
Abstract
A series of pyrazole integrated thiazolo[2,3-b]dihydropyrimidinone derivatives were synthesized as dual anti-inflammatory and antimicrobial agents. Among the compounds studied, 3-fluoro-4-methylphenyl analogues (3a, 3e, and 3i) are considered to be promising leads for novel anti-inflammatory agents compared with the standard drug. The superior antimicrobial property of the compounds 3a, 3b, and 3d indicates that 3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazole substitution is a favourable site for high activity. Molecular docking studies were carried out in order to predict the hypothetical binding mode of these compounds to the COX-2 isoenzyme. The results of the present study suggest that 1,3-diaryl pyrazole substitution on thiazolo[2,3-b]dihydropyrimidinone derivatives might potentially constitute a novel class of anti-inflammatory agents with antimicrobial property and could be an interesting approach for the design of new selective COX-2 inhibitory agents.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are commonly prescribed for the treatment of acute and chronic inflammation, pain, and fever (Allavena et al. 2008; Sorbera et al. 2001; Palomer et al. 2002). Most currently used NSAIDs have limitations for therapeutic use since they cause gastrointestinal and renal side effects, which are inseparable from their pharmacological activities (Smith et al. 2000; Dogne et al. 2005). In addition, inflammation is known not only as a symptom of a great deal of common diseases, but also as an early phase of some life-threatening diseases like cancer, heart vascular diseases, Alzheimer’s dementia, etc (Dillingham et al. 2002; Dekhane et al. 2011; Selkoe 2001). Therefore, investigations for the development of safer and more effective anti-inflammatory agents are of great interest for medicinal researchers. The rapidly increasing incidence of antimicrobial resistance also represents a serious problem worldwide. Therefore, the development of new and different antimicrobial drugs is a very important objective and many research programmes are directed towards the design of new antimicrobial agents (Li and Ma 2015; Beekmann et al. 2005; Chua et al. 2008). This situation highlights the need to identify lead molecules from newer classes of compounds, in order to develop novel and effective NSAIDs and antimicrobial agents in the future.
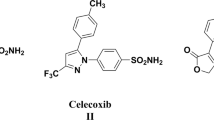
Among various classes of chemical moieties, pyrazole and its derivatives represent important building blocks in drug research (Kumar et al. 2015, 2013a). In addition, they are of interest in their own right due to their pharmacological properties. The changes in their structure offer a high degree of diversity, which is useful for the development of new therapeutic agents (Fustero et al. 2011, 2009). Antipyrine is the first pyrazoline derivative used as an anti-inflammatory agent. However, its use is restricted due to its GI side effect (Sivakumar et al. 2014). The literature survey reveals that many pyrazole and pyrazoline derivatives have been used for clinical application as NSAIDs, i.e., Phenylbutazone, Celecoxib, and Deracoxib (Fig. 1) (Capone et al. 2003; Kumar et al. 2013b). Furthermore, much attention was given to pyrazoles as antimicrobial agents after the discovery of the natural pyrazole C-glycosde pyrazofurin (Fig. 2), which demonstrated a broad spectrum of antimicrobial activity (Comber et al. 1991). The discovery of a dual anti-inflammatory-antimicrobial agent with potential activity and fewer adverse effects is a priority of the current global research, and consequently has occupied our prime interest in recent years (Sivakumar et al. 2014, Keche et al. 2012; Li and Zhao 2014; Bekhit et al. 2010).

Structures of some known pyrazole NSAIDs

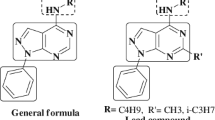
Structures of antibiotic pyrazofurin and structures of some reported thiazolo pyrimidinone (a) and pyrazole integrated thiazolo pyrimidinone derivatives (b and c) with anti-inflammatory and antimicrobial activity
Pyrimidines have a long distinguished history as important constituents of nucleic acids to their current use in chemotherapy for AIDS (Nagarapu et al. 2013). One possible reason for their activity is the presence of the pyrimidine base in thymine, cytosine, and uracil, which are essential building blocks of nucleic acids, DNA, and RNA (Sondhi et al. 2005). In addition, thiazolopyrimidines are pharmacological scaffolds that represent a wide range of biological activities such as antimicrobial, anti-inflammatory, analgesic, antioxidant, insecticides, antidepressant, antiviral, antitumour, calcium channel blocker, etc (Mohamed et al. 2010a, b; Abu-Hashem et al. 2011; Fatima et al. 2012; Khalifa et al. 2015). The reported significance of such synthons (A, Fig. 2) generated interest in the designing and synthesis of new thiazolo[3,2-a]pyrimidine-6-carboxylate derivatives (Ashok et al. 2007).
However, only a few approaches can be found in literature to synthesize these compounds. The major limitations of these reported methods are long reaction time, more solvent, and less yield due to multistep reaction condition. Multi-component reactions (MCRs) have emerged as an efficient and powerful tool in modern synthetic organic chemistry in which three or more different starting materials react to give a final product in a one pot procedure (Kiyani and Ghorbani 2015; Viveka et al. 2012, 2015a, d, 2014). Some of the reported pyrazole integrated thiazolo pyrimidinone derivatives (B and C) (Fig. 2) showed pronounced anti-inflammatory and antimicrobial activities (Bekhit et al. 2003). Prompted by these observations and in continuation of our research work related to pyrazole chemistry (Viveka et al. 2012, 2015a, b, c, d), we hereby report the synthesis of a new series of pyrazole embodied thiazolopyrimidinone in a single framework. All the compounds were synthesized via MCR approach and their anti-inflammatory and antimicrobial activities was evaluated.
Results and discussion
Chemistry
The key intermediates dihydropyrimidinones (DHPMs) 1(a–d) were obtained by the Biginelli reaction involving substituted benzaldehyde, ethyl acetoacetate, and thiourea in the presence of HCl catalyst according to the procedure reported in the literature (Patel and Patel 2013). The versatile Vilsmeier reaction was employed to synthesize a series of substituted 4-formyl pyrazole 2(a–d) in good yields from the corresponding phenylhydrazone, according to our previous reports (Viveka et al. 2012, 2015a, b, d). These highly activated intermediates, 1(a–d) and 2(a–d), were then reacted with monochloroacetic acid, anhydrous sodium acetate in acetic acid-acetic anhydride medium, which resulted in the formation of the target compounds 3(a–p). The synthesis and characterization of such compounds has been reported in our previous publication (Viveka et al. 2012, 2015d). The structures of these compounds were established on the basis of elemental analysis, IR, 1H NMR, 13C NMR, and mass spectral studies. The synthetic strategy to prepare the targeted compounds 3(a–p) is illustrated in Scheme 1.

Synthetic pathway for the synthesis of the targeted compounds 3(a–p)
The FTIR spectrum of pyrazoline analogue 3e showed absorption band at 1737 cm−1 for carbonyl stretching. Also, the appearance of the absorption band at 1682 and 1591 cm−1 due to C=N, C=C stretching further supported the proposed structure. The 1H NMR spectrum of 3e, appearing as a triplet and quartet at δ 1.10 and 4.02 ppm, clearly confirmed the presence of the ethyl carboxylate group. In addition, two CH3 protons appearing as singlets at δ 2.17 and 2.38 ppm, supported the proposed structure. A singlet at δ 5.97 ppm confirmed the presence of proton in the pyrimidine ring. Pyrazole H-5 proton appeared as a singlet at δ 8.79 ppm and all the other aromatic protons resonated as a complex multiplet in the region of δ 6.99–8.02 ppm. Additional evidence was obtained by its 13C NMR spectrum, where two characteristic signals appeared at δ 14.39 and δ 60.64 ppm corresponding to the OCH2CH3 carbons of the proposed structure. The carbons of two other methyl groups appeared at δ 14.10 and 22.81 ppm and peaked at δ 54.98 ppm corresponding to the CH of the pyrimidine ring. Furthermore, the 13C NMR spectrum showed two characteristic peaks at δ 165.3 and 164.5 ppm, which was attributed to the ester and cyclic carbonyl group of the target compound. The absence of C=S prominent peak at δ 174.5 ppm in the 13C NMR spectrum gave strong evidence to the formation of thiazolo[2,3-b]dihydropyrimidinone derivative 3e. Also, the disappearance of the NH absorption peak (3100–3400 cm−1) in the IR spectrum and the absence of the two singlets at higher δ values corresponding to the –NH of 1(a–d) clearly established the conversion of 1(a–d) to 3(a–p). The mass spectra of all the final derivatives showed comparable molecular ion peak with respect to the molecular formula. Similarly, all the target compounds were also well-characterized by FTIR, 1H NMR, 13C NMR, and mass spectral studies. The physical data of the target compounds is given in Table 1.
Pharmacological screening
Anti-inflammatory activity
The anti-inflammatory screening of the synthesized compounds was carried out using the carrageenan-induced paw edema method of inflammation in rats at successive intervals of 1, 2, and 4 h compared with that of the standard drug Indomethacin. The results of the anti-inflammatory analysis are reported in Table 2. The results are expressed as mean ± SEM. The percentage inhibition for the synthesized compounds as well as the standard drug was determined. The tested compounds exhibited moderate anti-inflammatory within 2 h; the activity increased and reached peak level at 4 h and declined after 4 h.
The activity data depicts the anti-inflammatory potential of the synthesized compounds, and majority of the analogues were found to be active. From the anti-inflammatory activity result analysis, it was observed that the compounds 3a, 3b, 3e, 3f, 3i, 3m, and 3n showed good activity with 67.61 to 85.33% inhibition of the edema. The compounds 3a (85.33), 3e (81.32), and 3i (80.75) showed potent anti-inflammatory activity compared with the other test compounds and are comparable with the standard Indomethacin (86.76) (Table 2). This emphasizes the presence of the 3F–4CH3– substituted phenyl ring on the 5th position of the 3-oxothiazolopyrimidine nucleus in this pyrazole series. The presence of electron donating methoxy groups at 2, 5 position on the same phenyl ring [3b (75.61), 3f (79.56), 3j (63.86), and 3n (67.61)] showed superior activity over the compounds containing p-methoxy electron donating substituted analogues [3d (65.11), 3h (67.14), 3l (63.68), and 3p (55.92)]. The phenyl ring without any substituent on pyrimidine 3c, 3g, 3k, and 3o did not show considerable influence on the activity (Fig. 3). It was also demonstrated from the data that there was increase in the activity profile when the dihalogen groups present in the phenyl ring attached to the 3rd position of the pyrazole moiety.

SAR analysis of synthesized compounds 3(a–p)
Antimicrobial activity
The synthesized compounds 3(a–p) were also evaluated for their in vitro antibacterial and antifungal activities against Gram-positive organism (Staphylococcus aureus), Gram-negative organisms (Klebsiella pneumoniae, Pseudomonas aerogenosa, and Escherichia coli), and fungi (Aspergillus flavus and Fusarium verticillioides), which was compared with that of the standard drugs Streptomycin and Nystain. The zone of inhibition was determined by the agar well diffusion method. The results of the tested compounds are listed in Table 3. The minimum inhibitory concentration (MIC) for the active compounds showing maximum activity was determined by the agar dilution method (Table 4).
As observed in the antibacterial activity, compounds 3a, 3b, and 3d show good activity for all the tested species at MICs 3.12–25 μg/mL that contained electron withdrawing 3,4-dichloro phenyl groups on the pyrazole ring. The compound 3a exhibited maximum activity among all the compounds with MIC values of 3.12 μg/mL (E. coli, S. aureus), 12.5 μg/mL (K. pneumoniae), and 25 μg/mL (P. aerogenosa). Similarly, 3b also showed appreciable result against E. coli and K. pneumoniae (MICs 3.12 μg/mL) and was significantly active against S.aureus (6.25 μg/mL) and P. aerogenosa (12.5 μg/mL). The compound 3d was found to be active against all bacterial strains with MICs 6.25–25 μg/mL. From the inhibition results (Table 3), it was observed that compounds with 3,4-dichlorophenyl at the 3-position of the pyrazole showed enhanced antibacterial activity compared with 3,4-difluoro, 4-chloro and 4-fluorophenyl substituents. Significant enhancement was also observed in the antibacterial activity with the introduction of electron donating methoxy (3b, 3d, 3f, 3h, 3j, 3l, 3n, and 3p) and methyl (3a, 3e, 3i, and 3m) substituted on the 5th position of the 3-oxothiazolopyrimidine observed against E. coli, S. aureus, and K. pneumoniae bacterial species. As a result, the introduction of 2,5-dimethoxyphenyl groups on the pyrimidine ring are promising candidates for antibacterial research in this series of compounds (Fig. 3). The results of the antifungal activity revealed that the compounds 3a, 3b, 3d, 3g, and 3m showed average antifungal activity against both the tested species. It was also observed that there was no significant change in the antifungal activity with the substitution on the phenyl ring attached to the pyrimidine. However, the dihalo substituted phenyl ring on the pyrazole ring was comparatively more effective than the 4-halo substituted phenyl group and none of the compounds was superior to the reference drug.
Molecular docking study
To understand the rationale behind the type of interactions between the compounds and the enzymes, molecular docking study was performed using AutoDock tools 1.5.6 (ADT 4.2). The synthesized compounds (3a–p) were docked successfully into the active sites of the COX-2 enzyme. The molecular docking studies revealed that the compounds 3a, 3e, 3f, and 3i were more selective towards the COX-2 enzyme. The calculated binding energy of the docked compounds towards COX-2 was found to be between −129.88 and −540.47 kcal/mol (Table 5). The binding mode of the most active inhibitors 3a and 3e to the COX-2 protein and the interactions are shown in Fig. 4. On observing the dock results, it can be ascertained that interaction with amino acid ARG 120 was common for the active compounds. The compound 3a is nicely bonded to COX-2 via two hydrogen bonds and two π-cation interaction. The nitrogen atom of the pyrimidine ring of the compound 3a forms hydrogen bonds (N···H–N: 2.21 Å) with the amino hydrogen atom of ARG 120, and the oxygen atom of the ester group of the compound 3a forms hydrogen bonds (O···H–N: 2.08 Å) with the amino hydrogen atom of the LYS 83 residue. The dichlorophenyl, thiazolidinone rings forms π-cation interaction with the amino cation ARG 513 and ARG 120. Compound 3e is satisfactorily bonded to COX-2 via three hydrogen bonds and three π-cation interaction. The fluorine atom of the phenyl ring, the sulphur atom of the thiazolidinone ring, and the nitrogen atom of the pyrimidine ring form hydrogen bonds (F···H–N: 2.26 Å), (S···H–O: 2.23 Å), (N···H–N: 2.45 Å) with the amino hydrogen atom of the ARG 120 residue. The difluorophenyl, pyrazole, thiazolidinone rings forms π-cation interaction with the amino cation ARG 120 and ARG 513. In general, the molecular docking results were in agreement with the pharmacological data in terms of evaluation of the binding energy and binding mode.

Docking figures of 3a and 3e in COX-2 protein cavity
Conclusion
In conclusion, in an attempt to develop a new class of anti-inflammatory and antimicrobial agents, a novel thiazolo[2,3-b]dihydropyrimidinone possessing pyrazole moiety has been synthesized. The target compounds are achieved by employing the Knoevenagel reaction approach. All the compounds were prepared using the convenient one pot MCR sequence with less reaction time and good yield. To the best of our knowledge, the anti-inflammatory activity and the antimicrobial data of the above mentioned derivatives has been firstly reported by our group. The results of the in vivo anti-inflammatory activity assays demonstrated that the compounds 3a, 3b, 3e, 3f, 3i, 3m, and 3n showed promising anti-inflammatory activity, and among these 3a, 3e, and 3i were found to be comparable with the standard Indomethacin. Again, the presence of the 3-fluoro-4-methylphenyl ring on the 3-oxothiazolopyrimidine nucleus is a favourable site for high activity, followed by 2,5-dimethoxyphenyl substituent and p-methoxyphenyl substituent. The structure activity relationships (SARs) revealed that the 3,4-dichlorophenyl substituted pyrazole analogues (3a, 3b, and 3d) were identified as compounds with potent antibacterial activity and showed moderate antifungal property. Also, the molecular docking studies revealed that thiazolo[2,3-b]dihydropyrimidinone possessing pyrazole analogues exhibited good interaction with the active site of the docked receptor cyclooxygenase-2. The results of the present study suggest that pyrazole–thiazolopyrimidine hybrid can be an interesting approach for the design of new selective COX-2 inhibitory agents.
Experimental section
Materials and methods
All the reagents were purchased from commercial suppliers Sigma-Aldrich, Spectrochem India and used without further purification. Melting points were determined in an open capillary tube and were uncorrected. The progress of each reaction was monitored by ascending thin layer chromatography (TLC) on silica gel G (Merck 1.05570.0001), visualized by UV light. The IR spectra (in KBr pellets) were recorded on a Shimadzu-FTIR spectrometer and the wave numbers were given in cm−1. The 1H NMR and 13C NMR spectra were recorded (CDCl3/DMSO-d 6 mixture) on a Bruker AMX-400 NMR spectrometer with 5 mm PABBO BB-1H TUBES with TMS as internal standard. Mass spectra were recorded in Agilent Technology LC-mass spectrometer. Elemental analyses were carried out using VARIO EL-III (Elementar Analysensysteme GmBH).
Procedure for the preparation of ethyl 4-(substitutedphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 1(a–d)
A mixture of simple or substituted benzaldehyde (0.05 mol), ethylacetoacetate (0.05 mol), thiourea (0.05 mol) and a few drops of HCl as catalyst was refluxed in ethanol (30 mL) for 1–4 h and the progress of the reaction was monitored by TLC. After completion of reaction, the resulting mixture was cooled and poured onto crushed ice. The solid was collected by filtration, washed several times with water to remove unreacted thiourea, and then dried. The product was purified by recrystallization from methanol.
Ethyl 4-(3-fluoro-4-methylphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1a)
Yield: 88%; m.p. 175–177 °C; FT-IR (KBr) νmax (cm−1): 3255, 3132 (N–H), 2986 (Ar C–H), 1738 (C=O), 1579 (C=C), 1279 (C–O), 1097 (C=S), 1024 (C–F). 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 1.08 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.24 (s, 3H, pyrimidine-CH3), 2.62 (s, 3H, Ar–CH3), 3.96 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 5.35 (s, 1H, pyrimidine-CH), 7.06–7.54 (m, 3H, Ar–H), 9.67 (s, 1H, pyrimidine-NH), 10.54 (s, 1H, pyrimidine-NH); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 14.23 (–CH2–CH 3 ), 14.75, 17.48 (CH3), 54.68 (pyrimidine-CH), 60.72 (–CH 2 –CH3), 102.34, 113.75, 122.04, 123.54, 130.53, 141.82, 159.52, 161.52, 165.41 (ester C=O), 174.53 (C=S); LCMS (m/z): 309.13 [M+H]. Anal. Calcd. for C15H17FN2O2S: C, 58.42; H, 5.56; N, 9.08. Found: C, 58.46; H, 5.53; N, 9.06.
Ethyl 4-(2,5-dimethoxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1b)
Yield: 56%; m.p. 203–205 °C; FT-IR (KBr) νmax (cm−1): 3286, 3110 (N–H), 2951 (Ar C–H), 1726 (C=O), 1566 (C=C), 1284 (C–O), 1137 (C=S); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 1.07 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.37 (s, 3H, pyrimidine-CH3), 3.75 (s, 6H, Ar–OCH3 × 2), 3.98 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 5.42 (s, 1H, pyrimidine-CH), 7.18–7.77 (m, 3H, Ar–H), 9.79 (s, 1H, pyrimidine-NH), 10.29 (s, 1H, pyrimidine-NH); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 14.31 (–CH2–CH 3 ), 17.85 (CH3), 54.13 (pyrimidine-CH), 55.12, 56.59 (–OCH3 × 2), 60.84 (–CH 2 –CH3), 102.48, 112.74, 113.68, 115.91, 122.18, 148.52, 152.32, 160.26, 165.94 (ester C=O), 174.64 (C=S); LCMS (m/z): 337.01 [M+H]. Anal. Calcd. for C16H20N2O4S: C, 57.12; H, 5.99; N, 8.33. Found: C, 57.16; H, 5.93; N, 8.36.
Ethyl 6-methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1c)
Yield: 66%; m.p. 178–180 °C; FT-IR (KBr) νmax (cm−1): 3236, 3097 (N–H), 2918 (Ar C–H), 1724 (C=O), 1564 (C=C), 1205 (C–O), 1089 (C=S); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 1.04 (t, 3H, J = 7.2 Hz, –CH2–CH 3 ), 2.26 (s, 3H, pyrimidine-CH3), 3.95 (q, 2H, J = 7.2 Hz, –CH 2 –CH3), 5.14 (s, 1H, pyrimidine-CH), 7.18–7.33 (m, 5H, Ar–H), 9.62(s, 1H, pyrimidine-NH), 10.30 (s, 1H, pyrimidine-NH); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 14.44 (–CH2–CH 3 ), 17.57 (CH3), 54.45 (pyrimidine-CH), 60.02 (–CH 2 –CH3), 101.16, 126.81, 128.09, 128.97, 143.93, 145.45, 165.56 (ester C=O), 174.69 (C=S); LCMS (m/z): 277.12 [M+H]. Anal. Calcd. for C14H16N2O2S: C, 60.85; H, 5.84; N, 10.14. Found: C, 60.86; H, 5.83; N, 10.16.
Ethyl 4-(4-methoxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (1d)
Yield: 78%; m.p. 150–152 °C; FT-IR (KBr) νmax (cm−1): 3236, 3143 (N–H), 2922 (Ar C–H), 1763 (C=O), 1523 (C=C), 1273 (C–O), 1174 (C=S); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 1.04 (t, 3H, J = 6.4 Hz, –CH2–CH 3 ), 2.26 (s, 3H, pyrimidine-CH3), 3.59 (s, 3H, Ar–OCH3), 3.96 (q, 2H, J = 6.4 Hz, –CH 2 –CH3), 5.14 (s, 1H, pyrimidine-CH), 6.69–6.99 (m, 4H, Ar–H), 9.63 (s, 1H, pyrimidine-NH), 10.35 (s, 1H, pyrimidine-NH); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 14.42 (–CH2–CH 3 ), 17.60 (CH3), 53.86 (pyrimidine-CH), 53.94 (–OCH3), 60.08 (–CH 2 –CH3), 100.74, 128.74, 129.00, 132.70, 142.82, 145.80, 165.42 (ester C=O), 174.70 (C=S); LCMS (m/z): 307.12 [M+H]. Anal. Calcd. for C15H18N2O3S: C, 58.80; H, 5.92; N, 9.14. Found: C, 58.86; H, 5.93; N, 9.16.
General procedure for the synthesis of 4-Formylpyrazole 2(a–d)
(Substituted phenyl) ethanone phenylhydrazone (0.01 mol) was added to the mixture of Vilsmeier-Haack reagent (prepared by drop-wise addition of 1.2 mL POCl3 in ice cooled 5 mL DMF) and refluxed for 6 h. The reaction mixture, after cooling, was poured onto crushed ice, followed by neutralization using sodium bicarbonate. The solid product thus obtained was collected by filtration, washed with water and recrystallized from ethanol (Viveka et al. 2015a, b, d).
3-(3,4-Dichlorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (2a)
Yield: 83%; m.p. 150–152 °C; FT-IR (KBr) νmax (cm−1): 3041 (Ar C–H), 1678 (C=O), 1571 (C=N), 1489 (C=C), 813 (C–Cl); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 7.42–7.83 (m, 9H, Ar–CH), 9.97 (s, 1H, CHO). 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 119.79, 122.75, 128.43, 129.10, 130.24, 130.61, 131.21, 131.77, 132.34, 137.15, 138.88, 149.98, 184.87 (aldehyde C=O); LCMS (m/z): 317.12 [M+H] (35Cl2), 319.14 [M+H+2] (35Cl37Cl), 321.24 [M+H+4] (37Cl2); Anal. Calcd. for C16H10Cl2N2O: C, 60.59; H, 3.18; N, 8.83. Found: C, 60.56; H, 3.13; N, 8.86.
3-(3,4-Difluorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (2b)
Yield: 76%; m.p. 95–98 °C; FT-IR (KBr) νmax (cm−1): 3056 (Ar C–H), 1675 (C=O), 1578 (C=N), 1453 (C=C), 1086 (C–F); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 7.44–9.37 (m, 9H, Ar–CH), 9.96 (s, 1H, CHO); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 117.99, 119.71, 122.58, 126.04, 128.33, 129.18, 130.19, 136.83, 138.88, 148.48, 149.19, 150.43, 150.91, 151.65, 184.87 (aldehyde C=O); LCMS (m/z): 285.2 [M+H]; Anal. Calcd. for C16H10F2N2O: C, 67.60; H, 3.55; N, 9.85. Found: C, 67.62; H, 3.53; N, 9.86.
3-(4-Chlorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (2c)
Yield: 82%; m.p. 140–142 °C; FT-IR (KBr) νmax (cm−1): 3022 (Ar C–H), 1698 (C=O), 1554 (C=N), 1496 (C=C), 789 (C–Cl); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 7.14–8.37 (m, 10H, Ar–CH), 9.65 (s, 1H, CHO); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 106.54, 120.32, 126.54, 128.53, 129.56, 130.04, 130.89, 131.43, 134.77, 139.78, 150.54 (Ar–C), 187.58 (aldehyde C=O); LCMS (m/z): 283.12 [M+H] (35Cl), 285.14 [M+H+2] (37Cl); Anal. Calcd. for C16H11ClN2O: C, 67.97; H, 3.92; N, 9.91. Found: C, 67.96; H, 3.93; N, 9.86.
3-(4-Fluorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (2d)
Yield: 64%; m.p. 110–112 °C; FT-IR (KBr) νmax (cm−1): 3011 (Ar C–H), 1694 (C=O), 1574 (C=N), 1463 (C=C), 1081 (C–F); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 7.16–8.16 (m, 10H, Ar–CH), 9.58 (s, 1H, CHO); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 106.28, 116.59, 120.70, 126.54, 127.93, 129.08, 129.96, 130.08, 139.74, 150.50, 160.78 (Ar–C), 187.61 (aldehyde C=O); LCMS (m/z): 267.34 [M+H]; Anal. Calcd. for C16H11FN2O: C, 72.17; H, 4.16; N, 10.52. Found: C, 72.21; H, 4.19; N, 10.56.
Procedure for the synthesis of ethyl 2-((3-(substitutedphenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(substituted phenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate 3(a–p)
A solution of ethyl 6-methyl-4-(substituted phenyl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 1 (10 mmol), 3-(substitutedphenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde 2 (10 mmol), monochloroacetic acid (15 mmol) and anhydrous sodium acetate (2 g) in glacial acetic acid (25 mL)-acetic anhydride (20 mL) mixture was refluxed for 2–6 h and monitored by TLC. The mixture was cooled to room temperature and then poured onto crushed ice with stirring. The precipitated solid was collected by filtration, washed with cold water and recrystallized from methanol (Viveka et al. 2012, 2015d).
Ethyl 2-((3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(3-fluoro-4-methylphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3a)
Yield: 91%; m.p. 127–130 °C; FT-IR (KBr) νmax (cm−1): 2966 (Ar C–H), 1726 (C=O), 1672 (C=N), 1587 (C=C), 1210 (C–O), 1031 (C–F) 761 (C-Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.19 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.21 (s, 3H, pyrimidine-CH3), 2.51 (s, 3H, Ar–CH3), 4.10 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.13 (s, 1H, pyrimidine-CH), 7.02–7.77 (m, 12H, Ar–H), 8.18 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.10 (–CH2–CH 3 ), 14.22, 22.70 (CH3), 55.11 (pyrimidine-CH), 60.53 (–CH 2 –CH3), 109.33, 113.92, 116.26, 119.24, 119.57, 123.46, 127.16, 127.84, 128.95, 129.14, 129.42, 129.74, 129.91, 130.11, 130.20, 132.33, 135.20, 139.10, 151.94, 153.58, 155.17, 159.74, 164.73 (cyclic C=O), 165.54 (ester C=O); LCMS (m/z): 647.24 [M+H] (35Cl2), 649.24 [M+H+2] (35Cl37Cl), 650.24 [M+H+4] (37Cl2); Anal. Calcd. for C33H25Cl2FN4O3S: C, 61.21; H, 3.89; N, 8.65. Found: C, 61.18; H, 3.92; N, 8.66.
Ethyl 2-((3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(2,5-dimethoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3 b)
Yield: 78%; m.p. 98–101 °C; FT-IR (KBr) νmax (cm−1): 2978 (Ar C–H), 1723 (C=O), 1653 (C=N), 1565 (C=C), 1226 (C–O), 786 (C–Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.23 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.45 (s, 3H, pyrimidine-CH3), 3.69 (s, 6H, Ar–OCH3 × 2), 4.01 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.20 (s, 1H, pyrimidine-CH), 6.88–7.79 (m, 12H, Ar–H), 8.21 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.21 (–CH2–CH 3 ), 22.69 (CH3), 53.79 (pyrimidine-CH), 55.79, 56.25 (–OCH3 × 2), 60.24 (–CH 2 –CH3), 108.21, 112.43, 114.13, 115.17, 116.15, 119.16, 120.23, 121.32, 127.02, 127.73, 128.32, 128.89, 129.32, 129.71, 130.26, 131.18, 133.20, 133.39, 139.72, 142.54, 152.81, 154.08, 159.74, 161.17, 164.51 (cyclic C=O), 165.21(ester C=O); LCMS (m/z): 675.21 [M+H] (35Cl2), 677.22 [M+H+2] (35Cl37Cl), 679.21 [M+H+4] (37Cl2); Anal. Calcd. for C34H28Cl2N4O5S: C, 60.45; H, 4.18; N, 8.29. Found: C, 60.49; H, 4.19; N, 8.26.
Ethyl 2-((3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-7-methyl-3-oxo-5-phenyl-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3c)
Yield: 67%; m.p. 135–137 °C; FT-IR (KBr) νmax (cm−1): 2994 (Ar C–H), 1736 (C=O), 1680 (C=N), 1589 (C=C), 1232 (C–O), 759 (C–Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.16 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.35 (s, 3H, pyrimidine-CH3), 4.09 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.18 (s, 1H, pyrimidine-CH), 7.26–7.78 (m, 14H, Ar–H), 8.19 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.21 (–CH2–CH 3 ), 22.32 (CH3), 55.73 (pyrimidine-CH), 61.69 (–CH 2 –CH3), 107.56, 120.12, 121.49, 122.31, 124.67, 125.13, 126.42, 127.23, 127.41, 128.42, 129.03, 129.92, 130.02, 130.81, 133.45, 134.32, 139.71, 142.52, 143.52, 150.31, 154.57, 160.41, 164.52 (cyclic C=O), 165.51(ester C=O); LCMS (m/z): 615.15 [M+H] (35Cl2), 617.15 [M+H+2] (35Cl37Cl), 619.15 [M+H+4] (37Cl2); Anal. Calcd. for C32H24Cl2N4O3S: C, 62.44; H, 3.93; N, 9.10. Found: C, 62.41; H, 3.95; N, 9.06.
Ethyl 2-((3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(4-methoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3d)
Yield: 88%; m.p. 112–114 °C; FT-IR (KBr) νmax (cm−1): 2989 (Ar C–H), 1742 (C=O), 1652 (C=N), 1567 (C=C), 1224 (C–O), 767 (C-Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.21 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.49 (s, 3H, pyrimidine-CH3), 3.75 (s, 3H, Ar–OCH3), 3.99 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.13 (s, 1H, pyrimidine-CH), 6.98–7.89 (m, 13H, Ar–H), 8.21 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.34 (–CH2–CH 3 ), 21.86 (CH3), 54.83 (pyrimidine-CH), 55.62 (–OCH3), 60.86 (–CH 2 –CH3), 107.53, 112.53, 120.91, 121.54, 122.76, 125.43, 126.34, 127.40, 128.81, 129.54, 130.41, 130.89, 132.50, 133.50, 133.72, 135.71, 139.72, 142.51, 150.12, 154.56, 158.71, 162.71, 164.71 (cyclic C=O), 165.79(ester C=O); LCMS (m/z): 645.12 [M+H] (35Cl2), 647.12 [M+H+2] (35Cl37Cl), 649.12 [M+H+4] (37Cl2); Anal. Calcd. for C33H26Cl2N4O4S: C, 61.40; H, 4.06; N, 8.68. Found: C, 61.34; H, 4.01; N, 8.70.
Ethyl 2-((3-(3,4-difluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(3-fluoro-4-methylphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3e)
Yield: 72%; m.p. 166–167 °C; FT-IR (KBr) νmax (cm−1): 2969 (Ar C–H), 1737 (C=O), 1682 (C=N), 1591 (C=C), 1224 (C–O), 1055 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.10 (t, 3H, J = 7.2 Hz, –CH2–CH 3 ), 2.17 (s, 3H, pyrimidine-CH3), 2.38 (s, 3H, Ar–CH3), 4.02 (q, 2H, J = 4.0 Hz, –CH 2 –CH3), 5.97 (s, 1H, pyrimidine-CH), 6.99–8.02 (m, 12H, Ar–H), 8.79 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.10 (–CH2–CH 3 ), 14.39, 22.81 (CH3), 54.98 (pyrimidine-CH), 60.64 (–CH 2 –CH3), 108.86, 114.70, 116.26, 119.54, 119.73, 123.04, 123.45, 125.48, 125.62, 127.25, 127.94, 128.00, 129.78, 130.48, 130.84, 131.43, 131.55, 133.26, 133.39, 138.96, 139.46, 139.52, 152.22, 152.45, 155.01, 160.23, 162.19, 164.51 (cyclic C=O), 165.34 (ester C=O); LCMS (m/z): 615.29 [M+H]; Anal. Calcd. for C33H25F3N4O3S: C, 64.69; H, 4.10; N, 9.12. Found: C, 64.72; H, 4.06; N, 9.06.
Ethyl 2-((3-(3,4-difluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(2,5-dimethoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3f)
Yield: 74%; m.p. 105–107 °C; FT-IR (KBr) νmax (cm−1): 2972 (Ar C–H), 1726 (C=O), 1676 (C=N), 1587 (C=C), 1249 (C–O), 1031 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.22 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.43 (s, 3H, pyrimidine-CH3), 3.75 (s, 6H, Ar–OCH3 × 2), 4.09 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.24 (s, 1H, pyrimidine-CH), 6.76–7.77 (m, 12H, Ar–H), 8.19 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.17 (–CH2–CH 3 ), 22.81 (CH3), 53.92 (pyrimidine-CH), 55.67, 56.14 (–OCH3 × 2), 60.35 (–CH 2 –CH3), 107.42, 112.62, 114.49, 115.85, 116.03, 116.37, 117.12, 119.15, 119.50, 122.79, 125.30, 127.02, 127.63, 128.22, 128.42, 129.03, 129.72, 130.67, 139.17, 151.67, 152.36, 153.13, 153.74, 155.07, 162.27, 164.63 (cyclic C=O), 165.83 (ester C=O); LCMS (m/z): 643.17 [M+H]; Anal. Calcd. for C34H28F2N4O5S: C, 63.54; H, 4.39; N, 8.72. Found: C, 63.49; H, 4.41; N, 8.69.
Ethyl 2-((3-(3,4-difluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-7-methyl-3-oxo-5-phenyl-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3g)
Yield: 81%; m.p. 134–136 °C; FT-IR (KBr) νmax (cm−1): 2978 (Ar C–H), 1734 (C=O), 1684 (C=N), 1569 (C=C), 1218 (C–O), 1054 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.22 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.41 (s, 3H, pyrimidine-CH3), 4.15 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.07 (s, 1H, pyrimidine-CH), 7.51–7.98 (m, 14H, Ar–H), 8.23 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.23 (–H2–CH 3 ), 22.31 (CH3), 54.82 (pyrimidine-CH), 61.06 (–CH 2 –CH3), 108.16, 119.98, 120.92, 121.86, 123.75, 125.22, 125.69, 126.54, 127.11, 128.31, 129.01, 129.36, 130.11, 130.99, 133.45, 134.89, 137.34, 142.17, 144.02, 150.17, 154.57, 160.38, 164.18 (cyclic C=O), 165.85 (ester C=O); LCMS (m/z): 583.16 [M+H]; Anal. Calcd. for C32H24F2N4O3S: C, 65.97; H, 4.15; N, 9.62. Found: C, 66.01; H, 4.10; N, 9.59.
Ethyl 2-((3-(3,4-difluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(4-methoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3h)
Yield: 73%; m.p. 167–169 °C; FT-IR (KBr) νmax (cm−1): 2984 (Ar C–H), 1734 (C=O), 1661 (C=N), 1558 (C=C), 1218 (C–O), 1045 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.34 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.56 (s, 3H, pyrimidine-CH3), 3.46 (s, 3H, Ar–OCH3), 4.13 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.18 (s, 1H, pyrimidine-CH), 6.18–7.92 (m, 13H, Ar–H), 8.33 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.18 (–CH2–CH 3 ), 22.37 (CH3), 54.74 (pyrimidine-CH), 55.96 (–OCH3), 61.57 (–CH 2 –CH3), 107.51, 113.72, 119.98, 121.38, 122.89, 125.12, 126.38, 128.12, 128.37, 129.19, 130.14, 131.01, 132.45, 133.18, 133.99, 135.17, 137.52, 142.71, 150.15, 153.16, 157.98, 161.91, 163.97 (cyclic C=O), 164.99 (ester C=O); LCMS (m/z): 613.16 [M+H]; Anal. Calcd. for C33H26F2N4O4S: C, 64.70; H, 4.28; N, 9.15. Found: C, 64.78; H, 4.23; N, 9.14.
Ethyl 2-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(3-fluoro-4-methylphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3i)
Yield: 59%; m.p. 90–93 °C; FT-IR (KBr) νmax (cm−1): 2967 (Ar C–H), 1716 (C=O), 1684 (C=N), 1572 (C=C), 1234 (C–O), 1041 (C–F), 758 (C-Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.29 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.16 (s, 3H, pyrimidine-CH3), 2.48 (s, 3H, Ar–CH3), 4.18 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 5.98 (s, 1H, pyrimidine-CH), 6.92–8.01 (m, 13H, Ar–H), 8.24 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.18 (–CH2–CH 3 ), 14.51, 22.18 (CH3), 56.13 (pyrimidine-CH), 61.28 (–CH 2 –CH3), 107.13, 113.78, 120.13, 121.52, 122.34, 122.78, 123.34, 126.57, 128.34, 129.54, 130.02, 130.84, 131.59, 132.76, 134.76, 138.76, 141.84, 142.54, 150.57, 154.79, 160.09, 161.2, 164.12 (cyclic C=O), 165.76 (ester C=O); LCMS (m/z): 613.12 [M+H] (35Cl), 615.12 [M+H+2] (37Cl); Anal. Calcd. for C33H26ClFN4O3S: C, 64.65; H, 4.27; N, 9.14. Found: C, 64.65; H, 4.31; N, 9.11.
Ethyl 2-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(2,5-dimethoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3j)
Yield: 72%; m.p. 103–105 °C; FT-IR (KBr) νmax (cm−1): 2978 (Ar C–H), 1724 (C=O), 1677 (C=N), 1598 (C=C), 1215 (C–O), 756 (C–Cl); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 1.10 (t, 3H, J = 7.2 Hz, –CH2–CH 3 ), 2.26 (s, 3H, pyrimidine-CH3), 3.61 (s, 6H, Ar–OCH3 × 2), 3.90 (q, 2H, J = 6.8 Hz, –CH 2 –CH3), 6.02 (s, 1H, pyrimidine-CH), 6.63–8.00 (m, 13H, Ar–H), 8.75 (s, 1H, pyrazole CH); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 14.36 (–CH2–CH 3 ), 22.91 (CH3), 53.93 (pyrimidine-CH), 55.89, 56.68 (–OCH3 × 2), 60.36 (–CH 2 –CH3), 107.39, 113.84, 114.52, 115.98, 117.43, 119.75, 119.78, 119.88, 122.13, 128.02, 128.65, 128.88, 129.01, 129.44, 130.05, 130.17, 130.61, 130.77, 134.33, 139.16, 151.06, 152.99, 153.98, 159.68, 161.120, 164.51 (cyclic C=O), 165.21(ester C=O); LCMS (m/z): 641.12 [M+H] (35Cl), 643.12 [M+H+2] (37Cl); Anal. Calcd. for C34H29ClN4O5S: C, 63.69; H, 4.56; N, 8.74. Found: C, 63.68; H, 4.63; N, 8.79.
Ethyl 2-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-7-methyl-3-oxo-5-phenyl-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3k)
Yield: 70%; m.p. 118–120 °C; FT-IR (KBr) νmax (cm−1): 2942 (Ar C–H), 1734 (C=O), 1682 (C=N), 1591 (C=C), 1234 (C–O), 768 (C–Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.31 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.46 (s, 3H, pyrimidine-CH3), 4.18 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.11 (s, 1H, pyrimidine-CH), 7.03–8.02 (m, 15H, Ar–H), 8.22 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.22 (–CH2–CH 3 ), 23.04 (CH3), 54.98 (pyrimidine-CH), 61.30 (–CH 2 –CH3), 106.37, 119.97, 121.43, 122.34, 126.38, 127.07, 127.89, 128.12, 128.94, 129.34, 129.88, 130.40, 131.34, 134.78, 139.86, 142.50, 143.86, 150.29, 155.73, 161.78, 164.57 (cyclic C=O), 165.39 (ester C=O); LCMS (m/z): 581.13 [M+H] (35Cl), 583.13 [M+H+2] (37Cl); Anal. Calcd. for C32H25ClN4O3S: C, 66.14; H, 4.34; N, 9.64. Found: C, 66.18; H, 4.33; N, 9.69.
Ethyl 2-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(4-methoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3l)
Yield: 49%; m.p. 182–185 °C; FT-IR (KBr) νmax (cm−1): 2981 (Ar C–H), 1734 (C=O), 1678 (C=N), 1541 (C=C), 1234 (C–O), 763 (C–Cl); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.17 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.51 (s, 3H, pyrimidine-CH3), 3.85 (s, 3H, Ar–OCH3), 4.09 (q, 2H, J = 4.4 Hz, –CH 2 –CH3), 6.13 (s, 1H, pyrimidine-CH), 6.82–7.83 (m, 14H, Ar–H), 8.18 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.16 (–CH2–CH 3 ), 21.72 (CH3), 53.93 (pyrimidine-CH), 56.12 (–OCH3), 61.50 (–CH 2 –CH3), 106.68, 113.76, 120.14, 121.36, 122.81, 126.54, 128.10, 128.96, 129.14, 129.89, 130.10, 131.84, 134.50, 135.56, 139.70, 141.68, 150.32, 154.27, 158.75, 160.72, 164.52 (cyclic C=O), 165.78 (ester C=O); LCMS (m/z): 611.28 [M+H] (35Cl), 613.27 [M+H+2] (37Cl); Anal. Calcd. for C33H27ClN4O4S: C, 64.86; H, 4.45; N, 9.17. Found: C, 64.85; H, 4.40; N, 9.16.
Ethyl 5-(3-fluoro-4-methylphenyl)-2-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3m)
Yield: 66%; m.p. 134–136 °C; FT-IR (KBr) νmax (cm−1): 2976 (Ar C–H), 1731 (C=O), 1654 (C=N), 1582 (C=C), 1238 (C–O), 1041 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.22 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.05 (s, 3H, pyrimidine-CH3), 2.55 (s, 3H, Ar–CH3), 4.37 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 5.69 (s, 1H, pyrimidine-CH), 6.87–8.05 (m, 13H, Ar–H), 8.16 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.36 (–CH2–CH 3 ), 14.84, 22.92 (CH3), 56.54 (pyrimidine-CH), 61.10 (–CH 2 –CH3), 107.28, 114.10, 120.84, 121.78, 122.57, 122.98, 124.01, 126.76, 128.34, 129.18, 130.13, 130.94, 131.68, 132.84, 134.86, 138.88, 141.42, 143.22, 150.17, 154.86, 161.19, 163.28, 165.24 (cyclic C=O), 166.15 (ester C=O); LCMS (m/z): 597.23 [M+H]; Anal. Calcd. for C33H26F2N4O3S: C, 66.43; H, 4.39; N, 9.39. Found: C, 66.41; H, 4.40; N, 9.40.
Ethyl 5-(2,5-dimethoxyphenyl)-2-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3n)
Yield: 75%; m.p. 99–101 °C; FT-IR (KBr) νmax (cm−1): 2973 (Ar C–H), 1759 (C=O), 1665 (C=N), 1563 (C=C), 1230 (C–O), 1054 (C–F); 1H NMR (DMSO-d 6, 400 MHz, δ ppm): 1.19 (t, 3H, J = 7.2 Hz, –CH2–CH 3 ), 2.38 (s, 3H, pyrimidine-CH3), 3.14 (s, 6H, Ar–OCH3 × 2), 3.95 (q, 2H, J = 6.8 Hz, –CH 2 –CH3), 6.12 (s, 1H, pyrimidine-CH), 6.89–8.03 (m, 13H, Ar–H), 8.69 (s, 1H, pyrazole CH); 13C NMR (DMSO-d 6, 100 MHz, δ ppm): 14.14 (–CH2–CH 3 ), 22.84 (CH3), 53.98 (pyrimidine-CH), 54.92, 56.33 (–OCH3 × 2), 60.14 (–CH 2 –CH3), 106.89, 112.16, 114.34, 115.78, 117.58, 118.87, 119.34, 119.78, 121.86, 127.56, 128.79, 128.96, 129.05, 129.86, 130.05, 130.18, 130.68, 131.24, 135.12, 139.84, 151.12, 152.96, 153.73, 161.12, 162.34, 164.86 (cyclic C=O), 165.86 (ester C=O); LCMS (m/z): 625.22 [M+H]; Anal. Calcd. for C34H29FN4O5S: C, 65.37; H, 4.68; N, 8.97. Found: C, 65.40; H, 4.70; N, 8.99.
Ethyl 2-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-7-methyl-3-oxo-5-phenyl-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3o)
Yield: 86%; m.p. 182–185 °C; FT-IR (KBr) νmax (cm−1): 2964 (Ar C–H), 1721 (C=O), 1674 (C=N), 1576 (C=C), 1231 (C–O), 1048 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.36 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.54 (s, 3H, pyrimidine-CH3), 4.38 (q, 2H, J = 5.6 Hz, –CH 2 –CH3), 6.74 (s, 1H, pyrimidine-CH), 7.02–8.04 (m, 15H, Ar–H), 8.31 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.24 (–CH2–CH 3 ), 23.86 (CH3), 54.50 (pyrimidine-CH), 61.98 (–CH 2 –CH3), 106.57, 119.68, 121.54, 122.64, 126.75, 127.79, 127.92, 128.23, 128.98, 129.53, 130.14, 130.96, 131.76, 134.56, 139.85, 142.16, 143.74, 150.85, 155.26, 161.58, 164.19 (cyclic C=O), 165.50 (ester C=O); LCMS (m/z): 565.26 [M+H]; Anal. Calcd. for C32H25FN4O3S: C, 68.07; H, 4.46; N, 9.92. Found: C, 68.01; H, 4.48; N, 9.89.
Ethyl 2-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-5-(4-methoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate (3p)
Yield: 76%; m.p. 163–165 °C; FT-IR (KBr) νmax (cm−1): 2975 (Ar C–H), 1728 (C=O), 1673 (C=N), 1586 (C=C), 1232 (C–O), 1038 (C–F); 1H NMR (CDCl3, 400 MHz, δ ppm): 1.23 (t, 3H, J = 5.6 Hz, –CH2–CH 3 ), 2.68 (s, 3H, pyrimidine-CH3), 3.68 (s, 3H, Ar–OCH3), 4.12 (q, 2H, J = 4.4 Hz, –CH 2 –CH3), 6.25 (s, 1H, pyrimidine-CH), 6.94–7.95 (m, 14H, Ar–H), 8.23 (s, 1H, pyrazole CH); 13C NMR (CDCl3, 100 MHz, δ ppm): 14.36 (–CH2–CH 3 ), 21.58 (CH3), 53.86 (pyrimidine-CH), 56.05 (–OCH3), 61.87 (–CH 2 –CH3), 106.14, 112.86, 119.86, 121.24, 122.68, 126.53, 128.94, 129.10, 129.75, 130.57, 131.48, 134.82, 135.32, 139.17, 141.35, 150.18, 154.57, 158.25, 160.68, 162.18, 164.58 (cyclic C=O), 164.64 (ester C=O); LCMS (m/z): 595.23 [M+H]; Anal. Calcd. for C33H27FN4O4S: C, 66.65; H, 4.58; N, 9.42. Found: C, 66.63; H, 4.60; N, 9.40.
Pharmacology
Anti-inflammatory activity
The newly synthesized compounds were evaluated for their anti-inflammatory activity against carrageenan-induced rat paw edema assay model (Winter et al. 1962). Male Wistar rats (200–250 g) were fasted with free access to water at least 12 h prior to the experiments and divided randomly into different groups (control, standard, and test groups) of five rats each. The first group of rats was treated with 1 mL of 1% gum acacia suspension orally (control), the second group was administered a dose of 20 mg/kg of the Indomethacin (standard) and the third group was treated with 50 mg/kg suspension of the test compounds. After 1 h, the animals were injected with 0.1 mL of 1% carrageenan in normal saline subcutaneously to the sub-planar region of the right hind paw. The paw volume was measured after 1, 2, and 4 h using the Plethysmometer.
Antimicrobial activity
Antibacterial activity
Newly synthesized pyrazoles were investigated for their antibacterial activity against Gram-positive organism (Staphylococcus aureus) and Gram-negative organisms (Klebsiella pneumoniae, Pseudomonas aerogenosa, and Escherichia coli) by the disc diffusion method (Viveka et al. 2015a; Nandakumar et al. 2010). The discs 6.25 mm in diameter were punched from Whatman No.1 filter paper. Batches of 100 discs were dispensed in each screw-capped bottle and sterilized by dry heat at 140 °C for an hour. The test compounds were prepared with different concentration using dimethylformamide (DMF). One millilitre containing 100 times the amount of chemical was added to each bottle, which contained 100 discs, which were then placed in triplicate in a nutrient agar medium separately seeded with fresh bacteria. The incubation was carried out at 37 °C for 24 h. Solvent and growth controls were kept, and the zones of inhibition were noted, and minimum inhibitory concentration (MIC) for the active compounds was determined. The results were compared with the standard Streptomycin.
Antifungal activity
The newly synthesized compounds were screened for their antifungal activity against Aspergillus flavus and Fusarium verticillioides in DMSO by serial plate dilution method (James et al. 1970; Fenlon and Cynamon 1986). Sabouraud’s agar media were prepared by dissolving peptone (1 g), D-glucose (4 g) and agar (2 g) in distilled water (100 mL) and the pH was adjusted to 5.7. Normal saline was used to make a suspension of the spores of the fungal strain for lawning. A loopful of a particular fungal strain was transferred to 3 mL of saline to get a suspension of the corresponding species. Agar media of 20 mL was poured into each petri dish. An excess of the suspension was decanted, and the plates were dried by placing them in an incubator at 37 °C for 1 h. Using an agar punch, wells were made on these seeded agar plates, and 10–50 mg/mL of the test compounds in DMSO was added into each well labelling. Similarly, a control was also prepared using the solvent DMSO. The petri dishes were prepared in triplicate and maintained at 37 °C for 3–4 days. Antifungal activity was determined by measuring the inhibition zone. The obtained data was compared with the standard Nystain.
Molecular docking study
Molecular docking study was performed using AutoDock tools 1.5.6 (ADT 4.2). The 3D structures of the compounds in pdb form were prepared using CS ChemDraw Ultra 12.0. The 3D crystal structure of COX-2 was downloaded from the Protein Data Bank (PDB ID: 1CX2). Hetero atoms and water molecules were removed and hydrogen was added to the protein for correct calculation of the partial atomic charges. The grid box was set at 64, 64, and 64 Å (x, y, and z) with centre x = 25.0, y = 23.0, and z = 19.0 for 1CX2.pdb. The Lamarckin genetic algorithm (LCA) was applied to search for conformers that possessed the lowest binding energy. The results of the molecular docking analysis were obtained as estimated free energy of binding in kcal/mol.
References
Abu-Hashem AA, Youssef MM, Hussein HAR (2011) Synthesis, antioxidant, antituomer activities of some new thiazolopyrimidines, pyrrolothiazolopyrimidines and triazolopyrrolothiazolopyrimidines derivatives. J Chin Chem Soc 58:1–8
Allavena P, Sica A, Solinas G, Porta C, Mantovanii A (2008) The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 66:1–9
Ashok M, Holla BS, Kumari NS (2007) Convenient one pot synthesis of some novel derivatives of thiazolo[2,3-b]dihydropyrimidinone possessing 4-methylthiophenyl moiety and evaluation of their antibacterial and antifungal activities. Eur J Med Chem 42:380–385
Beekmann SE, Heilmann KP, Richter SS, Garcia-de-Lomas J, Doern GV (2005) Antimicrobial resistance in Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis and group A β-haemolytic streptococci in 2002–2003: results of the multinational GRASP Surveillance Program. Int J Antimicrob Agents 25:148–156
Bekhit AA, Fahmy HTY, Rostom SAF, Baraka AM (2003) Design and synthesis of some substituted 1H-pyrazolyl-thiazolo[4,5-d]pyrimidines as anti-inflammatory-antimicrobial agents. Eur J Med Chem 38:27–36
Bekhit AA, Fahmy HTY, Rostom SAF, Bekhit AEDA (2010) Synthesis and biological evaluation of some thiazolylpyrazole derivatives as dual anti-inflammatory antimicrobial agents. Eur J Med Chem 45:6027–6038
Capone ML, Tacconelli S, Sciulli MG, Patrignani P (2003) Clinical pharmacology of selective COX-2 inhibitors. Int J Immunopathol Pharmacol 16:49–58
Chua T, Moore CL, Perri MB, Donabedian SM, Masch W, Vager D, Davis SL, Lulek K, Zimnicki B, Zervos MJ (2008) Molecular epidemiology of methicillin-resistant Staphylococcus aureus bloodstream isolates in urban detroit. J Clin Microbiol 46:2345–2352
Comber RN, Gray RJ, Secrist JA (1991) Acyclic analogues of pyrazofurin: syntheses and antiviral evaluation. Carbohydr Res 216:441–452
Dekhane DV, Pawar SS, Gupta S, Shingare MS, Patil CR, Thore SN (2011) Synthesis and anti-inflammatory activity of some new 4,5-dihydro-1,5-diaryl-1H-pyrazole-3-substituted-heteroazole derivatives. Bioorg Med Chem Lett 21:6527–6532
Dillingham RA, Lima AA, Guerrant RL (2002) Cryptosporidiosis: epidemiology and impact. Microbes Infect 4:1059–1066
Dogne JM, Supuran CT, Pratico D (2005) Adverse cardiovascular effects of the coxibs. J Med Chem 48:2251–2257
Fatima S, Sharma A, Saxena R, Tripathi R, Shukla SK, Pandey SK, Tripathi R, Tripathi RP (2012) One pot efficient diversity oriented synthesis of polyfunctional styryl thiazolopyrimidines and their bio-evaluation as antimalarial and anti-HIV agents. Eur J Med Chem 55:195–204
Fenlon CH, Cynamon MH (1986) Comparative in vitro activities of ciprofloxacin and other 4-quinolones against Mycobacterium tuberculosis and Mycobacterium intracellulare. Antimicrob Agents Chemother 29:386–388
Fustero S, Rosello SM, Barrio P, Fuentes AS (2011) From 2000 to Mid-2010: a fruitful decade for the synthesis of pyrazoles. Chem Rev 111:6984–7034
Fustero S, Simon-Fuentes A, Sanz-Cervera JF (2009) Recent advances in the synthesis of pyrazoles. A review. Org Prep Proced Int 41:253–290
James D, Lowry M, Jaqua MJ, Selepak ST (1970) Detailed methodology and implementation of a semi automated serial dilution micro technique for antimicrobial susceptibility testing. Appl Microbiol 20:46–53
Keche AP, Hatnapure GD, Tale RH, Rodge AH, Kamble VM (2012) Synthesis, anti-inflammatory and antimicrobial evaluation of novel 1-acetyl-3,5-diaryl-4,5-dihydro (1H) pyrazole derivatives bearing urea, thiourea and sulfonamide moieties. Bioorg Med Chem Lett 22:6611–6615
Khalifa NM, Al-Omar MA, Amr AE, Baiuomy AR, Abdel-Rahman RF (2015) Synthesis and biological evaluation of some novel fused thiazolo[3,2-a]pyrimidines as potential analgesic and anti-inflammatory agents. Russ J Bioorg Chem 41:192–200
Kiyani H, Ghorbani F (2015) Boric acid-catalyzed multi-component reaction for efficient synthesis of 4H-isoxazol-5-ones in aqueous medium. Res Chem Intermed 41:2653–2664
Kumar V, Kaur K, Gupta GK (2015) Trifluoromethylpyrazoles as anti-inflammatory and antibacterial agents: a review. J Fluorine Chem 178:306–326
Kumar V, Kaur K, Gupta GK, Sharma AK (2013a) Pyrazole containing natural products: synthetic preview and biological significance. Eur J Med Chem 69:735–753
Kumar V, Kaur K, Gupta GK, Gupta AK, Kumar S (2013b) Developments in synthesis of an anti-inflammatory drug, celecoxib: a review. Recent Pat Inflamm Allergy Drug Discov 7:124–134
Li X, Ma S (2015) Advances in the discovery of novel antimicrobials targeting the assembly of bacterial cell division protein FtsZ. Eur J Med Chem 95:1–15
Li M, Zhao BX (2014) Progress of the synthesis of condensed pyrazole derivatives (from 2010 to mid-2013). Eur J Med Chem 85:311–340
Mohamed SF, Flefel EM, Amr AE-GE, Abd El-Shafy DN (2010a) Anti-HSV-1 activity and mechanism of action of some new synthesized substituted pyrimidine, thiopyrimidine and thiazolopyrimidine derivatives. Eur J Med Chem 45:1494–1501
Mohamed MS, Kamel R, Fatahala SS (2010b) Synthesis and biological evaluation of some thio containing pyrrolo[2,3-d]pyrimidine derivatives for their anti-inflammatory and anti-microbial activities. Eur J Med Chem 45:2994–3004
Nagarapu L, Vanaparthi S, Bantu R, Kumar CG (2013) Synthesis of novel benzo[4,5]thiazolo[1,2-a]pyrimidine-3-carboxylate derivatives and biological evaluation as potential anticancer agents. Eur J Med Chem 69:817–822
Nandakumar A, Thirumurugan P, Perumal PT, Vembu P, Ponnuswamy MN, Ramesh P (2010) One-pot multicomponent synthesis and anti-microbial evaluation of 2′-(indol-3-yl)-2-oxospiro(indoline-3,4′-pyran) derivatives. Bioorg Med Chem Lett 20:4252–4258
Palomer A, Cabre F, Pascual J, Campos J, Trugillo MA, Entrena A, Gallo MA, Garcia L, Macleon D, Espinosa A (2002) Identification of novel cyclooxygenase-2 selective inhibitors using pharmacophore models. J Med Chem 45:1402–1411
Patel V, Patel V (2013) Synthesis, characterization and antifungal activity of substituted ethyl 5,7-dimethyl-3-oxo-2,3-dihydro-5H-[1,3]-thiazolo[3,2-a]pyrimidie-6-carboxylate derivatives. Der Pharmacia Sinica 4:72–78
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Sivakumar KK, Rajasekaran A, Senthilkumar P, Wattamwar PP (2014) Conventional and microwave assisted synthesis of pyrazolone Mannich bases possessing anti-inflammatory, analgesic, ulcerogenic effect and antimicrobial properties. Bioorg Med Chem Lett 24:2940–2944
Smith ML, Hawcroft G, Hull MA (2000) The effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: evidence of different mechanisms of action. Eur J Cancer 36:664–674
Sondhi SM, Goyal RN, Lahoti AM, Singh N, Shukla R, Raghubir R (2005) Synthesis and biological evaluation of 2-thiopyrimidine derivatives. Bioorg Med Chem 13:3185–3195
Sorbera LA, Lesson PA, Castanar J, Castanar RM (2001) Valdecoxib and Parecoxib sodium: analgesic, antiarthritic, cyclooxygenase-2 inhibitor. Drugs Future 26:133–140
Viveka S, Dinesha, Laxmeshwar SS, Nagaraja GK (2012) Ethyl 7-methyl-5-(4-methylphenyl)-3-oxo-2-{[3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-4-yl] methylidene}-2,3-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimidine-6-carboxylate. Molbank M776:1–5
Viveka S, Dinesha, Madhu LN, Nagaraja GK (2015a) Synthesis of new pyrazole derivatives via multicomponent reaction and evaluation of their antimicrobial and antioxidant activities. Monatsh Chem 146:1547–1555
Viveka S, Dinesha, Shama P, Nagaraja GK, Ballav S, Kerkar S (2015b) Design and synthesis of some new pyrazolyl-pyrazolines as potential anti-inflammatory, analgesic and antibacterial agents. Eur J Med Chem 101:442–451
Viveka S, Dinesha, Shama P, Nagaraja GK, Deepa N, Sreenivasa MY (2015c) Design, synthesis, and pharmacological studies of some new Mannich bases and S-alkylated analogs of pyrazole integrated 1,3,4-oxadiazole. Res Chem Intermed https://doi.org/10.1007/s11164-015-2170-7
Viveka S, Dinesha, Shama P, Naveen S, Lokanath NK, Nagaraja GK (2015d) Design, synthesis, anticonvulsant and analgesic studies of new pyrazole analogues: a Knoevenagel reaction approach. RSC Adv 5:94786–94795
Viveka S, Prabhuswamy M, Dinesha, Lokanath NK, Nagaraja GK (2014) Synthesis, crystal structure, and characterization of new 2,4,5-triphenyl imidazole: 4,5-diphenyl-2-(3,4,5-trimethoxyphenyl)-1H-imidazole. Mol Cryst Liq Cryst 588:83–94
Winter CA, Risley EA, Nuss GW (1962) Carrageenin-induced edema in hind paws of the rat as an assay for anti-inflammatory drug. Proc Soc Exp Biol Med 111:544–547
Acknowledgements
The authors gratefully acknowledge the UGC, SAP, and DST-PURSE for the financial assistance. We are grateful to IISC Bangalore, Mysore University and USIC Mangalore University for providing the spectral analysis. We gratefully acknowledge Prasanna Shama, N.G.S.M. Institute of Pharmaceutical Sciences, Paneer Deralakatte, Mangalore, for anti-inflammatory activity and Guru Basavarajaswamy from Acharya & B.M. Reddy College of Pharmacy for molecular docking study. Also, thankful to Poornachandra Rao K and Sreenivasa MY from the Department of Microbiology Manasagangothri, Mysore University, Mysore, for antimicrobial study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Viveka, S., Dinesha, Nagaraja, G.K. et al. One pot synthesis of thiazolo[2,3-b]dihydropyrimidinone possessing pyrazole moiety and evaluation of their anti-inflammatory and antimicrobial activities. Med Chem Res 27, 171–185 (2018). https://doi.org/10.1007/s00044-017-2058-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-2058-8