
Abstract
A series of thirty-one new compounds were synthesized and evaluated for their anti-HIV-1 and cytotoxicity activity. Of these, twelve were found to be inhibitors of HIV replications in primary human lymphocytes with median effective concentration (EC50) values < 20 µM. However, most of the compounds demonstrated cytotoxicity in different cells. Our structure activity relationship study identified different patterns. In the series of 2-aryl pyrrolidines, comparing the activity of the compounds containing 2-aryl substituents we observed that compounds 1c, 1f–j, 2f,g with benzyloxyphenyl and isopropoxy groups were more potent. Compounds 1g–j, 2f,g, in which the 1-aryl moiety contained a methyl group in 3,5- or 4-positions also showed high activity. In the series of compounds containing the amide, aminomethyl and nitrile groups we observed an increase in activity with C(O)NH2 < CH2NH2 < CN. In the series of 2-pyrimidinyl pyrrolidines, the best results were demonstrated with derivatives 5e and 5f, in which the presence of a benzyl fragment in 1st and aniline fragment in 6th positions of pyrimidine ring we observed an increase in anti-HIV activity. Molecular docking studies of synthesized compounds with HIV-1 reverse transcriptase enzyme were performed. Binding energies of ligands were estimated, and the interacting amino acids of HIV-1 reverse transcriptase protein were shown. Based on corroborative results of the molecular docking studies and in vitro experiments, we suggest that three groups of synthesized ligands (1c, 1f–i), (2f,g), (5e,f, 7) are of high interest for further research on new drugs against HIV.
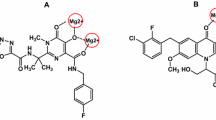
Graphic abstract
General structure of synthesized 2-aryl and 2-pyrimidinyl pyrrolidines.

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus type 1 (HIV-1) is the etiological agent of acquired immunodeficiency syndrome (AIDS). HIV undergoes rapid genetic variation caused primarily by the enormous number of viruses produced daily in an infected individual and genetic variability due to the lack of DNA proofreading ability. The variation within individuals combined with the historic lack of human migration has led to the development of diverse HIV-1 subtypes. When combined with the selection of resistant viruses under drug treatment, these factors complicate the development of effective drugs.
More than half of the currently approved anti-AIDS drugs target the reverse transcriptase (RT) enzyme. The RT associated with HIV is actually the target for three FDA approved classes of inhibitors: nucleoside RT inhibitors (NRTIs), nucleotide RT inhibitors (NtRTIs) and non-nucleoside RT inhibitors (NNRTIs). The NRTIs and NtRTIs interact with the catalytic site (that is the substrate-binding site) of the enzyme, whereas the NNRTIs interact with an allosteric (that is non-catalytic) site located at a short distance from the catalytic site.
NNRTIs gained the greatest importance because of their specificity and low cytotoxicity. All non-nucleoside inhibitors bind to a hydrophobic pocket near the polymerase active site. NNRTIs were found to have more potential as a class of anti-HIV agents versus the NRTIs and nucleotide reverse transcriptase inhibitors (NtRTIs) because they differ structurally from the nucleoside analogues. NNRTIs usually do not interfere with the human cell cycle and are specific inhibitors of RT enzyme of HIV-1.
NNRTIs bind to HIV-1 RT in a hydrophobic pocket (NNIBP) that contains side chains of aromatic amino acid residues Tyr181, Tyr188, Phe227, Trp229 and Tyr318 and of hydrophobic amino acid residues Pro95, Leu100, Val106, Val108, Val179, Leu234 and Pro236 from the p66 subunit. Glu138 is the only amino acid residue of the p51 subunit that interacts with NNRTIs; Glu138 does not directly interact with all NNRTIs [1].
The era of NNRTIs began with the discovery of HEPT and TIBO as specific inhibitors of RT. With the discovery of α-anilinophenylacetamides (α-APAs), the era of flexible derivatives started and the most active compound in this series found is Loviride, the simplicity of its structure and the relative ease of its synthesis made the α-APA series attractive for lead optimization (Fig. 1) [2, 3].

Non-nucleoside reverse transcriptase inhibitors
Consequently, studies on the synthesis and biological activity of new NNRTIs are worth pursuing. Herein, we report our molecular docking and biological studies of new synthesized derivatives of 2-aryl and 2-pyrimidinyl pyrrolidines, which contain fragments of known reverse transcriptase inhibitors Loviride and HEPT. The molecular docking studies were performed in order to estimate binding energies of synthesized compounds and interacting amino acids of HIV-1 reverse transcriptase enzyme, and the obtained data were compared with the biological results of anti-HIV in vitro studies.
The purpose of our studies was to efficiently predict the potent anti-HIV activity via modeling, construction of new compounds and determination of the fragments responsible for the observed activity.
Experimental work
Anti-HIV-1 assay
Human peripheral blood mononuclear (PBM) cells were stimulated with phytohemagglutinin A for 2–3 days prior to use. HIV-1/LAI obtained from the Centers for Disease Control and Prevention (Atlanta, GA) was used as the standard reference virus for the antiviral assays. The antiviral median effective concentration (EC50) and 90% effective concentration (EC90) were determined from the concentration–response curve using the median effect method [4].
Cytotoxicity assay
Compounds were evaluated for their potential toxic effects on uninfected PHA-stimulated human PBM cells, in CEM (T-lymphoblastoid cell line obtained from American Type Culture Collection, Rockville, MD) and Vero (African green monkey kidney) cells. The 50% inhibition concentration (IC50) was determined from the concentration–response curve using the median effect method [5].
Molecular docking and ADME-Tox studies
Three-dimensional crystal structures with resolution < 2 Å of HIV-1 reverse transcriptase (RT) in complex with NNRTI compounds were downloaded from Protein Data Bank [6]. Therefore, four structures were chosen: 2ZD1, 6C0J, 4G1Q and 4KO0 [7,8,9,10]. The preparation of structures (addition of hydrogens, removal of water and co-crystallized compounds) and ligand files for docking were done with AutoDockTools4 and Avogadro [11, 12]. The docking of twenty-eight ligands with four structures of HIV-1 RT was performed with AutoDock Vina molecular docking software [13]. Grid boxes were generated using the co-crystallized ligands. Diagrams of ligand–protein interactions of docking results were drawn with LigPlot + software [14]. Figures were obtained using PyMOL 2.3 (The PyMOL Molecular Graphics System, version 2.0 Schrödinger, LLC).
ADME-Tox evaluation was carried out using admetSar [15] and SwissADME [16] web tools.
Results and discussion
Chemistry
2-Aryl pyrrolidine derivatives
In the course of our studies directed to search for new NNRTI inhibitors, we have synthesized 2-arylpyrrolidines with different groups in various positions of the heterocyclic ring.
By acylation, corresponding 2-aryl-2-(arylamino)acetonitriles with 3-chloropropanoyl chloride and further intramolecular cyclization under phase transfer catalytic conditions were prepared 1,2-diaryl substituted pyrrolidine carbonitriles 1a–j (Fig. 2) [17,18,19,20,21].

Aryl pyrrolidine derivatives
Different substituted pyrrolidine carbonitriles were reduced to corresponding aminomethyl pyrrolidines 2a–g according to the method developed by us, using NaBH4/PEG-400/CoCl2 system [19, 21]. The pyrrolidine carboxamides 3a and 3b were synthesized from corresponding pyrrolidine carbonitriles via reaction with conc. H2SO4 (Fig. 2) [20].
2-Pyrimidinyl pyrrolidine derivatives
The next series of synthesized pyrrolidine derivatives refers to pyrimidinyl pyrrolidines. By condensation of 6-amino pyrimidinediones 4a–c with pyroglutamic acid methyl ester or 2-pyrrolidone, in the presence of PCl3 under Vilsmeier reaction conditions, 6-imino-5-tetrahydro-1H-2-pyrrolylidenehexahydro-2,4-pyrimidinediones 5a–g were synthesized. For the reduction of 6-imino group of 5d, we used the reducing system NaBH4/1,4-dioxane/CoCl2/PEG-400. The ester fragment was reduced to form hydroxymethylderivative 6. Compound 7 was synthesized by acylation of 5c with benzoyl chloride (Fig. 3) [22, 23].

Pyrimidinyl pyrrolidine derivatives
Biology
Anti-HIV activity
The synthesized compounds appeared to have good anti-HIV activity, and among thirty-one compounds in this series, twelve exhibited micromolar activity with EC50 values < 20 µM. Compounds 1h,i and 5f proved to be the most potent compounds with EC50 value of 5.3, 2.0 and 0.48 µM, respectively; however, they were cytotoxic to all three cell lines and had narrow therapeutic windows. Only compound 1c was non-toxic to PBM, CEM and Vero cells while displaying a reasonable EC50 value of 5.2 µM (Table 1) [21].
Cytotoxicity studies
Compounds were evaluated for their cytotoxicity in human PBM, CEM and Vero cells to determine their spectrum of toxicity. CEM cells are a line of lymphoblastic cells originally derived from a child with acute lymphoblastic leukemia, whereas Vero cells are derived from African green monkey kidney cells.
Compounds 2b,c, 3a,b, 4a, 4c, 5a,b, 5d and 6 were non-toxic to all the cell systems used; however, they were also inactive or largely inactive versus HIV-1 (Table 1).
Molecular docking studies
Results of performed molecular docking experiments in the form of binding energy and interacting amino acids in a hydrophobic pocket of reverse transcriptase enzyme HIV-1 for each ligands of synthesized compounds and the biological results of anti-HIV studies are presented in Table 2.
From the data obtained, it follows that for most derivatives of 2-aryl and 2-pyrimidinyl pyrrolidines, a direct correlation is observed between low binding energies and biological results of anti-HIV in vitro studies. As expected, the predicted and actually determined biological activity is directly related to the greatest decrease in binding energy. However, for some of the compounds studied, we did not observe a correlation between the decrease in energy and anti-HIV activity.
Pyrrolidine carbonitriles 1a–j interact with HIV-1 reverse transcriptase enzyme with high binding energies at (− 11.8 to − 9.70 kcal/mol). From this group, compounds 1c (Fig. 4a), 1f–h (Fig. 4b), 1i (Fig. 4c) showed low values of EC50 (Table 2) and compound 1h showed the highest docking binding energy among all studied ligands (− 11.8 kcal/mol). Binding of this group of compounds to the NNRTI-binding site is driven by hydrophobic interactions with Leu234, Tyr181, Val106, Trp229, Tyr188, Val179, Lys103 and Leu100. Only compound 1h in the group showed the possibility of the formation of two weak electrostatic hydrogen bonds with Lys103 (distance of the bond 3.27 Å) and Tyr188 (distance of the bond 3.20 Å).

Interaction of pyrrolidine carbonitriles 1a–j and aminomethyl pyrrolidines 2a–g with NNRTI-binding pocket of HIV-1 RT enzyme. For each compound, only interacting amino acids are presented. a 3D structure of the compound 1c; b 3D structure of the compound 1h (dotted line colored in yellow represents possible hydrogen bonding with Tyr188 and Lys103); c 3D structure of the compound 1i; d 3D structure of the compound 2g
Aminomethyl pyrrolidines 2a–g had good binding energies (from − 9.50 to − 11.0 kcal/mol) for HIV-1 reverse transcriptase enzyme. Compounds 2f and 2g (Fig. 4d) showed high binding energies to NNRTI-binding site of HIV-1 RT protein and low values of EC50. These two compounds have similar binding energies − 10.4 kcal/mol (compound 2f) and − 10.5 kcal/mol (compound 2g). Studied compounds 2a–g interact with Glu138, Tyr181, Tyr188, Trp229, Leu234, Leu100, Lys103, Val106 and Val179.
Pyrrolidine carboxamides 3a,b showed good binding energies; they are non-toxic to all the cell systems used, but inactive against HIV-1.
Within pyrimidinyl pyrrolidines 5a–g, 6, 7 with binding energies at − 11.8 to − 7.30 kcal/mol with HIV-1 reverse transcriptase, compounds 5e,f and 7 showed good binding energies and low values of EC50 (Table 2).
Compound 5f (Fig. 5a) binds with the NNRTI-binding site of HIV-1 RT protein with the binding energy of -9.90 kcal/mol and showed lowest EC50 values of 0.48 µM. Compound 7 (Fig. 5b) forms one weak hydrogen bond with Lys103 with the bond distance 3.03 Å. Common interacting amino acids of this group are Tyr188, Tyr181, Glu138, Val179, Lys103, Leu100, Tyr318, Leu234, Phe227 and Val106.

Interaction of pyrimidinyl pyrrolidines 5f and 7 with NNRTI-binding pocket of HIV-1 RT enzyme. a 3D structure of the compound 5f; b 3D structure of the compound 7 (dotted line colored in yellow represents possible hydrogen bonding with Lys101)
Based on the results of molecular docking, the following interacting amino acids: Leu234, Tyr181, Val106, Tyr188, Val179, Lys103 and Leu100 are common for all analyzed compounds and therefore considered important for interaction between NNRTIs and HIV-1 RT protein. Interaction of the all studied compounds with HIV-1 RT protein is mostly driven by hydrophobic forces.
In silico analysis of the compounds of three analyzed groups of synthesized ligands (1c, 1f–i), (2f,g), (5e,f, 7) showed their high affinity to HIV-1 RT and low values of EC50 protein and therefore are of high interest for further research on synthesis of new anti-HIV drugs. ADMET pharmacokinetic properties of mentioned compounds, were evaluated using admetSAR web tool and presented in table of supplementary materials. Based on SwissADME druglikeness evaluation, all mentioned compounds, except 1h and 1i, meet required properties, including Lipinski, Ghose, Veber, Egan and Muegge requirements. Compounds 1h and 1i have WLOGP > 5.6 and XLOGP3 > 5, which violates Ghose and Muegge requirements, respectively.
SAR studies
In the study of the relationship between chemical structure and biological activity, there were some patterns. In the series of 2-arylpyrrolidines, comparing the activity of the compounds containing 2-aryl substituents it can be seen that compounds 1c, 1f–j, 2f,g with benzyloxyphenyl and isopropoxy groups are more active. Compounds 1g–j, 2f,g in which the 1-aryl moiety contained methyl group in 3,5- or 4-positions also showed high activity. In the series of compounds containing amide, aminomethyl and nitrile groups observed an increase in activity with C(O)NH2 < CH2NH2 < CN (Table 1) [21].
In the series of 2-pyrimidinyl pyrrolidines, the best results were demonstrated derivatives 5e,f, in which the presence of a benzyl fragment in 1st, anilino fragment in 6th and pyrrolidine fragment in 5th positions of pyrimidine ring increases anti-HIV activity (Table 1).
Further structural modifications could be performed on the basis of the data obtained for these compounds to get worthwhile molecules with greater potential as anti-HIV-1 agents.
Conclusions
In summary, we herein report the anti-HIV-1 activity of new 2-aryl and 2-pyrimidinyl pyrrolidine derivatives. Twelve compounds were active against HIV-1 with EC50 values less than 20 µM, which indicates the potential of these compounds as anti-HIV-1 agents. Among these thirty-one compounds, 1h,i and 5f were the most potent anti-HIV-1 agents with EC50 values < 5 µM. They are weakly active compared to the AZT control, and the activity may be due to the toxicity observed in primary human lymphocytes. On these grounds, the structural modification of these compounds is expected to result in more potent candidates of anti-HIV drugs.
References
Prajapati DG, Ramajayam R, Yadav MR, Giridhar R (2009) The search for potent, small molecule NNRTIs. Bioorg Med Chem 17:5744–5762. https://doi.org/10.1016/j.bmc.2009.06.060
De Corte BL (2005) From 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk](1,4)benzodiazepin-2(1H)-one (TIBO) to Etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Med Chem 48:1689–1696. https://doi.org/10.1021/jm040127p
Pauwels R, Andries K, Debyser Z, Van Daele P, Schols D, Stoffels P, De Vreese K, Woestenborghs R, Vandamme AM, Janssen CGM, Anne J, Cauwenbergh G, Desmyter J, Heykants J, Janssen MAC, De Clercq E, Janssen PAJ (1993) Potent and highly selective human immunodeficiency virus type 1 (HIV-1) inhibition by a series of a-anilinophenylacetamide derivatives targeted at HIV-1 reverse transcriptase. Proc Natl Acad Sci USA 90:1711–1715. https://doi.org/10.1073/pnas.90.5.1711
Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie M-W, Hart GC, Smith GA, Hahn EF (1990) Activities of 3′-azido-3′-deoxythymidine nucleotide dimers in primary lymphocytes infected with human immunodeficiency virus type 1. Antimicrob Agents Chemother 34(6):1061–1067. https://doi.org/10.1128/aac.34.6.1061
Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF (2002) Antiviral activities and cellular toxicities of modified 2′,3′-dideoxy-2′,3′-didehydrocytidine analogues. Antimicrob Agents Chemother 46(12):3854–3860. https://doi.org/10.1128/AAC.46.12.3854-3860.2002
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242. https://doi.org/10.1093/nar/28.1.235
Das K, Bauman JD, Clark AD, Frenkel YV, Lewi PJ, Shatkin AJ, Arnold E (2008) High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci USA 105(5):1466–1471. https://doi.org/10.1073/pnas.0711209105
Yang Y, Kang D, Nguyen LA, Smithline ZB, Pannecouque C, Zhan P, Steitz TA (2018) Structural basis for potent and broad inhibition of HIV-1 RT by thiophene[3,2-d]pyrimidine non-nucleoside inhibitors. Elife. https://doi.org/10.7554/eLife.36340
Kuroda DG, Bauman JD, Challa JR, Patel D, Troxler T, Das K, Hochstrasser RM (2013) Snapshot of the equilibrium dynamics of a drug bound to HIV-1 reverse transcriptase. Nat Chem 5(3):174–181. https://doi.org/10.1038/nchem.1559
Bollini M, Frey KM, Cisneros JA, Spasov KA, Das K, Bauman JD, Jorgensen WL (2013) Extension into the entrance channel of HIV-1 reverse transcriptase–crystallography and enhanced solubility. Bioorg Med Chem Lett 23(18):5209–5212. https://doi.org/10.1016/j.bmcl.2013.06.093
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791. https://doi.org/10.1002/jcc.21256
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. https://doi.org/10.1186/1758-2946-4-17
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51(10):2778–2786. https://doi.org/10.1021/ci200227u
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52(11):3099–3105. https://doi.org/10.1021/ci300367a
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
Gasparyan SP (2011) Synthesis of new analogs of 2-arylpyrrolidinecarbonitriles. Chem J Armenia 64(1):117–122
Gasparyan SP, Alexanyan MV, Harutyunyan GK, Hovhannesyan VE, Martirosyan VV, Paronikyan RV, Stepanyan HM, Martirosyan AH (2012) Synthesis and biological properties of new analogs of 2-arylpyrrolidinecarbonitriles and pyrrolidinecarboxamides. Pharm Chem J 46(6):331–333. https://doi.org/10.1007/s11094-012-0792-2
Gasparyan SP, Alexanyan MV, Harutyunyan GK, Hovhannesyan VE, Martirosyan AH, Panosyan HA (2014) Metal-complex reduction of nitrile groupe in substituted pyrrolidinecarbonitriles. Chem J Armenia 67(2–3):239–246
Gasparyan SP (2016) Synthesis of new derivatives of 2-arylpyrrolidinecarbonitriles and pyrrolidinecarboxamides. Chem J Armenia 69(3):333–340
Martirosyan AH, Gasparyan SP, Alexanyan MV, Harutyunyan GK, Panosyan HA, Schinazi RF (2017) Anti-human immunodeficiency activity of novel 2-arylpyrrolidine analogs. Med Chem Res 26(1):101–108. https://doi.org/10.1007/s00044-016-1731-7
Martirosyan A, Tamazyan R, Gasparyan S, Alexanyan M, Panosyan H, Martirosyan V, Schinazi R (2010) Synthesis of 6-imino-5-tetrahydro-1H-2-pyrrolylidenhexahydro-2,4-pyrimidinediones as intermediates for the synthesis of C-azanucleosides. Tetrahedron Lett 51(2):231–233. https://doi.org/10.1016/j.tetlet.2009.11.01
Gasparyan SP, Alexanyan MV, Harutyunyan GK, Kocharov SL, Martirosyan AH, Tamazyan RA, Ayvazyan AG, Panosyan HA, Danagulyan GG (2016) Synthesis of new derivatives of 5-(3,4-dihydro-2H-pyrrol-5-yl)pyrimidine. Russ J Org Chem 52(11):1646–1653. https://doi.org/10.1134/S1070428016110166
Acknowledgements
The authors are grateful to the Civilian Research and Development Foundation (CRDF) (Grant No. ARB2-2701-YE-05). This work was also facilitated in part by the NIH Grant P30-AI-050409 (to RFS). This paper is dedicated to the memory of our friend and colleague Dr. Ashot H. Martirosyan who had a huge role in implementation of this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gasparyan, S.P., Martirosyan, A.H., Alexanyan, M.V. et al. Design, antihuman immunodeficiency activity and molecular docking studies of synthesized 2-aryl and 2-pyrimidinyl pyrrolidines. Mol Divers 25, 2045–2052 (2021). https://doi.org/10.1007/s11030-020-10095-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-020-10095-1