
Abstract
A series of new class of P-heterocycle encompassing urea and thiourea derivatives, N-(substitutedphenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)ureas 11a–e/thioureas 11f–k, was accomplished from the precursor intermediate, 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-one, 9. The compound 9 was obtained by cyclization of pyridine-2,3-diamine, 6 with POCl3 followed by amidation with NaNH2. The products were tested for their in vitro and in vivo anti-inflammatory activity, and in vitro antimicrobial activity including minimum inhibitory concentration. Compounds 11a, 11d and 11j exhibited comparable anti-inflammatory activity to the standard drug, diclofenac, both in in vitro and in vivo assays, which might be due to the presence of lipophilic functional groups, F, NO2 and CF3. The compounds 11c and 11j exhibited potential growth of inhibition against selected bacterial and fungal strains at lower minimum inhibitory concentrations, while most of the thiourea-linked analogues exhibited good antimicrobial activity. A molecular modelling study was performed on cyclooxygenase isoenzyme (COX-2) to investigate the hypothetical binding mode of the most active anti-inflammatory agents, and binding conformers were proposed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Prostaglandins are the modulators and play an essential role in inflammation. The foremost cause of inflammatory syndromes is chronic and acute inflammation and various kinds of arthritis; it makes an immense trouble to humanity such as loss of working hours, stress and costly treatment. The diverse heterogeneous and chemically unrelated agents called non-steroidal anti-inflammatory drugs (NSAIDs) exhibited their effect in the interruption of biosynthesis of prostaglandins and thromboxanes by inhibiting COX enzymes (Vane and Botting, 1996; Green, 2001). Hence, they exhibited common therapeutic actions such as analgesic, antipyretic and anti-inflammatory effects and are usually indicated for the treatment of pain, fever and acute or chronic inflammatory diseases (Bennet et al., 2005). However, tNSAIDs agents have been challenged against some side effects such as gastric bleeding, gastrointestinal ulcers and suppression of the renal functions (Vonkeman and Van de Laar, 2010; Allison et al., 1992). Therefore, the researchers have placed considerable attention towards the discovery of potent anti-inflammatory agents with lack of undesirable side effects.
The phosphorus cyclic chemistry is promptly growing field in recent times since they are exhibiting variety of pharmacological and biological activities such as antimicrobial (Holla and Ashok, 2007), insecticidal (Eugenia et al., 2006), herbicidal (He et al., 1999) and anticancer (Bull and Naidu, 2000). Predominantly, phosphorus heterocycles accompanying with heteroatoms like N and O are attractive molecules because of their diversity of structures found in various biological properties (Ashley and Bartlett, 1982; Karp, 1999; Hewitt and Newland, 1977). In few reports, six-membered heterocyclic compounds containing two nitrogen and two phosphorus atoms have displayed good biological activities, for example phosphorus analogue of mimic thymine 1 is a promising chemotherapeutic anticancer agent. Recently, our group developed pyrido-based [1,3,2]oxazaphosphol-2-yl)guanidine derivatives that exhibited potent anti-inflammatory activity (Ramana et al., 2013). The outstanding key role of the cyclic phosphorus molecules in medicinal and agricultural fields, and the development of new biologically active popular targets are essential.
In few fields like medicine, food flavouring, agrochemicals, rubber chemicals, dyes and adhesives, and pyridine derivatives are one of the most considerable frameworks (Pozharskii et al., 1997). The most molecular scaffolds possessing pyridine structure have been exhibiting a broad spectrum of pharmacological activities, antimicrobial (Patel et al., 2011), antiviral (Bernardino et al., 2007), anti-inflammatory (Liu et al., 2012), anticonvulsant (Paronikyan et al., 2002), and anti-HIV and anticancer (Tucker et al., 2008; Srivastava and Pandeya, 2011). Also, certain peptides with pyridine ligand act as anti-HIV metal chelators (Kurosaki et al., 2001), bis[di-1,1-(2-pyridyl)ethyl]amine metal complex has been used in DNA cleavage studies (Hemmert et al., 2001), nicotinamide adenine dinucleotide (NAD) and phosphorylated nicotinamide adenine dinucleotide (NADP) are involved in several biological processes (Kapinos and Sigel, 2002), and some of pyridine derivatives are known to be suitable ligands for many of transition metal ions.
Further, urea and thiourea derivatives are the most multipurpose bioactive molecules and have been reported as antibacterial, antifungal, antitubercular, anti-inflammatory, antithyroid, antihelmintic, rodenticidal, insecticidal, herbicidal and plant growth regulatory properties (Yuan et al., 2001; Zhang et al., 2004, 1998; Zhou et al., 2004; Eweis et al., 2006; Saeed et al., 2008). Benzoylphenyl urea compounds like diflubenzuron, 2 and penfluzuron, 3 are one class of the insect growth regulators (IGR), which inhibit chitin synthesis and are responsible for the formation of insect cuticle (Fournet et al., 1993), N-(1,2,4-triazol-3-yl)-N′-arylthiourea acts as effective uncoupler of oxidative phosphorylation in mitochondria (Kubota et al., 1985), benzyl pyridylthiourea derivatives 4 and 5 (Fig. 1) were found to be non-nucleoside inhibitor of the reverse transcriptase enzyme of HIV (Venkatachalam et al., 2004), and few studies have been concerning on acyl urea(thiourea), and 2H-1,24-thiadiazole [2,3-a] pyrimidines assessed antiviral activity. Hence, the scientists are interested in developing the possibility of next-generation urea, and thiourea molecules could be more potent as chemotherapeutic agents as well as new generation of library of hybrid molecules for dual mode of biological activity.

Some biologically active phosphorus, urea and thiourea molecules
Considering the ubiquitous biological activities of pyridine, phosphorus molecules, urea and thiourea derivatives, and as in the part of our continuing research on the development of new bio-active phosphorus molecules (Koteswara Rao et al., 2010; Subba Rao et al., 2013), we designed a series of new compounds by incorporating above moieties together in one scaffold with good hope that these molecules will enhance the biological activity. Anti-inflammatory and antimicrobial activities were evaluated for the synthesized compounds. To the best of our knowledge, there have been no reports found on anti-inflammatory and antimicrobial activities of urea and thiourea derivatives of precursor, 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-one. Among the title products, three compounds have effectively worked as anti-inflammatory agents both in vitro and in vivo and a few compounds exhibited potent antimicrobial activity.
Materials and methods
Chemistry
All the starting materials and solvents were procured from Aldrich and SD Fine-Chem Limited, and solvents used are distilled and preserved under N2 atmosphere. The reactions were monitored by TLC (Merck silica plates), and the compounds are purified by column chromatography using Merck 120 mesh silica gel. Melting points were recorded with open capillary tube on Guna melting point apparatus and are uncorrected. IR spectra were recorded on Perkin-Elmer spectrophotometer using KBr discs. NMR spectra were recorded with BRUKER-400 MHz (400.13 MHz for 1H NMR, 100.62 MHz for 13C NMR and 161.9 MHz for 31P) spectrometer. Tetramethylsilane was used as internal standard in DMSO-d 6 for recording 1H and 13C NMR spectra. 31P NMR spectra were recorded using H3PO4 (85 %) as external reference. Results are presented as chemical shift δ in ppm, multiplicity, J values in Hertz (Hz), number of protons and proton’s position. Multiplicities are shown as the abbreviations: s (singlet), brs (broad singlet), d (doublet), t (triplet) and m (multiplet). International principles and regulations are concerned during the biological activity screening. Numbering was given to the title compounds for assigning the proper spectral characterization (Fig. 1).
Synthetic procedure for the synthesis of compound 9
Pyridine-2,3-diamine, 6 (2.5 g, 0.023 mol, 1 equiv) was dissolved in 45 mL of THF/pyridine (1:2) containing dimethylpiperazine (DMPipz) (7.6 mL, 0.056 mol, 2 equiv), and the reaction mixture was cooled to 0 °C. To this cold solution was added POCl3, 7 (2.6 mL, 0.028 mol, 1.2 equiv) in 6 mL of THF dropwise for 45 min through dropping funnel under stirring by maintaining temperature at 0-5 oC. The reaction mixture temperature was raised to 55 °C and stirred for 4.0 h to afford 2-chloro-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo [4,5-b]pyridin-2-one, 8. After completion of the reaction as checked by TLC, the hot reaction mixture was cooled to 30 °C, filtered off and washed the residual salt, DMPipz.HCl with 10 mL of THF/pyridine (1:2) solvent. The compound, NaNH2 (1.17 g, 0.029 mol, 1.3 equiv), was added to combined filtrate and stirred the reaction mixture at 45 °C for 3.0 h. The progress of reaction was monitored by TLC; the salt (NaCl) was removed by filtration as residue and concentrated the filtrate under vacuum. The crude product was washed with 10 % ethyl acetate/n-hexane (five times) and recrystallized with methanol to obtain pure brown colour 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-one (9) (2.97 g, 76.5 %).
2-Amino-2,3-dihydro-1H-2λ 5 -[1,3,2] diazaphospholo[4,5-b]pyridin-2-one ( 9 )
Brown colour solid (this compound was attained by the reaction of pyridine-2,3-diamine with POCl3 followed by treating with sodamide (NaNH2). The compound was obtained as a brown colour solid); IR (KBr) νmax 3416, 1243 cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 7.50 (1H, d, J = 7.6 Hz, H-7), 7.10–7.21 (2H, m, H-8, H-9), 5.92 (1H, s, H-1), 5.26 (1H, s, H-3), 3.14 (2H, s, H-10); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 148.9 (–N–C–N–, C-5), 136.7 (–C–N–, C-4), 131.9 (–N–CH, C-7), 123.9 (–CH, C-9), 112.9 (–CH, C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −8.47; ESI-MS (pos) m/z = 171 (M + H+) (100), 153 (M + H+–H2O) (38).
General synthetic procedure for the synthesis of urea and thiourea derivatives 11(a–k)
The mixture of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one, 9 (1.17 mmol), substituted phenyl isocyanates/isothiocyanates, 10(a–k) (1.2 mmol) and DMPipz (1.2 mmol) was dissolved in 15 mL of THF/pyridine (2:1) in a flask. The reaction content was heated to 55-60 oC and stirred for 3.0-4.5 h. After completion of the reaction as checked by TLC, the reaction mixture was cooled to room temperature and concentrated under vacuum at 50 oC. The crude product was washed with 20 % DCM (DCM and n-Hexane) (5 mL × 5 times) followed by recrystallization with methanol to get pure title products, and a few molecules were purified by column chromatography using 5 % methanol/dichloromethane as an eluent.
N-(4-Fluorophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)urea (11a)
Light brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1-fluoro-4-isocyanatobenzene (10a), and it was obtained as a light brown solid); m.p. 258–260 °C; IR (KBr) νmax 3395, 3270, 3028 (-N-H, str), 1658 (–C=O, str), 1534 (–C=N, str), 1242 (–P=O, str), 1118 (–C–F, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 10.52 (s, 1H, –P–NH–CO–, H-14), 9.48 (s, –CO–NH–, H-10), 8.56 (d, 1H, J = 6.8 Hz, H-7), 7.86 (d, 2H, J = 7.6 Hz, H-16, H-20), 7.67–7.77 (m, 2H, H-8, H-9), 7.64 (d, 2H, J = 7.2 Hz, H-17, H-19), 6.53 (s, 1H, H-1), 5.78 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 158.9 (d, J = 22.6 Hz, C-18), 154.7 (C-12), 152.7 (C-5), 146.0 (C-15), 144.3 (C-7), 140.9 (C-4), 124.1 (C-9), 121.9 (C-16, C-20), 118.3 (C-17, C-19), 116.1 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −11.8; ESI-MS (pos) m/z: 308 (M + H+) (100 %), 198 (M + H+ –C6H5FN) (28 %), 171 (M + H+ –C7H4FNO) (19 %), 139 (M + H+ –C5H6N4OP) (56 %).
N-(4-Nitrophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)urea ( 11b )
Light green solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1-isocyanato-4-nitrobenzene (10b), and it was obtained as a light green solid); m.p. 272–274 °C; IR (KBr) νmax 3382, 3236, 3099 (–N–H, str), 1672 (–C=O, str), 1564 (–N=O, str), 1532 (–C=N, str), 1236 (–P=O, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 10.23 (s, 1H, –P–NH–CO–, H-14), 9.53 (s, 1H, –CO–NH–, H-10), 8.24 (d, 2H, J = 9.2 Hz, H-17, H-19), 8.17 (d, 1H, J = 8.8 Hz, H-7), 7.76 (d, 2H, J = 9.2 Hz, H-16, H-20), 7.68–7.73 (m, 2H, H-8, H-9), 6.71 (s, 1H, H-1), 6.31 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 155.3 (C-12), 151.7 (C-5), 147.3 (C-15), 145.8 (C-18), 141.4 (C-7), 140.4 (C-4), 126.3 (C-17, C-19), 124.9 (C-9), 117.8 (C-16, C-20), 113.4 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −12.9; ESI-MS (pos) m/z: 335 (M + H+) (100 %), 170 (M + H+ –C7H5N2O3) (26 %), 198 (M + H+ –C6H5N2O2) (43 %); Anal. Calcd. for C12H11N6O4P: C, 43.12; H, 3.32; N, 25.14; Found: C, 43.08; H, 3.32; N, 25.04 %.
N-(3,5-Difluorophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b] pyridin-2-yl)urea ( 11c )
Brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1,3-difluoro-5-isocyanatobenzene (10c), and it was obtained as a brown solid); m.p. 250–253 °C; IR (KBr) νmax 3396, 3291 (br) (–N–H, str), 1649 (–C=O, str), 1509 (–C=N, str), 1259 (–P=O, str), 1143, 1107 (–C–F, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 10.84 (s, 1H, –P–NH–CO–, H-14), 9.01 (s, 1H, –CO–NH–, H-10), 8.12 (d, 1H, J = 6.8 Hz, H-7), 7.33 (s, 2H, H-16, H-20), 7.24 (s, 1H, H-18), 6.98–7.10 (m, 2H, H-8, H-9), 6.74 (s, 1H, H-1), 6.19 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 154.4 (C-12), 153.4 (d, J = 28.9 Hz, C-17, C-19), 152.2 (C-5), 150.9 (C-15), 144.8 (C-7), 139.5 (C-4), 123.9 (C-9), 119.5 (C-8), 113.4 (C-16, C-20), 103.5 (C-18); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −13.6; ESI-MS (pos) m/z: 326 (M + H+) (100 %), 198 (M + H+ –C6H4F2N) (32 %), 171 (M + H+ –C7H3F2NO) (59 %), 157 (M + H+ –C5H6N4OP) (26 %).
N-(2-Fluoro-5-nitrophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)urea ( 11d )
Dark brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1-fluoro-2-isocyanato-4-nitrobenzene (10d), and it was obtained as a dark brown solid); m.p. 266–268 °C; IR (KBr) νmax 3420, 3272 (br, –N–H, str),1640 (–C=O, str), 1567 (–N=O), 1530 (–C=N, str), 1236 (–P=O, str), 1163 (–C–F, str) cm-1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 10.88 (s, 1H, –P–NH–CO–, H-14), 9.32 (s, 1H, –CO–NH–, H-10), 8.74 (s, 1H, H-20), 8.32 (d, 1H, J = 6.4 Hz, H-7), 8.09 (d, 1H, J = 6.8 Hz, H-18), 7.81 (d, 1H, J = 6.4 Hz, H-17), 7.23–7.29 (m, 2H, H-8, H-9), 6.81 (s, 1H, H-1), 6.26 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 159.7 (C-16), 155.6 (C-12), 153.1 (C-5), 146.1 (C-19), 143.6 (C-7), 140.7 (C-4), 123.8 (C-9), 123.4 (C-18), 123.9 (C-15), 119.2 (C-17), 118.8 (C-20), 116.7 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −8.7; ESI-MS (pos) m/z: 353 (M + H+) (100 %), 198 (M + H+ –C6H4FN2O2) (27 %), 184 (M + H+ –C5H6N4OP) (37 %), 170 (M + H+ –C7H4FN2O3) (74 %); Anal. Calcd. for C12H10FN6O4P: C, 40.92; H, 2.86; N, 23.86; Found: C, 40.89; H, 2.81; N, 23.73 %.
N-(3,4-Dichlorophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b] pyridin-2-yl)urea ( 11e )
Dark brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1,2-dichloro-4-isocyanatobenzene (10e), and it was obtained as a dark brown solid); m.p. 245–247 °C; IR (KBr) νmax 3416, 3272, 3056 (–N–H, str), 1642 (–C=O, str), 1510 (–C=N, str), 1228 (–P=O, str), 826 (–C–Cl, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 10.76 (s, 1H, –P–NH–CO–, H-14), 9.22 (s, 1H, –CO–NH–, H-10), 8.14 (d, 1H, J = 6.0 Hz, H-7), 7.91 (s, 1H, H-16), 7.72 (d, 1H, J = 6.8 Hz, H-19), 7.37 (d, 1H, J = 6.8 Hz, H-20), 7.13–7.18 (m, 2H, H-8, H-9), 6.41 (s, 1H, H-1), 6.04 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 154.7 (C-12), 152.3 (C-5), 144.9 (C-15), 143.1 (C-7), 140.7 (C-4), 136.2 (C-17), 133.7 (C-18), 132.7 (C-16), 128.5 (C-20), 124.6 (C-9), 123.9 (C-19), 116.2 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −4.6, −12.5; ESI-MS (pos) m/z: 358 (M + H+) (100 %), 360 (M + H++ 2) (32 %), 198 (M + H+ –C6H4Cl2N) (28 %).
N-(2-Oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)-N′-phenyl-thiourea ( 11f )
Brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with isothiocyanatobenzene (10f), and it was obtained as a brown solid); m.p. 310–312 °C; IR (KBr) νmax 3394, 3205, 3035 (–N–H, str), 1530 (–C=N, str), 1249 (–P=O, str), 1193 (–C=S, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 12.70 (brs, 1H, –P–NH–CS–, H-14), 9.81 (brs, 1H, –CS-NH-, H-10), 8.10 (d, 1H, J = 4.8 Hz, H-7), 7.46–7.51 (m, 2H, H-16, H-20), 7.26–7.35 (m, 3H, H-17, H-18, H-19), 7.07–7.14 (m, 2H, H-8, H-9), 6.98 (s, 1H, H-1), 6.17 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ 179.7 (C-12), 169.9 (C-5), 146.5 (C-15), 142.3 (C-7), 139.5 (C-4), 128.8 (C-17, C-19), 128.3 (C-18), 124.4 (C-16, C-20), 123.6 (C-9), 119.2 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = -0.34, -16.46; ESI-MS (pos) m/z: 306 (M + H+) (100 %), 214 (M + H+ - C6H6N) (30 %), 152 (M + H+ - C5H5N3OP) (19 %), 137 (M + H+ –C5H6N4OP) (45 %).
N-(4-Fluorophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)thiourea ( 11g )
Light brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with i1-fluoro-4-isocyanatobenzene (10 g), and it was obtained as a light brown solid); m.p. 168–170 °C; IR (KBr) νmax 3385, 3273, 3177, 3033 (–N–H, str), 1508 (–C=N, str), 1207 (–P=O, str), 1150 (–C=S, str), 1115 (–C–F, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 12.74 (brs, 1H, –P–NH–CS–, H-14), 9.78 (brs, 1H, –CS–NH–, H-10), 8.14 (d, 1H, J = 5.2 Hz, H-7), 7.53 (d, 2H, J = 8.4 Hz, H-17, H-19), 7.46 (d, 2H, J = 8.0 Hz, H-16, H-20), 7.17–7.21 (m, 2H, H-8, H-9), 6.54 (s, 1H, H-1), 5.98 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 181.5 (C-12), 169.9 (C-5), 160.2 (d, J = 31.2 Hz, C-18), 146.5 (C-7), 142.3 (C-15), 135.6 (C-4), 125.5 (C-16, C-20), 118.0 (C-9), 116.1 (C-17, C-19), 115.2 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = -1.1, -9.5; ESI-MS (pos) m/z: 324 (M + H+) (100 %), 229 (M + H+ - C6H4F) (42 %), 170 (M + H+ - C7H5FNS) (36 %), 152 (M + H+ - C5H3N3OP) (15 %); Anal. Calcd. for C12H11FN5OPS: C, 44.58; H, 3.43; N, 21.66; Found: C, 44.53; H, 3.41; N, 21.60 %.
N-(4-Chlorophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)thiourea ( 11h )
Light yellow solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1-isothiocyanato-4-chlorobenzene (10 h), and it was obtained as a light yellow solid); m.p. 131–133 °C; IR (KBr) νmax 3406, 3262, 3054 (-N-H, str), 1525 (–C=N, str), 1232 (–P=O, str), 1194 (–C=S, str), 823 (–C–Cl, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 12.23 (brs, 1H, –P–NH–CS–, H-14), 9.30 (brs, 1H, –CS–NH–, H-10), 8.22 (d, 1H, J = 5.2 Hz, H-7), 7.86 (d, 2H, J = 6.0 Hz, H-16, H-20), 7.40 (d, 2H, J = 6.4 Hz, H-17, H-19), 7.12–7.18 (m, 2H, H-8, H-9), 6.33 (s, 1H, H-1), 5.96 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 180.9 (C-12), 166.4 (C-5), 146.1 (C-7), 143.5 (C-15), 140.1 (C-18), 137.1 (C-16, C-20), 135.2 (C-4), 132.6 (C-17, C-19), 125.3 (C-9), 116.8 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −13.1; ESI-MS (pos) m/z: 340 (M + H+) (100 %), 342 (M + H++ 2) (33 %), 213 (M + H+ - C6H6ClN) (51 %), 170 (M + H+ - C7H5ClNS) (39 %).
N-(4-Nitrophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)thiourea ( 11i )
Yellow solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1-isothiocyanato-4-nitrobenzene (10i), and it was obtained as a yellow solid); m.p. 146–148 °C; IR (KBr) νmax 3422, 3254, 3042 (–N–H, str), 1532 (–N=O, str), 1504 (–C=N, str), 1242 (–P=O, str), 1208 (–C=S, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 12.43 (brs, 1H, –P–NH–CS–, H-14), 9.36 (brs, 1H, –CS–NH–, H-10), 8.69 (d, 2H, J=6.4 Hz, H-17, H-19), 8.10 (d, 1H, J=5.6 Hz, H-7), 7.48 (d, 2H, J=6.4 Hz, H-16, H-20), 7.21–7.28 (m, 2H, H-8, H-9), 6.42 (s, 1H, H-1), 6.08 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 180.1 (C-12), 164.2 (C-5), 147.6 (C-15), 146.9 (C-18), 145.8 (C-7), 134.6 (C-4), 130.7 (C-16, C-20), 128.6 (C-17, C-19), 124.4 (C-9), 117.0 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = −11.3; ESI-MS (pos) m/z: 351 (M + H+) (100 %), 213 (M + H+ - C6H6N2O2) (58 %), 195 (M + H+ - C5H7N3OP) (19 %), 182 (M + H+ - C5H6N4OP) (42 %).
N-(2-Oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)-N′-[3-(trifluoromethyl) phenyl]thiourea ( 11j )
Brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1-isothiocyanato-3-(trifluoromethyl)benzene (10j), and it was obtained as a brown solid); m.p. 154–156 °C; IR (KBr) νmax 3423, 3209, 3046 (–N–H, str), 1526 (–C=N, str), 1278 ((CF3) –C–F, str), 1228 (–P = O, str), 1190 (–C=S, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 12.66 (brs, 1H, –P–NH–CS–, H-14), 10.07 (brs, 1H, –CS-NH-, H-10), 8.08 (d, 1H, J = 5.2 Hz, H-7), 8.04 (s, 1H, H-16), 7.53 (t, 1H, J = 8.0 Hz, H-18), 7.45 (d, 1H, J = 7.6 Hz, H-18), 7.40 (d, 1H, J = 7.6 Hz, H-20), 7.10–7.21 (m, 2H, H-8, H-9), 6.83 (s, 1H, H-1), 6.74 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 181.5 (C-12), 169.8 (C-5), 149.4 (C-7), 146.4 (C-15), 142.3 (C-17), 140.4 (C-4), 129.9 (C-16), 129.2 (C-20), 128.9 (C-19), 126.1 (C-17, –CF3), 125.4 (C-9), 120.2 (C-18), 116.1 (C-8); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = -11.3; ESI-MS (pos) m/z: 374 (M + H+) (100 %), 356 (M + H+ - H2O) (74 %), 229 (M + H+ - C7H4F3) (16 %); Anal. Calcd. for C13H11F3N5OPS: C, 41.83; H, 2.97; N, 18.76; Found: C, 41.73; H, 2.95; N, 18.72 %.
N-(2,6-Difluorophenyl)-N′-(2-oxo-2,3-dihydro-1H-2λ 5 -[1,3,2]diazaphospholo[4,5-b]pyridin-2-yl)thiourea ( 11k )
Dark brown solid (this compound was prepared by the reaction of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b] pyridin-2-one (9) with 1,3-difluoro-2-isothiocyanatobenzene (10 k), and it was obtained as a dark brown solid); m.p. 187–189 °C; IR (KBr) νmax 3396, 3225, 3032 (-N-H, str), 1520 (–C = N, str), 1226 (-P = O, str), 1199 (–C = S, str), 1124 (–C–F, str) cm−1; 1H NMR (DMSO-d 6 , 400.13 MHz): δ = 12.49 (brs, 1H, -P-NH–CS–, H-14), 9.42 (brs, 1H, –CS-NH-, H-10), 8.14 (d, 1H, J = 5.6 Hz, H-7), 7.78 (d, 2H, J = 6.0 Hz, H-17, H-19), 7.52 (t, 1H, J = 6.4 Hz, H-18), 7.09–7.19 (m, 2H, H-8, H-9), 6.73 (s, 1H, H-1), 5.98 (s, 1H, H-3); 13C NMR (DMSO-d 6 , 100.62 MHz): δ = 179.8 (C-12), 169.8 (d, J = 29.5 Hz, C-16, C-20), 160.1 (C-5), 146.1 (C-7), 139.7 (C-4), 126.9 (C-18), 124.3 (C-9), 116.5 (C-8), 114.8 (C-15), 111.5 (C-17, C-19); 31P NMR (DMSO-d 6 , 161.9 MHz): δ = -11.9; ESI-MS (pos) m/z: 342 (M + H+) (100 %), 213 (M + H+ - C6H5F2N) (42 %), 173 (M + H+ - C5H6N4OP) (28 %).
Anti-inflammatory activity
In vitro anti-inflammatory activity
Anti-inflammatory activity in vitro was investigated using inhibition of albumin denaturation technique according to methods reported by Mizushima and Kobayashi 1968 and Sakat et al., 2010 with minor modifications. The test compounds and standard drug, diclofenac, were dissolved in minimum amount of dimethylformamide (DMF) and diluted with phosphate buffer to make the final concentration of 200 μg/mL. For in vitro assay, the mixture was prepared consisting of tested compounds (0.05 mL) and 0.45 mL of 1 % aqueous solution of bovine albumin fraction in phosphate buffer. The pH of test samples was adjusted to 6.8 by adding 1 N HCl with small trembling. After, the test samples were incubated at 37 °C for 20 min and heated at 57 °C for 20 min in water bath to induce the denaturation of protein. The samples were cooled, and measured their turbidity spectrophotometrically at 660 nm. Per cent inhibition of denaturation was calculated using the following equation. The experiments were performed in triplicate, and average results are summarized in Fig. 2.

In vitro anti-inflammatory activity of the title urea and thiourea derivatives 11(a–k)
In vivo anti-inflammatory activity
Animals
Albino Wistar rats of either sex (150–200 g) were obtained from Central Animal House, Sri Venkateswara University, Tirupati. The animals were housed in cages at room temperature of 23 ± 2 °C with 12 h of light and dark cycles and fed with free access of food and water, ad libitum. The animals are fetched to the laboratory 12 h before starting the experiment and fed with only water, ad libitum. The experiments on animals were performed according to the protocol, which has been approved by Institutional Animal Ethics Committee.
Carrageenan-induced Paw Oedema method
Anti-inflammatory activity was assessed in vivo using the most conventional carrageenan-induced hind paw oedema method. Albino Wistar rats were divided into 7 groups containing 6 rats for each one and were fasted for 12 h before commencement of the experiment. A mark was made on left and right side of the hind paws just beyond tibio-tarsal junction to ensure constant paw one. Freshly prepared 1 % of 0.1 mL saline solution of carrageenan was inducted into right hind paw of rats by subcutaneous injection. The first group was administered orally with control (0.9 % of 0.1 mL saline solution), and the second group was treated with standard drug, diclofenac sodium 20 mg/kg body weight (positive control). The remaining groups were administered with synthesized compounds at the same dosage of the standard drug, 1 h before the administration of carrageenan. The paw volumes were measured immediately (initial volume) using plethysmometer, and there after, the paw volumes were noted at 1-, 2-, 3- and 4-h intervals of time. The difference between initial and subsequent reading concerned the oedema volume for the corresponding time. The percentage of inhibition was calculated using the following formula.
where V t is the oedema volume after treatment with synthesized compounds/drugs and V c is the oedema volume after treatment with negative control.
Cotton pellet-induced granuloma method
In vivo anti-inflammatory activity was also evaluated using cotton pellet-induced granuloma method. Albino Wistar rats were selected and divided into 7 groups (6 animals each one). Group-1 was administered with control (saline), and Group-2 received standard drug, diclofenac sodium (5 mg/kg body weight). Groups 3 to 7 were treated with synthesized compounds with equal dosage of standard. Accurately, 35 ± 1 mg of cotton swabs was cut from dental rolls and sterilized in a hot air oven at 90 °C for 2.0 h. The selected animals’ abdomen was shaved cleanly and washed with 70 % ethanol. Two sterilized pellets were implanted into subcutaneous tissue on either side of axilla and sterile grass pith in groin region of the rat through a single incision under mild general ether anaesthesia. The administrations of drug candidates were repeated regularly for each 24 h until 7 days. On eighth day, the rats were killed and cotton pellets accompanying granulomatous tissues were removed with a pair of forceps. The pellets were freed from extraneous tissue and dried in an oven at 60 °C for 30 h. The dried pellets were weighted, and difference between the initial and final weight was taken as a measure of granuloma formation. The percentage of inhibition was calculated using the formula given below.
where W t is a granuloma weight of the tested compound and W c is a granuloma weight of the control.
Acute toxicity
Acute toxicity was performed according to the Organization for Economic Cooperation and Development (OECD) guidelines. Albino female mice (25–30 g wt) were used to carry out the experiment. The groups of female mice each consisting of 6 mice were kept into cage at 27 ± 2 °C with 60 % relative humidity, and 12-h light/dark cycle was maintained. A specified fixed dose of selected compounds in 50, 100, 150, 200 and 300 mg/kg was administered orally as a single dose as fine suspension prepared in saline using gum acacia powder. The acute toxicity symptoms and the behavioural changes like dullness, piloerection and recumbency produced by test compounds were observed continuously for 4-, 8-, 12- and 24-h intervals of time, and the behavioural changes were recorded. Acute toxicity data revealed that none of the test compounds exhibited any toxicity up to 400 mg/kg and no animal death was observed. Further, no abnormalities were detected in tested animals during the treatment of test compounds.
Molecular modelling study
Refinement of COX-2
The 3.0 Å crystal structure of cyclooxygenase-2 (COX-2) (PDB ID: 1CX2) was derived from protein data bank (PDB) (Rajiv et al., 2012). The 1CX2 protein was prepared by removing water molecules and bound ligands. Further, the COX-2 protein was refined with MD simulation, which was carried out with Visual Molecular Dynamics (VMD) tool. The CHARMM 27 field was used for parameterization, and the program NAMD was used for energy minimization and molecular dynamics (MD) simulations. All of MD simulations were carried out in explicit water, employing periodic boundary conditions. The system was first energy minimized for 1000 steps with atoms of COX-2 (1CX2) fixed, and then, energy minimization was performed for 2500 steps.
Simulation parameters
The MD simulations system was equilibrated at 300 k for 10 ps with COX-2 (1CX2) atoms fixed followed by 20 ps MD without restraints. The system was subsequently simulated for 100 ps at 350 k with the following parameters. The classical equations of motion were integrated by a leap-frog integrator using a time step at 1 fs. The impulse-based ver let-I/r-RESPA method was used to perform multiple time stepping: 4 fs long-range electrostatic, 2 fs for short-range non-bonded forces, and 1 fs for bonded force. The swift function was used to cut off the Lennard-Jones potential, with the first cut-off at 10 Å and the second cut-off at 12 Å. Short-range interactions were calculated at intervals of 4 fs. All bonds involving hydrogen atoms were constrained to their equilibrium bond parameters using the SHAKE along them. Langevin dynamics were employed to maintain the pressure at 1 atm with a Langevin piston period of 100 fs and oscillation decay time of 50 fs. Trajectories were recorded every 200 fs. Subsequently, the dynamics behaviour and structural changes of the receptor were analysed by calculation of energy and the root-mean-square deviation (RMSD), and the graph is shown in Fig. S1 (See Supplementary Material).
Docking Studies
Docking experiments were performed using AutoDock v. 4.2 tool (Madhu Sudhana and Usha Rani, 2013) in order to find the preferred binding conformations of ligand in the ICX2. The analysis of binding conformation using a scoring function was based on the free energy of binding. For the ligands, conjugate gradient minimizations with CHARMM force field were performed. The grid parameter file of 1CX2 was generated using AutoDock v. 4.2 (Grott and Olson, 2010). A grid box was generated; it was large enough to cover the entire receptor-binding site. The number of grid points in x-, y- and z-axes was 60 × 60 × 60. The distance between two connecting grid points was 0.375. AutoDock 4.2 and a Lamarckian genetic algorithm were used for receptor-fixed ligand-rigid docking calculations. Ten search attempts (GA run parameter) were performed for ligand. The maximum number of energy evaluations before the termination of LGA run was 2,500,000, and the maximum number of generations of the LGA run before termination was 27,000. Other docking parameters were set to the software’s default values. Ten conformations of ligand in complex with the receptor were obtained, which were finally ranked on the basis of binding energy. The resulting conformations were visualized in the PyMol Viewer tool (https://www.pymol.org/).
QSAR studies
The synthesized compounds were predicted for quantitative structure–activity relationship (QSAR) using Osiris and Molinspiration servers.
Antimicrobial activity
Two gram-positive bacteria, Staphylococcus faecalis (MTCC-0459), Bacillus cereus (ATCC-11778), two gram-negative bacteria, Escherichia coli (ATCC-9637), Pseudomonas marginalis (MTCC-2758), and three fungi such as Aspergillus niger (MTCC-1881), Fusarium oxysporum (MTCC-1755)and Penicillium chrysogenum (MTCC-1996) were selected to investigate antimicrobial activity of the synthesized urea and thiourea products at two different concentrations, 50 and 100 μg/disc using disc diffusion method approved by the guidelines of National Committee for Clinical Laboratory Standards (NCCLS). Stock solutions of the synthesized compounds were prepared in DMSO, and it was used as negative control. The standard antibiotics, ciprofloxacin for bacteria and ketoconazole for fungi were used as positive controls.
Approximately, 24-/48-h old culture of selected bacteria/fungi was mixed with sterile physiological saline, and the turbidity was adjusted to the standard inoculum of McFarland scale 0.5 ≈ 106 colony-forming unit (CFU) per mL. Petri plates containing 20 mL of Muller-Hinton agar (Hi-media) and potato dextrose agar (Hi-media) were used for examining antibacterial and antifungal activities, respectively. The culture was spread on surface of the solidified media, and a sterile glass spreader was used for even distribution of the inoculum. Sterile discs of Whatman No. 1 filter paper of about 8 mm diameter were impregnated in the test samples; positive control and negative control were then placed on the culture plates. The bacteria-inoculated plates were incubated for 24 h at 37 °C, and fungal-inoculated plates were incubated for 72 h at 25 °C. The zone of inhibition of bacteria/fungi around the disc was calculated edge-to-edge zone of the confluent growth which corresponds to the sharpest edge of the zone and was measured in millimetres. The tests were repeated in triplicate, and average value was taken as final reading.
Determination of the minimum inhibitory concentration (MIC)
Minimum inhibitory concentration (MIC) was determined by micro-broth dilution method. The minimum concentration, at which there was no visually detectable bacteria/fungal growth, was taken as MIC. To examine MICs of the test solutions, various serial concentrations 50, 45, 40, 35, 30, …, 1 μg/mL of the test solutions were prepared from the stock solution. Specifically, 0.1 mL of standardized inoculum (1–2 × 107 CFU/mL) was added to each test tube. The bacterial tubes were incubated aerobically for 24 h at 37 °C, and fungal tubes were incubated for 72 h at 25 °C. Control was maintained for each test sample. The lowest concentration (highest dilution) of test compound that produced no visible signs of bacterial/fungal growth (no turbidity) when compared to the control tubes was regarded as MIC.
Results and discussion
Chemistry
The synthetic strategy for the synthesis of title urea and thiourea derivatives 11a–k is illustrated in Scheme 1.

Preparation of urea and thiourea compounds of 2-amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-one
At first, the key intermediate, 2-amino-2,3-dihydro-1H-2λ5 [1,3,2]diazaphospholo[4,5-b]pyridin-2-one, 9 was prepared in two steps for the synthesis of target scaffolds. Pyridine-2,3-diamine, 6 was cyclized with slow addition of phosphoryl chloride (POCl3) 7 in the presence of catalytic amount of base, dimethyl piperazine (DMPipz), to afford the cyclized monochloride derivative, 2-chloro-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-one, 8. Subsequently, the cyclized monochloride 8 was involved in amination by the replacement of ‘Cl’ with –NH2 using NaNH2 to get precursor compound 9 by filtering the salt, NaCl, and the evaporation of solvent from the filtrate followed by recrystallization of the crude product from methanol. Finally, the desired target urea 11a–e/thiourea 11f–k derivatives were achieved by the reaction of 9 with various functionalized phenylisocynates 10a–e/ phenylisothiocynates 10f–k in the presence of base. All the reactions were performed as solution in THF/Py (1:2), and the reaction mixtures were stirred for 3.0–4.5 h at 55 °C to form the final products. The final pure urea and thiourea derivatives 11a–k were obtained by washing the crude product with 20 % DCM (DCM and n-Hexane) (10 mL × 5 times) followed by recrystallization from methanol. Few molecules are purified by column chromatography using dichloromethane and methanol (9.5:0.5) as an eluent (Table 1).
The structures of all newly synthesized products were confirmed by IR, NMR (1H, 13C and 31P) and mass spectral data. The possible absorption bands in IR spectra in the region of 3470–3010 cm−1 are assigned for –NH stretching in urea and thiourea as well as present in diazaphosphole moiety. The appearance of bands in the region of 1625–1685 cm−1 confirmed –C=O stretching in urea derivatives, 1190–1235 cm−1 corresponding to –C=S stretching in thiourea derivatives and 1205–1260 cm−1 relating to –P=O stretching present in the synthesized compounds. In 1H NMR spectra, appearance of signals as singlets in the ranges of 10.20–10.96 ppm and 9.01–9.70 ppm confirmed the protons attached to nitrogen of –P(O)–NH– and –C(O)–NH–, respectively, in urea derivatives, and chemical shift values in the region of 12.20–12.86 and 9.24–9.84 ppm confirmed the protons attached to nitrogen of –P(O)–NH– and –C(S)–NH–, respectively, in thiourea derivatives. Two signals separated as singlets at 5.86–6.28 ppm and 6.30–6.98 ppm were assigned for protons attached to nitrogen in the diazaphosphole moiety. In 13C NMR spectra, the chemical shift values in the region of 153–156 and 178–184 ppm confirmed the carbons of –C=O and –C=S in urea and thiourea derivatives, respectively. The appearance of signals in 31P NMR spectra in the range of −0.34 to −13.8 ppm confirmed the presence of ‘P’ atom in all the title products. Molecular ion peaks and fragmented ions in the mass spectra of the products have given additional assistance in the structural elucidation of the products.
Anti-inflammatory activity
In vitro anti-inflammatory activity
The anti-inflammatory activity of the newly synthesized urea and thiourea derivatives, 11a–k was tested by in vitro method and inhibition of albumin denaturation technique (Mizushima and Kobayashi, 1968; Sakat et al., 2010). In vitro activity of the compounds was screened at the concentration of 200 µg/mL oral dose and compared with the same dose of standard drug, diclofenac, and results are presented in Fig. 2. The bio-screening results disclosed that all the compounds exhibited inhibitions of albumin denaturation at the concentration of 200 µg/mL, and most of the compounds showed more than to half maximal (50 %) inhibition except compound 11f that showed 44.73 %. Compound 11f did not have any substituted functional group on phenyl ring, which might be the cause for low inhibition of albumin denaturation. Promptly, urea compounds, 11a bonded with 4-fluorophenyl ring (70.35 %), 11b connected with 4-nitrophenyl ring (78.95 %) and 11d having 2-fluoro-5-nitrophenyl ring (78.14 %), and thiourea derivatives, 11i containing 4-nitrophenyl ring (70.36 %) and 11j having with 3-trifluoromethylphenyl ring (78.39 %), exhibited potent inhibition of albumin denaturation closer to the standard drug, diclofenac (89.64 %). It was observed from the biological activity that more electronegative functional groups like F-, NO2- and –CF3-substituted compounds showed potential activity as compared to less electronegative groups such as –Cl-substituted compounds, 11h. In particular, the electronegative functional groups present on para or ortho position exhibited promising activity except –CF3 present on meta position. The potent in vitro anti-inflammatory activity results of the synthesized compounds have encouraged performing in vivo study for these potent active compounds.
In vivo anti-inflammatory activity
Consequently, to examine the efficacy of the active synthesized compounds (11a, 11b, 11d, 11i and 11j) found in in vitro study, in vivo anti-inflammatory activity was screened using carrageenan-induced paw oedema (Winter et al., 1962) and cotton pellet-induced granuloma (Goldstein et al., 1967) methods. The potential action of test samples was compared with the reference anti-inflammatory drug, diclofenac. The obtained results were expressed as mean ± SE. Statistical analysis was carried out using one-way analysis of variance (ANOVA) with Dennett’s post-test followed by the significant difference [P < 0.05].
In carrageenan-induced paw oedema method, 1.0 h before carrageenan administration Albino Wistar rats were treated with the active synthesized compounds at the same dosage (20 mg/kg) of the standard drug, diclofenac. The paw volumes were measured at 1.0-, 2.0-, 3.0- and 4.0-h intervals of time, and calculated % inhibition of oedema is given in Table 2. As seen in Table 2, all the tested compounds showed potent activity after 1.0-h treatment and gradually reduced for subsequent hours, whereas urea compounds 11a bearing 4-fluorophenyl ring and 11d bonding with 2-fluoro-5-nitrophenyl ring exhibited potential inhibition of oedema 22.02 and 22.93 %, respectively, and it was closer to the standard, diclofenac (22.48 %) after 1.0-h treatment. One of the thiourea derivatives 11j bearing 3-trifluromethylphenyl ring showed more inhibition of oedema (21.67 %) than that of standard, diclofenac (20.18 %) after 2.0-h treatment. Compound 11i showed moderate inhibition of oedema and did not show any effect even after 4 h of post-administration. Additionally, to distinguish in vivo anti-inflammatory effect of the active synthesized compounds, cotton pellet-induced granuloma method was performed at the concentration of 5 mg/kg. Weights of granuloma of the treated rats and % inhibition of the granuloma were calculated, and the results are tabulated in Table 3. The compounds 11a (48.53 %), 11d (52.61 %) and 11j (44.41 %) exhibited potent granuloma inhibition activity approximately closer to standard drug, diclofenac (56.14 %). It is interesting to note that among all the newly synthesized compounds 11a, 11d and 11j showed best anti-inflammatory activity both in in vitro and in vivo, but the compound 11i showed promising in vitro anti-inflammatory activity and low activity in in vivo study.
Acute toxicology study
Acute toxicity of the synthesized compounds 11a, 11b, 11d, 11i and 11j was performed in single dosage administration according to the Organisation for Economic Cooperation and Development (OECD) guidelines (Pahari et al., 2010; Jaouhari et al., 1999). Albino female mice (25–30 g wt) were used to carry out the experiment. The experimental results exposed that the tested compounds did not exhibit any abnormalities (toxicity) until the compound concentration of 300 mg/kg within 24-h observation.
Molecular docking study
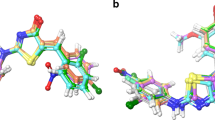
In order to interpret the binding mode of the most active anti-inflammatory synthesized derivatives, 11a, 11b, 11d, 11i and 11j with COX-2 isoenzyme, in comparison with the standard anti-inflammatory agent, diclofenac, the molecular docking study was subjected using the AutoDock Vina tools (Madhu Sudhana and Usha Rani, 2013). Three-dimensional structure of COX-2 complex with a selective inhibitor, SC-558, as the target protein (Fig. 3) was taken from the PDB (Protein Data Bank) entry 1CX2. The COX-2 protein was refined with MD simulation which was carried out with the Visual Molecular Dynamics (VMD) tool (Humphrey et al., 1996). The CHARMM 27 field was used for parameterization, and the program NAMD was used for energy minimization and molecular dynamics (MD) simulations (MacKerell et al., 1998; Phillips et al., 2005). The diclofenac and selected ligands were redocked at the crystal enzyme structure of the 1CX2, and the best energy conformations of 1CX2-ligand were studied. To evaluate the docking accuracy of AutoDock 4.2, the cocrystallized ligand diclofenac was redocked within the active site of 1CX2. AutoDock was successful in reproducing the binding position for diclofenac, showing a RMSD of 3.4 Å for all atoms in comparison with original poses of X-ray structure complexes. The analysis of the binding conformation using a scoring function was based on the free energy of binding (Huey et al., 2007). The individual energies of the 1CX2 and the ligands are tabulated in Table 4.

The co-crystalized structure of COX-2
In the docked conformers, the compounds showing the best binding energies (scores) in order are 11a (−9.9) = 11b (−9.9) > 11d (−9.7) > 11i (−9.2) = 11j (−9.2) > diclofenac (−7.8). Interestingly, it was observed that all the active molecules showed the best energies than that of anti-inflammatory standard drug, diclofenac.
The compound 11a was bound to the binding energy of −9.9 kcal/mol by the formation of five hydrogen bonds with Gly45, Cys41, Gln42, Glu465 and Glu465 of active site of 1CX2 protein, and the compound 11b was bound to the binding energy of −9.9 kcal/mol by the formation of six stable hydrogen bonds with Cys41, Gly45, Arg44, Gln461, Asn39 and Ala156 residues of target protein within the active site. Compound 11d was bound to the binding energy of −9.7 kcal/mol by the formation of seven hydrogen bonds with Pro154, Ala156, Asn34, Arg44, Gly45, Cys41 and Cys36 residues, compound 11i was bound to the binding energy of −9.2 kcal/mol by the formation of seven hydrogen bonds with Ala156, Gln461, Asn39, Arg44, Gly45, Gly45 and Cys 47 residues, and the compound 11j was bound to the binding energy of −9.2 kcal/mol by the formation of two hydrogen bonds with Glu465 and Gly45 residues within the active site of 1CX2 protein. The standard compound diclofenac was bound to the binding energy of −7.8 kcal/mol by hydrophobic and electrostatic interactions among active site residues such as Glu465, Ser471, Gly45, Asn43 and Gln461 of 1CX2 protein (Fig. 4).

Interaction mode of 11a, 11b, 11d, 11i, 11j and diclofenac docked and minimized in the COX-2 enzyme
QSAR studies
QSAR studies employ statistical techniques for correlating physical and chemical properties of molecules with their observed biological activities. We performed QSAR studies on the active synthesized compounds using Osiris and Molinspiration servers, and the obtained results are given in Table 5. The data displayed that all the tested compounds satisfied the Lipinski’s rule of five with zero violations and also the octanol/water partition coefficient (miLogp). In addition, it was predicted that the drug transport properties such as bio-availability, solubility and topological molecular polar surface area (TPSA) and toxicity properties such as mutagenicity, tumorigenicity, irritant and reproductive effect were calculated. The tested compounds did not exhibit any toxicity, whereas all the compounds exhibited good drug transport properties. However, compound 11j showed better drug transport properties as compared to the remaining tested compounds.
Antimicrobial activity
It is well known that pyridine (El-Sayed Ali et al., 2009; Khidre et al., 2011), urea and thiourea (Saeed et al., 2010; Siwek et al., 2011) derivatives have unveiled promising antimicrobial activity. Therefore, all the newly synthesized compounds were evaluated for their antibacterial and antifungal activities for probing the potent compounds from the synthesized compounds using disc diffusion method approved by the guidelines of National Committee for Clinical Laboratory Standards (NCCLS) (National Committee et al., 1997). The activities were screened at two different concentrations 50 and 100 μg/disc in DMSO of the synthesized compounds.
The bacterial cultures such as S. faecalis (MTCC-0459), B. cereus (ATCC-11778) (Gram-positive) and E. coli (ATCC-9637), P. marginalis (MTCC-2758) (gram-negative) were used for evaluating the antibacterial activity, and ciprofloxacin was used as the standard for comparing the activity. The antibacterial bio-screening data given in Table 6 displayed that urea derivative 11c bearing 2,4-difluorophenyl ring and thiourea derivatives 11f bonded with simple phenyl ring, 11j attached with 3-trifluoromethylphenyl ring and 11k linked with 2,6-diflurophenyl ring exhibited promising antibacterial activity against all the tested bacterial strains which are closer to the standard, ciprofloxacin. However, compounds 11d against P. marginalis (MTCC-2758), 11e against E. coli (ATCC-9637) and 11h against B. cereus (ATCC-11778) showed potent activity; nevertheless, the remaining compounds exhibited moderate activity.
Three fungal strains such as A. niger (MTCC-1881), F. oxysporum (MTCC-1755) and P. chrysogenum (MTCC-1996) were selected to investigate the antifungal activity, and ketoconazole was used as the standard drug (Table 7). Among all the synthesized compounds, 11c having 2,4-difluorophenyl ring, 11e possessing 3,4-dichlorophenyl ring, 11h bonding 4-chlorophenyl ring and 11j bearing with 3-trifluoromethylphenyl ring against all the tested fungal strains and, particularly, the compounds 11a against F. oxysporum, 11d, 11i, and 11k against P. chrysogenum exhibited comparable potential growth of fungal inhibition with the standard, ketoconazole. The other synthesized compounds exhibited moderate activity on tested microorganisms. However, the compounds, 11c, 11f, 11j and 11k exhibited potent antibacterial activity and 11c, 11e, 11h and 11j showed promising antifungal activity.
The minimum inhibitory concentration was further investigated using micro-broth dilution method (Bonjar Shahidi et al., 2004) for the potent compounds 11a, 11c, 11e, 11f, 11g, 11h, 11j and 11k which were selected based on the performance of the zone of bacterial/fungal inhibition at 50 and 100 μg/disc concentrations. The MIC data (Table 8) revealed that all the tested compounds showed low MIC values in the range of 8.0–31.0 μg/mL, whereas compounds 11a (13.0 μg/mL) against B. cereus, 11c against B. cereus (8.0 μg/mL) and E. coli (10.0 μg/mL), 11e (11.0 μg/mL) and 11j (11.0 μg/mL) against A. niger and 11e (14.0 μg/mL) against F. oxysporum exhibited lower MIC values.
Conclusion
Based on the biological importance of P-heterocyclic, urea and thiourea derivatives and pyridine frameworks, we designed new compounds by incorporating above moieties together in one scaffold and synthesized a series of [1,3,2]diazaphospholo[4,5-b]pyridin-2-one-based urea and thiourea derivatives 11a–k. All the newly synthesized compounds were evaluated for their in vitro and in vivo anti-inflammatory and in vitro antimicrobial activities as well as a molecular docking study at COX-2 isoenzyme. The compounds showed significant anti-inflammatory activity both in vitro and in vivo, whereas compounds 11a, 11d and 11j exhibited better inhibition of oedema than that of the remaining compounds and approximate to the standard, diclofenac at the administration of 1 and 2 h, respectively. Among the synthesized compounds, urea derivative 11c and thiourea derivatives 11f, 11j and 11k showed better bacterial growth of inhibition against all the tested bacterial strains, and urea derivatives 11c and 11e, and thiourea derivatives 11h and 11j exhibited promising zone of inhibition against tested fungal strains. Interestingly, compounds 11c against B. cereusand E. coli, and 11e and 11j against A. niger showed potential activity at lower minimum inhibitory concentrations in the range of 8–11 µg/mL. In the docking study, the active synthesized compounds 11a, 11b, 11d, 11i and 11j showed good binding profile (binding affinities in the range of −9.2 to −9.9 Å) with COX-2 isoenzyme than that of the standard, diclofenac (binding affinity −7.8 Å), and these compounds exhibited good drug transport properties and no toxicity properties.
Overall, the compounds bearing lipophilic functional groups (F, NO2 and CF3) exhibited potential activities, and the present series of compounds could be developed as new class of dual anti-inflammatory and antimicrobial agents. Therefore, structural modification of this kind of molecules would deserve as worthy of new chemotherapeutic agents in future.
References
Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI (1992) Gastrointestinal damage associated with the use of nonsteroidal anti-inflammatory drugs. N Engl J Med 327:749–754
Ashley GW, Bartlett PA (1982) A Phosphorus-containing pyrimidine analog as a potent inhibitor of cytidine deaminase. Biochim Biophys Acta 108:1467–1474
Bennett JS, Daugherty A, Herrington D, Greenland P, Roberts H, Taubert KA (2005) The use of nonsteroidal anti-inflammatory drugs (NSAIDs): a Science Advisory from the American Heart Association. Circulation 111:1713–1716
Bernardino AMR, De Azevedo AR, Pinheiro LCD, Borges JC, Carvalho VL, Miranda MD, De Meneses MDF, Nascimento M, Ferreira D, Rebello MA (2007) Synthesis and antiviral activity of new 4-(phenylamino)/4-[(methylpyridin-2-yl)amino]-1-phenyl-1Hpyrazolo[3,4-b]pyridine-4-carboxylic acids derivatives. Med Chem Res 16:352–369
Bonjar Shahidi GH (2004) Evaluation of antibacterial properties of iranian medicinal-plants against Micrococcus luteus, Serratia marcescens, Klebsiella pneumoniae and Bordetella bronchoseptica. Asian J Plant Sci 3:82–86
Bull EOJ, Naidu MSR (2000) Isoquino[2,1][1,3,2]-benzodiazaphosphrine derivatives: new potential agents for cancer chemotherapy. Phosphorus, Sulfur Silicon Relat Elem 162:231–243
El-Sayed Ali T (2009) Synthesis of some novel pyrazolo[3,4-b]pyridine and pyrazolo[3,4-d]pyrimidine derivatives bearing 5,6-diphenyl-1,2,4-triazine moiety as potential antimicrobial agents. Eur J Med Chem 44:4385–4392
Eugenia FC, Maria L, Gheorghe FC, Dana V (2006) Synthesis, characterization and correlative biological effects in wheat of a benzoxaza- and a diaza-phosphorus(V) heterocycles. J Serb Chem Soc 71:1031–1038
Eweis M, Elkholy SS, Elsabee MZ (2006) Antifungal efficacy of chitosan and its thiourea derivatives upon the growth of some sugar-beet pathogens. Int J Biol Macromol 38:1–8
Fournet F, Sannier C, Monteny N (1993) Effects of the insect growth regulators OMS 2017 and diflubenzuron on the reproductive potential of Aedes aegypti. J Am Mosq Control Assoc 9(4):426–430
Goldstein SA, Shemano L, Demer R, Beiler JM (1967) Cotton pellet granuloma pouch method for evaluation of anti-inflammatory activity. Arch Int Pharmacodyn Ther 165:294–301
Green GA (2001) Understanding NSAIDs: from aspirin to COX-2. Clin Cornerstone 3:50–60
Grott O, Olson J (2010) AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
He LN, Zhuo RX, Chen RY, Li K, Zhang YJ (1999) Synthesis of biologically active phosphorus heterocycles via cyclization reactions of Lawesson’s Reagent. Heteroat Chem 10:105–111
Hemmert C, Pitie M, Renz M, Gorintzka H, Soulet S, Meunier B (2001) Preparation characterization and crystal structure of manganese(III), Iron(III) and Cupper(III) complexes of the bis [di-1,1-(2-pyridyl)ethyl] amine (BDPEA) ligand; evaluation of their DNA cleavage activities. J Biol Inorg Chem 6:14–22
Hewitt DG, Newland GL (1977) Organophosphorus compounds. P-Arylated perhydro-1,2-azaphosphorines. Aust J Chem 30:579–587
Holla BS, Ashok M (2007) Convenient synthesis of some thiadiazolotriazinones carrying 4-methylthiobenzyl moieties as possible antimicrobial agents. Phosphorus Sulfur Silicon Relat Elem 182:981–991
Huey R, Morris GM, Olson AJ, Goodsell DS (2007) Software news and update a semiempirical free energy force field with charge-based desolvation. J Comput Chem 28:1145–1152
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Jaouhari JT, Lazrek HB, Seddik A, Jana M (1999) J Ethnopharmacol 64:211–217
Kapinos LE, Sigel H (2002) Acid/base and metal ion binding properties of pyridine-type ligands in aqueous solution. Effect of ortho substituents and interrelation between complex stability and ligand basicity. Inorg Chim Acta 337:131–142
Karp GM (1999) An expeditious route to novel 1,4,2-benzodiazaphosphepin-5-one 2-oxide analogues. J Org Chem 64:8156–8160
Khidre RE, Abu-Hashem AA, El-Shazly M (2011) Synthesis and anti-microbial activity of some 1- substituted amino-4, 6-dimethyl-2-oxo-pyridine-3-carbonitrile derivatives. Eur J Med Chem 46:5057–5064
Koteswara Rao V, JanardhanRao A, Subba Reddy S, Naga Raju C, VisweswaraRao P, Ghosh SK (2010) Synthesis, spectral characterization and biological evaluation of phosphorylated derivatives of galanthamine. Eur J Med Chem 45:203–209
Kubota S, Horie K, Misra HK, Toyooka K, Uda M, Shibuya M, Terada H (1985) Synthesis and uncoupling activities of hydrophobic thioureas. Chem Pharm Bull 33:662–666
Kurosaki H, Sharma RK, Aoki S, Inoue T, Okamoto Y, Sigiura Y, Doi M, Ishida T, Oysuka M, Goto M (2001) Synthesis, characterization, and spectroscopic properties of three novel pentadentate copper (II) complexes related to the metalchelating inhibitors against DNA binding with HIV-EP1. J Chem Soc Dalton Trans 1:441–447
Liu W, Zhou J, Zhang T, Zhu H, Qian H, Zhang H, Huang W, Gust R (2012) Design and synthesis of thiourea derivatives containing a benzo[5,6]cyclohepta[1,2-b]pyridine moiety as potential antitumor and anti-inflammatory agents. Bioorg Med Chem Lett 22:2701–2704
MacKerell AD Jr, Bashford D, Bellott M, Dunbrack RL Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wio´rkiewicz-Kuczera J, Yin D, Karplus M (1998) All atom empirical potential for Molecular Modeling and Dynamics studies of proteins. J Phys Chem. B 102:3586–3616
Madhu Sudhana S, Usha Rani A (2013) Identification of a potent inhibitor against Cathepsin-K for osteoporosis: a structure based virtual screening approach. Int J Pharm Bio Sci 4(B):927–943
Mizushima Y, Kobayashi M (1968) Interaction of anti-inflammatory drugs with serum proteins, especially with some biologically active proteins. J Pharma Pharmacol 20:169–173
National Committee for Clinical Laboratory Standards (1997) NCCLS approved standards M27-A. Wayne, USA
Pahari N, Saha D, Jain VK, Jain B, Mridha D (2010) Synthesis and evaluation of acute toxicity studies and analgesic characters of some novel indole derivatives. Int J Pharma Sci Res 1:399–408
Paronikyan EG, Noravyan AS, Dzhagatspany IA, Nazaryan IM, Paronikyan RG (2002) Synthesis and anticonvulsant activity of isothiazolo[5,4-b]pyrano(thiopyrano)[4,3-d]pyridine and isothiazolo[4,5-b]-2,7-naphthyridine derivatives. Pharm Chem J 36:465–467
Patel NB, Agravat SN, Shaikh FM (2011) Synthesis and anti- microbial activity of new pyridine derivatives-I. Med Chem Res 20:1033–1041
Phillips JC, Braun R, Wang W, Gumbart J, TajKhorshid E, Villa E, Chipot C, Skell RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Pozharskii AF, Soldatenkov AT, Katritzky AR (1997) Heterocycles in Life and Society. Wiley, UK
Rajiv KT, Sandhya B, Gita C, Girdhar SD, Suresh K, Vandana R, Naveen M, Azad R, Aruna Sree MK, Obaid A (2012) Synthesis and pharmacological evaluation of pyrazole [4,3-c] cinnoline derivatives as potential anti-inflammatory and antibacterial agents. Eur J Med Chem 57:176–184
Ramana KV, Rasheed S, Sekhar KC, Kumar KH, Raju CN (2013) Synthesis of novel phosphorylated guanidine derivatives from cyanamide and their anti-inflammatory activity. Chem Pharm Bull 61:25–32
Saeed S, Bhatti MH, Tahir MK, Jones PG (2008) Ethyl 4-(3-butyrylthioureido)benzoate. Acta Crystallogr Sect E 64:o1369
Saeed S, Rashid N, Jones PG, Ali M, Hussain R (2010) Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents. Eur J Med Chem 45:1323–1331
Sakat S, Juvekar AR, Gambhire MN (2010) In vitro antioxidant and anti-inflammatory activity of methanol extract of Oxalis corniculata Linn. Int J Pharma Pharmaco Sci. 2:146–155
Siwek A, Staczek P, Stafenska J (2011) Synthesis and structure-activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with Gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur J Med Chem 46:5717–5726
Srivastava A, Pandeya SN (2011) Indole a versatile nucleuse in pharmaceutical field. Int J Curr Pharm Rev Res 4:5–8
Subba Rao D, Srinivasulu D, Rajsekhar D, Naga Raju C (2013) CeCl3 7H2O-SiO2: catalyst promoted microwave assisted neat synthesis, antifungal and antioxidant activities of a-diaminophosphonates. Chin Chem Lett 24:759–763
Tucker TJ, Sisko JT, Tynebor RM, Williams TM, Felock PJ, Flynn JA, Lai M, Laing Y, McGaughey G, Liu M (2008) Discovery of 3-{5-[(6-amino-1H-pyrazolo[3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}-5-chlorobenzonitrile (MK-4965): a potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J Med Chem 51:6503–6511
Vane JR, Botting RM (1996) Mechanism of action of anti-inflammatory drugs. Scand J Rheumatol Suppl 102:9–21
Venkatachalam TK, Mao C, Uckun FM (2004) Effect of stereochemistry on the anti-HIV activity of chiral thiourea compounds. Bioorg Med Chem 12:4275–4284
Vonkeman HE, Van de Laar MA (2010) Nonsteroidal anti-inflammatory drugs: adverse effects and their prevention. Semin Arthritis Rheum 39:294–312
Winter CA, Risly EA, Nuss GW (1962) Carrageenan induced edema in the hind paw of the rat as an assay for anti-inflammatory drugs. Proc Soc Exp Biol Med 111:544–547
Yuan YF, Weang JT, Gimeno MC, Laguna A, Jones PG (2001) Synthesis and characterization of copper complexes with N-ferrocenoyl-N′(alkyl)thioureas. Inorg Chim Acta 324:309–317
Zhang YM, Wei TB, Wang XC, Yang SY (1998) Synthesis and biological activity of N-aroyl-N’-carboxyalkyl thiourea derivatives. Indian J Chem Sect B 37:604–606
Zhang YM, Wei TB, Xian L, Gao LM (2004) An efficient synthesis of polymethylene-bis-aroyl thiourea derivatives under the condition of phase-transfer catalysis. Phosphorus Sulfer Silicon Relat Elem 179:2007–2013
Zhou WQ, Li BL, Zhu LM, Ding JG, Yong Z, Lu L, Yang XJ (2004) Structural and spectral studies on N-(4-chloro)benzoyl-N′-(4-tolyl)thiourea. J Mol Struct 690:145–150
Acknowledgments
The authors DSR, GM and SMR gratefully acknowledge the financial support from the University Grant Commission (UGC), Government of India, by awarding the Senior Research Fellowship. The authors also express thanks to Hyderabad Central University, Osmania University and K. Naresh, Department of Biochemistry, S. V. University, for providing instrumentation facilities to characterize the compounds and biological data, respectively.
Conflict of interest
The authors have declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Devineni, S.R., Golla, M., Chamarthi, N.R. et al. 2-Amino-2,3-dihydro-1H-2λ5-[1,3,2]diazaphospholo[4,5-b]pyridin-2-one-based urea and thiourea derivatives: synthesis, molecular docking study and evaluation of anti-inflammatory and antimicrobial activities. Med Chem Res 25, 751–768 (2016). https://doi.org/10.1007/s00044-016-1518-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1518-x