of compound 10 showed π interaction with Tyr181. The stabilization energy of the protein-ligand complex in the case of compound 10 was (−) 69.20 kcal/mol.
Compounds 11 and 12 contain hydrogen and nitro group, respectively, at 4-position of benzimidazole nucleus as heterocyclic part, which during complexation with HIV-RT is surrounded by the β5–β6 loop. In both compounds benzimidazole nucleus showed π-cation interaction with Lys103 (6.5 Å and 6.0 Å). In compound 11, –NH– part of amide linkage formed one hydrogen bond with Tyr181 (2.8 Å). The stabilization energy of compounds 11 and 12 was (−) 64.79 and (−) 73.68 kcal/mol, respectively. Thus, nitro group at 4-position of benzimidazole nucleus better stabilized the RT-ligand complex.
In compound 13, –SO– group of sulphonamide moiety formed one H-bond with Val189 (2.9 Å), –NH– group of amide linkage formed another H-bond with H is235 (2.9 Å) and benzene ring of arylsulphonamide unit showed π-interaction with Tyr181 (5.0 Å). The stabilization energy of the protein-ligand complex in this case was (−) 61.29 kcal/mol, very close to that of etravirine [(−) 61.43 kcal/mol].
In all molecules, H-bonds were formed between –SO– and –NH– of the sulphonamide moiety and different amino acid residues, like Lys101, Lys 103 and Tyr188, Tyr318, respectively, present in the NNIBP of HIV-1 RT. In addition to this, in the case of compounds 4, 11, and 13, H-bonds were formed between carbonyl group of benzamide moiety and different amino acid residues, like Tyr181, Gly190, and His235 present in the NNIBP.
The proposed molecules interacted with the aromatic amino acids Tyr181, Tyr188, Tyr318, and Trp229, and the hydrophobic amino acid Lys103, present in the NNIBP, through favorable π-stacking and aliphatic interactions, whereas the benzene ring of arylsulphonamide and heterocyclic amine cores acted as wing I and II, respectively. The SAR data, as shown in Table 3 revealed that the arylsulphonamide core played an important role in the stabilization of these compounds within HIV-1 RT. These compounds interacted with residues Lys101, Lys103, Gly99, Gly190, and Tyr318 through H-bonds and with residues Lys103, Trp229, Tyr181, Tyr188, and Tyr318 through π-interactions, whereas in the case of nevirapine, H-bonding was absent. The ligands interacted through π-π interactions with Tyr181 and Trp188. In the case of etravirine Lys101 was involved in H-bonding and Tyr181 in π–π interactions. SAR studies on all inhibitors revealed that sulphonamide linkage played a crucial role in the stabilization of protein-ligand complex. As discussed previously, the sulphonamides containing H-bond acceptors directly interacted with Lys103 backbone of HIV-1 RT, which was similar to interactions shown by many “next generation inhibitors”. In the case of compounds 4, 6, 7, 8, 9, and 11, the sulphonamide linkage directly interacted with Lys103 backbone of HIV-1 RT. Superimposition of all minimized frames showed that the proposed inhibitors did bind to the HIV-1 RT inside the NNIBP in a common mode as proposed for second generation NNRTI—etravirine.
Scoring
Ligand scoring is a method to rapidly estimate the binding affinity of a ligand and is based on ligand pose geometry docked into a target receptor structure. To define binding affinity of ligands with receptor in a better way, hybrid title analogs and reference etravirine and nevirapine (for comparison) were rigorously analyzed through scoring functions, such as PLP1, PLP2, PMF, Lig_Internal_Energy, binding energy, dock score, and ultimately Ludi3.
Piecewise Linear Potential is a fast and simple docking function that has been shown to explain well the protein-ligand binding affinities. PLP scores are measured in arbitrary units and reported as negative values. Higher PLP scores indicate stronger receptor-ligand binding. Two versions of the PLP function available are: PLP1 and PLP2 (Gehlhaar et al., 1995). In the PLP1 function, each non-hydrogen ligand or non-hydrogen receptor atom is assigned a PLP atom type. Hydrogen atoms are excluded from consideration. In PLP2 function, PLP atom typing remains the same as in PLP1. In addition, an atomic radius is assigned to each atom except for hydrogen. It is worthwhile to mention that majority of title compounds showed comparable PLP scores to the reference ligands and compounds 4 (107.3), 8 (109.06), 9 (108.5), and 13 (109.7) exhibited PLP1 score very close to etravirine (107.62).
The PMF scoring functions were developed on the basis of statistical analysis of the 3D structure of protein-ligand complexes and were found to correlate well with protein-ligand binding free energies. The scores are calculated by summing pair wise interaction terms over all interatomic pairs of the receptor-ligand complex (Muegge and Martin, 1999). In a comparison test, it was found that all title compounds showed significant PMF scores with reference to standard ligand. The internal nonbonded ligand energy is calculated for each new conformation that is generated. The unsuitable conformations with high internal ligand energies (typically resulting from internal close contacts) are discarded. The internal ligand energy consists of a vdW term and an optional electrostatic term. The nonbonded vdW energy is computed using a standard 9–6 (unsoftened) potential using force field parameters consistent with the force field employed. Efficient ligand is classified with high negative ligand internal energy. All title hybrid analogs demonstrated significantly high negative values as compared to that of the reference.
All the scoring data for the title hybrid compounds along with reference are presented in Table 2. Candidate ligand poses were evaluated and prioritized according to the Dock Score function. It was clear from Table 2 that the reference ligand etravirine exhibited dock score of 55.05 and the hybrid compounds 4–13 showed dock score ranging from 48.21 to 59.91, which suggested that all molecules have similar mode of binding and significant binding affinity toward NNIBP receptor domain as that of the reference ligand. Further, the docking energy as shown in Table 2 reflected the stability of RT-ligand complexes, which showed that majority of the ligands moved into a stable position comparable to nevirapine and etravirine. Binding energy of the title molecules as well as of reference molecules was analyzed from their corresponding docked conformations in receptor. Results illustrated that binding energy of compounds 6 (−5.58), 10 (−5.27), 11 (−6.0), and 12 (−5.80) little bit similar to that of reference nevirapine (−5.57), whereas in the case of compounds 4 (−6.86), 7 (−7.17), 8 (−7.48), 9 (−7.54), and 13 (−7.47), the binding energy was similar to that of reference etravirine (−7.10). The binding free energy (∆G) and Predicted EC50 for each derivative were predicted using the Ludi_2 and Ludi_3 scoring functions, respectively (Lagos et al., 2008; Kumar et al., 2010). The present study on all these compounds proved that higher binding energy (∆G) was associated with higher affinity, which corresponded to lower EC50 value as shown in Table 2.
Molecular dynamic analysis
Further, the stability of the compound 10-RT complex obtained from the docking calculations was assessed via MD simulations. The RMSD values for the protein Cα atoms, side chain as well as heavy atoms were calculated by aligning the MD production phase trajectories to their initial structures. The RMSD plots show that 10-RT complex was stable during the production phase of the MD simulations (Fig. 5). The summary of standard dynamic cascade as presented in Table 6 further supported that 10-RT complex demonstrated higher stability as compared to that of the compound 10 in the unbound state.

Molecular dynamics trajectory plots correlating RMSD deviation from the compound 10 heavy atoms (1) coordinates, 10-HIV-RT Cα atoms (2) and side chain (3) over a simulation time of 50 ns
On the basis of extensive docking experiments using the DS 3.0software, ten promising compounds, 4–13, out of 55 initially taken for this study, were finally synthesized and screened for their anti-HIV activity.
Biological evaluation
Anti-HIV activity
All compounds were evaluated for their biological activity against WT HIV-1 strain III-B tusing TZM-b1 cells and The results expressed as EC50, CC50, and selectivity index (SI) values in Table 4. Etravirine and nevirapine were used as reference drugs. The result of anti-HIV assay demonstrated that the molecules did inhibit HIV expression, albeit, to a very low degree. The compound 10, nevertheless, exhibited high EC50 value in the range of 4.89 × 10−5 nm but, unfortunately, a low SI value—2.45, in comparison to etravirine and nevirapine lowered its usefulness. Analysis of the anti-HIV screening results (Table 4) revealed that all molecules 4, 5, 6, 7, 10, 11, and 12 showed very low level of in vitro anti-HIV activity in contrast to the predicted EC50 values (Table 2) on the basis of docking studies with HIV-1 RT.
Inhibition of HIV-1 RTWT
In order to substantiate that compound 10 targeted HIV-1 RT, its HIV-1 RT inhibitory activity was evaluated using an ELISA for quantifying expression of HIV-1 RTWT in culture medium and nevirapine as a reference compound (Table 5). The results showed that compound 10 inhibited the activity of HIV-1RTWT. Based on the chemical structure, general characteristics and RT assay, it was verified that compound 10 targeted the HIV-1 RT, and thus acted as an NNRTI. Its HIV-1 RT inhibitory pattern matched exactly to that of nevirpine (Table 6). Thus, the result clearly established the veracity of our concept of structural composition of the molecules for developing them as NNRTIs.
Conclusions
On the basis of extensive docking experiments, ten promising compounds out of 55 initially taken for this study, were synthesized using green protocols and screened for their anti-HIV activity. The compounds showed very promising in silico results as reflected by their high ∆G values, high binding affinity, significant scoring functions and high RT-ligand stabilization energy, which all together lowered the predicted EC50 values for the compounds. However, the molecules did not show the expected inhibitory action against HIV-1 under in vitro conditions. Only the compound 10 showed significant anti-HIV activity (4.89 × 10−5 nm), nevertheless, the low SI (2.45) marred its usefulness. The encouraging part of this study was that the compound 10 behaved as an NNRTI as per our expectations, on the basis of results obtained during HIV-1 RT assay.
The contrary results obtained from docking and experimental studies can be explained on the basis that the parent molecules used for docking studies might have got metabolized during in vitro cell experiments.
References
Bohm HJ (1994a) The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J Comput Aided Mol Des 8:243–256
Bohm HJ (1994b) On the use of LUDI to search the Fine Chemicals Directory for ligands of proteins of known three-dimensional structure. J Comput Aided Mol Des 8:623–632
Bohm HJ (1998) Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des 12:309–323
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217.
Carlsson J, Boukharta L, Aqvist J (2008) Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to HIV-1 reverse transcriptase. J Med Chem 51:2648–2656
Chen H, Lyne PD, Giordanetto F, Lovell T, Li J (2006) On evaluating molecular-docking Methods for pose prediction and enrichment factors. J Chem Inf Model 46:401–415
Deng X, Mani NS (2006) A facile, environmentally benign sulfonamide synthesis in water. Green Chem 8:835–838
Esposito F, Corona A, Tramontano E (2012) HIV-1 reverse transcriptase still remains a new drug target: Structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol Biol Int 2012:1–23
Gallivan JP, Dougherty DA (1999) Cation-pi interactions in structural biology. Proc Natl Acad Sci USA 96:9459–9464
Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DB, Fogel LJ, Freer ST (1995) Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally Flexible docking by evolutionary programming. Chem Biol 2:317–324
Hamdouchi C et al. (2005) Structure-based design of a new class of highly selective aminoimidazo [1, 2-a] pyridine-based inhibitors of cyclin dependent kinases. Bioorg Med Chem Lett 15:1943–1947
Jonckheere H, Anne J, De Clercq E (2000) The HIV-1 Reverse Transcriptase (RT) Process as Target For RT Inhibitors. Med Res Rev 20:129–154
Kumari G, Singh RK (2012) Highly active antiretroviral therapy for treatment of HIV/AIDS patients: current status and future prospects and the Indian scenario. HIV & AIDS Review 11:5–14
Kumari G, Singh RK (2013) Anti-HIV drug development: Structural features and limitations of Present day drugs and future challenges in the successful HIV/AIDS treatment. Curr Pharm Des 19:1767–1783
Kumari G, Nutan, Modi M, Gupta SK, Singh RK (2011) Rhodium (II) acetate-catalyzed stereoselective synthesis, SAR and anti-HIV activity of novel oxindoles bearing cyclopropane ring. Eur J Med Chem 46:1181–1188
Kumar M, Vijayakrishnan R, Subba Rao G (2010) In silico structure-based design of a novel class of potent and selective small peptide inhibitor of Mycobacterium tuberculosis Dihydrofolate reductase, a potential target for anti-TB drug discovery. Mol Divers 14:595–604
Lagos CF, Caballero J, Gonzalez-Nilo FD, Mahana DPC, Perez-Acle T (2008) Docking and quantative structure-activity relationship studies for the Bisphenylbenzimidazole family of non-nucleoside inhibitors of HIV-1 reverse transcriptase. Chem Biol Drug Des 72:360–369
Martins S, Ramos MJ, Fernandes PA (2008) The current status of the NNRTI family of Antiretroviral used in the HAART regime against HIV infection. Curr Med Chem 15:1083–1095
Miller BR, Parish CA, Wu EY (2014) Molecular dynamics study of the opening mechanism for DNA polymerase I. PLoS Comput Biol 10(12):e1003961.doi: 10.1371/journal.pcbi.1003961
Mosmann T (1983) Rapid colorimteric assay for cellular growth and survival application to proliferation and cytotoxicity assays. J Immun Methods 65:55–63
Muegge I, Martin YC (1999) A general and fast scoring func-tion for protein- ligand interactions: a simplified potential approach. J Med Chem 42:791–804
Parang K, Knaus EE, Wiebe LI (1998) Synthesis, in vitro anti-HIV activity, and biological stability of 5′-O-myristoyl analogue derivatives of 3′-fluoro-2′,3′-dideoxythymidine (FLT) as potential bifunctional prodrugs of FLT. Nucleos Nucleot 17:987–1008
Prathipati P, Saxena AK (2006) Evaluation of binary QSAR models derived from LUDI and MOE scoring functions for structure based virtual screening. J Chem Inf Model 46:39–51.
Rai D, Singh RK (2011) Synthesis and antibacterial activity of benzamides and sulfonamide derived from 2-amino-5-bromo/nitropyridine against bacterial strains isolated from clinical patients. Indian J Chem 50B:931–936
Singh A, Yadav D, Yadav M, Dhamanage A, Kulkarni S, Singh RK (2015) Molecular modeling, synthesis and biological evaluation of N-heteroaryl compounds as reverse transcriptase inhibitors against HIV-1. Chem Biol Drug Des 85:336–347
Singh RK, Yadav D, Rai D, Kumari G, Pannecouque C (2010) Synthesis, structure- activity Relationship and Antiviral activity of 3′-N, N-dimethylamino-2′,3′-dideoxythymidine and its prodrugs. Eur J Med Chem 45:3787–3793
Singh UP, Singh RK (2011) Molecular docking analysis of novel non nucleoside reverse transcriptase inhibitor in development: Implication for rational drug design retrovirology. Retrovirology 8:82
Tronchet JM, Seman M (2003) Nonnucleoside inhibitors of HIV-1 Reverse Transcriptase: From the Biology of reverse transcription to molecular design. Curr Top Med Chem 3:1496–1511
Venkatachalam CM, Jiang X, Oldfield T, Waldman M (2003) LigandFit: a novel method for the shape- directed rapid docking of ligands to protein active sites. J Mol Graph Model 21:289–307
Wang L et al. (2014) Fused heterocycles bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 2: Discovery of novel [1, 2, 4] Triazolo [1, 5-a] pyrimidines using a structure-guided core-refining approach. Eur J Med Chem 85:293–303
Wang R, Wang Y, Lu S (2003) Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem 46:2287–2303
Zhan P, Chen X, Li D, Fang Z, De Clercq E, Liu X (2013) HIV-1 NNRTIs: Structural diversity, pharmacophore similarity, and implications for drug design. Med Res Rev 33:E1–E72. doi:10.1002/med.20241
Zhan P, Pannecouque C, De Clercq E, Xinyong Liu (2016) Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J Med Chem 59:2849–2878
Zhou Z, Lin X, Madura JD (2006) HIV-1 RT nonnucleoside inhibitors and their interaction with RT for antiviral drug development. Infect Disord Drug Targets 6:391–413
Acknowledgments
Financial assistance in the form of WOS-A Fellowship (SR/WOS-A/CS-1053/2014) to Anuradha Singh by the DST, New Delhi is sincerely acknowledged. SK Gupta would like to acknowledge JC Bose Fellowship by Science and Engineering Research Board, DST, Govt. of India. The authors are thankful to IISc, Banglore for spectral characterization of compounds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Singh, A., Yadav, M., Srivastava, R. et al. Design and anti-HIV activity of arylsulphonamides as non-nucleoside reverse transcriptase inhibitors. Med Chem Res 25, 2842–2859 (2016). https://doi.org/10.1007/s00044-016-1707-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1707-7