
Abstract
The German wasp (Vespula germanica) is a highly successful invader on a global scale. These wasps were first observed in the Western Cape region in South Africa in 1972, and they have the potential to expand their range and cause significant damage to the native biodiversity. Our study used nuclear (DNA microsatellites) and mitochondrial DNA (mtDNA) from 42 wasp colonies to analyse the population genetics of V. germanica in their invaded South African range. We sequenced three mitochondrial genes; cytochrome c oxidase I, cytochrome b and cytochrome c oxidase II. We found six mtDNA haplotypes present in South Africa. Although only a single fertilized queen is sufficient for the establishment of a wasp nest, the probability of a single introduced queen to successfully establish a nest and a population is very small. If multiple queens were introduced at the same time, the probability for more than one haplotype being transferred to the new population increases. Therefore, the true number of queen introductions occurred in South Africa can be inferred to be between two and six. We examined nine microsatellite loci and found weak-to-no genetic sub-structuring, likely due to high dispersal rates. We concluded that German wasps in South Africa maintain a homogenous population with movement of individuals between localities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The successful establishment of invasive species can result in significant damage to their introduced ranges, including damage to economies, native biodiversity and human health (Pimentel et al. 2005; Kenis et al. 2009; Pyšek and Richardson 2010). Social insects, predominantly Hymenoptera, are classed globally as one of the most infamous invasive groups (Moller 1996; Chapman and Bourke 2001). Several social insect species are listed in the IUCN’s list of the most invasive species worldwide (Lowe et al. 2004). Factors said to enhance invasive success of social insects include their high reproductive rate and adept dispersal abilities, both through self- and human-mediated dispersal (Passera 1994; Moller 1996; Kenis et al. 2009). One key characteristic in the successful establishment of social insects is their eusocial nature which has been linked to their ecological dominance (Wilson and Hölldobler 2005). Social insects typically start a new colony from a single inseminated queen (Chapman and Bourke 2001). When establishing new populations, the wasp’s sex determination system drives the colony fate. Diploid individuals heterozygous at a single locus, complementary sex determiner (csd), develop as females, while haploid individuals develop as males. Diploid individuals homozygotes at the csd locus develop as diploid males, but these do not survive to adulthood or cannot sire offspring (Beye et al. 2003). This costly production of diploid males is the result of inbreeding depression caused by reduced genetic variation post-bottleneck. Thus, introductions by a single monandrous queen that has mated with a male with a matching allele in the csd locus will not succeed (Ding et al. 2017). Polyandry can be beneficial and facilitate the successful establishment in a new location (Crozier and Page 1985; Keller and Reeve 1994; Moller 1996; Goodisman et al. 2002). The invasion of Australia by the Asian honey bee, Apis cerana, occurred from a single introductory event in Cairns (Gloag et al. 2017). Balancing selection maintained heterozygosity at the csd locus by retaining rare csd alleles following the invasion, rescuing the population from a founder effect (Gloag et al. 2017).
Population genetics has become a useful tool in the investigation of invasive populations. It provides background information to the invasion process and assists in deciphering invasion colonization processes and demographics (Sakai et al. 2001). This background knowledge of the invasive process is important as it assists in the design of mitigation strategies to prevent spread (Sakai et al. 2001; Beggs et al. 2011; Masciocchi and Corley 2013). Geographical data alone are an inadequate means to determine the scale of eradication as there are multiple additional factors that influence invasion success including dispersal abilities, reproductive capabilities, enemy presence and genetic variation (Moller 1996). During the initial colonization of introduced species, a typical introduction will consist of a small number of individuals. Due to this small number of individuals, a genetic bottleneck may occur (Ellstrand and Elam 1993; Sakai et al. 2001). In order for a population to successfully persist and expand, the invasive propagule must overcome this bottleneck. Genetic variation has been observed to influence fitness in introduced ranges (Vandewoestijne et al. 2008; Markert et al. 2010; Dobelmann et al. 2017). The lack of genetic variation can result in inbreeding depression which can limit the populations’ adaptive evolution capabilities and in turn limit population growth (Ellstrand and Elam 1993; Nieminen et al. 2001). In some cases, the genetic constraints present after a bottleneck could also positively influence the rate of expansion through novel selective regimes as observed in invasive populations of the Argentine ant (Linepithema humile, Tsutsui et al. 2000).
Genetic variation is also a key component in adaptive evolution. Genetically diverse populations of invasive species are typically more prevalent when multiple introductions have occurred (Sakai et al. 2001; Dlugosch and Parker 2008). Some phenotypical changes due to genetic variation may encourage invasiveness, whereas some variations may be deleterious (Marsden et al. 2016). Gene flow between populations from multiple introductions could exacerbate the spread of these characteristics (Marsden et al. 2016). An examination of the genetics of invasive populations can assist in the understanding of the spread of an invasive species and in its future conservation and management (Estoup and Guillemaud 2010).
Vespula wasps are renowned invasive species and have successfully been established in several countries (Lester and Beggs 2019). The German wasp (Vespula germanica) is one notorious example. This species is native to the Palearctic region, its distribution encompasses Europe up to 62° N, the Mediterranean including Algeria and Morocco, and eastwards to northern India, China and Korea to 23° N (Spradbery and Maywald 1992). However, due to accidental human transport it has since been established itself in Australia, New Zealand, North America, South Africa and South America (Thomas 1960; Archer 1998; Sackmann et al. 2001; D’adamo et al. 2002; Beggs et al. 2011; de Villiers et al. 2017). Most Vespula species mate multiple times obligatorily in their native and invasive ranges (Loope et al. 2014). This is the case for V. germanica in their native range (Bonckaert et al. 2008) and in an introduced population in Australia (Goodisman et al. 2002).
Overall in South Africa, biological invasions have been calculated to cost around USD 600 million to the country (van Wilgen and Lange 2011). The Cape Floristic Region of South Africa (Fynbos biome) has an exceptionally high level of plant endemicity (Myers et al. 2000). The indigenous plant species richness in this region has been reduced by the introduction of invasive plants (Richardson et al. 1989). It is therefore of great concern that in addition to plant invaders, V. germanica (and other invasive invertebrates) has also been established in this region, further transforming its highly biodiverse ecosystem (Giliomee 2011). This wasp is known to have economic, social and environmental impacts in this invaded range (Haupt 2015). There has been, however, little genetic research on V. germanica in South Africa since its initial introduction which was reported in Cape Town in 1972 (Whitehead and Prins 1975; Veldtman et al. 2012). Vespula germanica is currently only documented in the Western Cape region of South Africa (Veldtman et al. 2012; de Villiers et al. 2017). However, recent modelling studies show that this wasp has the potential to expand to more climatically suitable areas including the vast majority of the Eastern part of South Africa (de Villiers et al. 2017).
Studies using DNA molecular markers can determine whether multiple introductions have occurred and give an estimate of the genetic variation present after an introduction event. Brenton-Rule et al. (2018) examined mitochondrial DNA (mtDNA) variation in the native and invaded ranges of V. germanica to determine the source of origin and intra-specific variation. Most of the invasions in the Southern Hemisphere appeared to originate from multiple introductions. A comparison of haplotypes from the invasive and native ranges of this wasp found haplotype matches suggesting the source of origin of those invasions, with the exception of South Africa. None of the mtDNA haplotypes surveyed from the South African populations of V. germanica matched a known haplotype from part of the wasp native range in Europe. The objectives of our study were first to estimate how many successful V. germanica wasp establishments have occurred in South Africa using mtDNA. Second, we sought to establish whether there is population structure among V. germanica sampled from different localities in South Africa, using DNA microsatellites. High diversity among locations would be indicative of population differentiation and limited dispersal, whereas a lack of genetic structure would suggest a homogeneous population with movement of individuals between localities.
Materials and methods
Sample collection
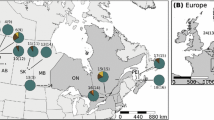
Vespula germanica wasps were collected from 42 nests across the invaded range in South Africa from 2016 to 2019 (Fig. 1; Supplementary Table 1). We extracted DNA from a total of 62 worker wasps from the 42 nests (Twenty wasps were replicates from some of the nests genotyped to ensure consistency but not included in the analyses as nestmates are related and not independent from one another; Goodisman et al. 2001). All samples were immediately preserved in 99% ethanol and frozen at − 20 °C once received in the laboratory.

Map showing the Vespula germanica sample locations across the invaded range in South Africa
DNA extraction
DNA was extracted from individual wasps by homogenizing the entire wasp, using a mixture 1 mL of GENEzol plant DNA reagent (Geneaid Biotech, Taiwan) and 5 μL of β-mercaptoethanol (Sigma-Aldrich, Michigan, USA) following Brenton-Rule et al. (2018). The extraction was then purified using standard chloroform/isopropanol protocols. DNA concentrations were determined using a NanoPhotometer (Implen, Germany).
Mitochondrial DNA
Polymerase chain reaction (PCR) was used to amplify portions of cytochrome b (cytb), cytochrome c oxidase I (CO1) and cytochrome c oxidase II (CO2) genes. CO2 incorporates the intergenic region between CO2 and CO1 that codes for tRNA–Leu. Primer sequences are listed in Supplementary Table 2. Each 25 μl PCR mix comprised 1 μl template DNA, 1 μl each of the forward and reverse primers, 9.5 μl of milliQ H2O and 12.5 μl MyTaq Mix (Bioline, London, UK). The PCR cycling conditions for each mitochondrial gene are shown in Supplementary Table 3. Samples were visualized using gel electrophoresis. The PCR product was then purified using ExoSAP-IT (Affymetrix, USA). Sequencing was performed on an ABI 3730xl DNA Sequencer (Macrogen, Seoul, Republic of Korea). Sequences were manually trimmed in Geneious v.8.0.5 (Kearse et al. 2012). The sequences were aligned using the ClustalW algorithm (Thompson et al. 1994) in Geneious. Sequences of one nest were excluded due to their low quality, thus a total of 41 nests were included in the mtDNA analyses. Mitochondrial DNA analyses also included five individuals, one each from Spain, Portugal, Austria, Belgium and France, respectively, from V. germanica’s native range in mainland Europe. All sequences were archived in GenBank (accession numbers MN727980-MN728042, MN736210-MN736272 and MN736273-MN736335). See Supplementary Table 1 for detailed accession number information relating loci with specimens.
For the phylogenetic analysis, we concatenated the three mtDNA genes (1518 base pairs) and homologous sequences for one Vespula vulgaris individual were downloaded from GenBank to be used as the outgroup. The most suitable model of sequence evolution was determined using BIC (Bayesian Information Criterion) scores in MEGA7 (Kumar et al. 2016). A phylogenetic tree was inferred using the MrBayes v.3.2.6 (Huelsenbeck and Ronquist 2001) plug-in for Geneious. Clade probabilities were obtained from the posterior distribution. Bayesian analyses were replicated twice, each with four Markov chains of 2 million generations. Trees were sampled every 2500 generations, of which the first 0.5 million generations were discarded as burnin.
A mitochondrial haplotype network of the South African and five European samples was inferred using the median-joining algorithm in NETWORK v.5 (http://www.fluxus-en- gineering.com; Bandelt et al. 1999).
A haplotype accumulation curve was constructed using the R package vegan (Oksanen et al. 2019). This curve was used to estimate whether the full haplotype diversity of South African wasps was sampled. Should the curve reach a horizontal asymptote, then it can be inferred that the haplotype diversity has been sampled adequately.
DNA microsatellites
Thirteen microsatellite loci previously designed for Vespula rufa (Thorén et al. 1995; Foster et al. 2001), Dolichovespula spp., Vespa cabro and Vespula spp. (Daly et al. 2002), Vespa mandarinia (Hasegawa and Takahasi 2002) and Vespa velutina (Arca et al. 2012) were assayed. Of these 13, ten loci amplified successfully (Rufa 5, Rufa 19, List 2003, List 2004, List 2007, List 2011, List 2019, VMA-6, VMA-7, R4-114; Supplementary Table 4). We genotyped these ten loci for each of the 62 V. germanica individuals from South Africa following Schmack et al. (2019). At the 5′-end of each forward primer, a M13-tag (TGTAAAACGACGGCCAGT) was added. Each of the 10 loci was amplified in a 10 µl PCR per individual containing 5 µl MyTaq Mix (Bioline, London, UK), 0.2 µl forward primer, 0.8 µl reverse primer, 0.8 µl M13-primer (labelled with one of the four dyes; 6-FAM, HEX, PET, NED), 0.8 µl bovine serum albumin (Sigma-Aldrich, New Zealand), 1.4 µl milliQ H2O and 1 µl of the template DNA. Each locus was assigned a specific dye and where possible completed in a single run to avoid dye shifts (Sutton et al. 2011). PCR thermocycling conditions, primer annealing temperatures and mixture groups are included in Supplementary Tables 4 and 5. PCR products were visualized using agarose gel electrophoresis. Genotyping was performed using an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) at Massey Genome Service (Massey University, Palmerston North, New Zealand). Alleles were sized using GeneScan 500 LIZ (Applied Biosystems) ladder and scored by hand using Geneious. The allelic scores obtained for the South African V. germanica wasps in this study are presented in Supplementary Table 6. To assess any possible scoring errors that may have occurred due to the presence of null alleles, stuttering and allelic dropout, we used Microchecker v.2.2.3 (van Oosterhout et al. 2004).
The South African wasp population descriptive statistics were calculated using GenAlEx v.6.5 (Peakall and Smouse 2012). We determined the number of alleles (Na), observed heterozygosity (Ho), and expected heterozygosity (He). Potential deviations from Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were estimated with Genepop v.4.2 (Rousset 2008) with the following Markov chain parameters: 1000 dememorizations, 1000 batches and 10,000 iterations. Significance levels (p = 0.05) for departure from HWE and LD were corrected for multiple comparisons.
To designate individuals into admixture proportions based on their allele frequencies, we used the program STRUCTURE v.2.3.4 (Pritchard et al. 2000). The data set was analysed applying the admixture model with the assumption of correlated frequencies, 100,000 run burn-in period, 1000,000 Markov Chain Monte Carlo iterations, and the possible number of South African V. germanica populations (k) ranged from 1 to 7. The analysis was repeated ten times across runs in order to ensure consistency. The Evanno et al.’s (2005) method was used to determine the most plausible number of clusters in our data set as implemented in STRUCTURE HARVESTER web v.0.6.94 (Earl and vonHoldt 2012). We also re-run the STRUCTURE analysis as described above including our samples, plus 20 European and three South African wasps genotyped by Schmack et al. (2019). Allelic scores from Schmack et al. (2019) were matched to ours by hand, as both studies were conducted in the same laboratory. Therefore, for the second STRUCTURE run we only included the seven loci in our study (Rufa 5, Rufa 19, List 2004, List 2007, List 2011, List 2019, R4-114) that were also genotyped by Schmack et al. (2019). Sample sizes were n = 45 for South Africa and n = 20 for Europe.
We carried out a factorial correspondence analysis (FCA) of multilocus genotypes to visually represent genetic variation at the individual level with GENETIX v.4.05 (Belkhir et al. 1998). Factorial correspondence analyses are computed in the bivariate space defined by the first two factorial components. The FCA incorporated data of V. germanica from Europe (n = 20) from Schmack et al. (2019). The overall FCA sample sizes were 45 South African nests and 20 European wasp nests.
Results
Number of successful wasp establishments in South Africa
The mitochondrial data showed six unique haplotypes in our data set from South Africa (H1–H6). Two haplotypes were more common; H1 was the most common haplotype, found in 31 wasps from South Africa and one wasp from Austria. The second most common South African haplotype (H2) was represented by five wasps from five different locations. These two haplotypes were three nucleotide substitutions apart. Two samples from Jonkershoek share the same haplotype (H3), and the remaining three haplotypes were represented by an individual sample from Kleinmond (H4), Worchester (H5) and Stellenbosch (H6), respectively (Figs. 1, 2).

Unrooted network showing the relationship of mtDNA haplotypes sampled from South African and European V. germanica. Each of the circles represents a unique haplotype. The size of the circles is proportional to the number of alleles found. Only South African haplotypes have been named (H1–H6)
The rarefaction analysis showed that the haplotype discovery curve is beginning to plateau, indicating that much of the South African genetic diversity has been sampled in our study (Fig. 3). It is possible, however, that additional genetic diversity would be found with additional sampling.

Bayesian phylogeny of V. germanica based on 1518 bp of concatenated CO1, CO2 and CytB genes. Sample sizes are n = 41 nests from South Africa and n = 5 nests from Europe. Support values above branches correspond to Bayesian posterior probabilities. The scale bar indicates nucleotide substitutions per site. Number of haplotypes per locality is shown between parentheses
The best-fitting model of sequence evolution was Hasegawa–Kishino–Yano (HKY) model (Hasegawa et al. 1985). The Bayesian phylogeny shows that all six haplotypes sampled are closely related. The number of nucleotide substitutions between haplotypes varied from one to five (Fig. 4). The percentage of sequence similarity for the 1518 bp data set was 99.5% including the five European samples.

Haplotype accumulation curve showing the six haplotypes sampled in South Africa. The vertical lines represent 95% confidence intervals
Genetic structure in South African wasps
No variation was observed in the microsatellite locus VMA7, and thus, it was excluded in all subsequent analyses. Following sequential Bonferroni correction, no significant linkage disequilibrium was detected among paired loci comparisons. Significant deviation from Hardy–Weinberg equilibrium was observed for locus List2004, following Bonferroni correction. No evidence of scoring errors due to stuttering, null alleles or large allele dropout was detected in our data set. We found an average expected (He) of 0.482 and an observed heterozygosity (Ho) of 0.489. The He ranged from 0.024 to 0.661, and Ho ranged from 0.024 to 0.976 (Table 1).
The largest number of alleles was 6, and the smallest number of alleles was 2 (Table 1). The average number of alleles across all loci was 3.333. This small number of effective alleles in South Africa is in agreement with low genetic diversity.
The deltaK (Δk) method suggested that the optimal number of genetic clusters was k = 2, Δk = 7.98 (Supplementary Figure 1). After visually inspecting the assignment plot, it was clear that the individuals were assigned to each of the two groups relatively equally (Supplementary Figure 2), which indicates that no substructure could be observed based on geography in our data set. Secondary optima at k = 3, Δk = 1.51 and at k = 6, Δk = 0.66 show the same pattern of individuals assigned equally to each cluster in the assignment plots, further suggesting population homogeneity with no substructure (Supplementary Figure 2). Thus, this result is highly indicative that there is a single, homogenous South African population.
We therefore run STRUCTURE a second time for an extended data set containing additional European samples; we found that the most likely number of admixture proportions was still 2, Δk = 85.66. The South African samples appeared to cluster with one sample from Switzerland in higher proportion (Fig. 5), while showing no admixture similarity with any other European country sampled.

STRUCTURE admixture proportion outputs for V. germanica in South Africa and Europe inferred from 7 microsatellite loci. We incorporated data from 45 South African individuals plus 20 European samples. The most likely k value was k = 2. Secondary optima identified at k = 3 and k = 6 are also shown
Due to the lack of substructure in the South African samples, we included 20 European samples from Schmack et al. (2019) in the FCA analyses. The first two axes of the FCA explain 14.8% and 10.8% of the respective variance observed (Fig. 6). Two European samples grouped with the South African samples and two of the South African samples plotted within the European samples suggesting shared genetic materials.

Factorial correspondence analysis of individual multilocus genotypes. Purple squares represent South African wasps, and green squares represent European wasps (color figure online)
Discussion
Invasive species resulting from human-mediated introductions represent a major threat to biodiversity, agriculture and the economies of invaded regions (Kenis et al. 2009; Pyšek and Richardson 2010; Lester and Beggs 2019). Population genetics is important in biological invasion studies and assists in the design of mitigation strategies to prevent spread (Sakai et al. 2001; Beggs et al. 2011; Masciocchi and Corley 2013). Using genetic markers as a preliminary step provides a deeper understanding of the background of the invasion. Knowing the demographics, history and colonization process of the invasion can assist in localizing eradication efforts, therefore minimizing associated costs and potential environmental impacts (Abdelkrim et al. 2005). This localization effort is important in the V. germanica population of South Africa, as they are still confined to a small area and range expansion has been slow; therefore, preventing further spread is of great importance (Veldtman et al. 2012; Haupt 2015; de Villiers et al. 2017; Brenton-Rule et al. 2018).
In this study, we uncovered six mtDNA haplotypes. Only a single fertilized queen is required for the establishment of a wasp nest and mtDNA is maternally inherited. However, the probability of a single introduced queen to successfully establish a nest and a population is very small. There needs to be more than one nest producing the next generation of queens and males, otherwise the queens and males produced by that one nest would need to inbreed, resulting in diploid male production. If multiple queens were introduced at the same time, the probability for more than one haplotype being transferred to the new population increases. Therefore, the true number of queen introductions occurred in South Africa can be inferred to be between two and six (the maximum number of haplotypes). Given that V. germanica has been documented in South Africa for approximately 47 years (Veldtman et al. 2012), it is highly unlikely that some of the haplotypes have arisen via recent mutations from the main haplotypes sampled in such short evolutionary time. Previous research outlining the origins of global invasions suggested that multiple introductions have occurred in South Africa; however the full haplotype diversity was not sampled (Brenton-Rule et al. 2018). Our larger sample size allowed for most mtDNA haplotype diversity to be sampled and confirmed multiple queen introductions.
Vespula germanica in South Africa presents low genetic diversity. The phylogenetic tree shows a homogenous population with closely related mtDNA haplotypes. The analyses of the nuclear data also showed no evidence of population sub-structuring within South Africa. This overall lack of genetic diversity could be due to a genetic bottleneck that might have occurred following each introduction with genetic drift acting to decimate variation or high dispersal rates by queens. However, we do not know the levels of genetic variation present in V. germanica’s native range; therefore, another explanation might be that the species as a whole harbours little genetic variability. We think this latter explanation unlikely due to the extensive geographic distribution of native V. germanica populations and the results from a recent study that compared samples of V. germanica from Argentina, Australia, New Zealand and several European countries (Brenton-Rule et al. 2018). The authors found levels of genetic variation in the native range much higher than what we found within South Africa suggesting that bottlenecks might have likely occurred.
Previous studies have utilized mtDNA to determine the origin of invasive wasp populations (Lester et al. 2014; Brenton-Rule et al. 2018). Brenton-Rule et al. (2018) incorporated nine South African samples in their analyses and suggested mainland Europe as the source of the South African introduction. However, their study lacked any samples from the extensive native range of V. germanica outside of Europe. In our mitochondrial data, an Austrian sample showed an exact match with the South African wasps we sampled but the second most common mitochondrial haplotype present in South Africa was not detected in the other four European samples we compared them to. These findings suggest that an increased number of native range samples would be required to make a solid conclusion on the origin of the South African V. germanica population.
There are various factors that could have facilitated the V. germanica invasion into South Africa. Sometimes invasive success could be due to phenotypical changes and reduced genetic diversity in Hymenoptera invasions (Chapman and Bourke 2001). Invasive Vespula wasp colonies possess an increased number of workers produced by multiple-mated queens (Goodisman et al. 2002). Polyandry is suggested to have helped Vespula wasps in other invaded regions (Goodisman et al. 2002; Dobelmann et al. 2017; Schmack et al. 2019), and could also be the case for the South African V. germanica population. Further research could be conducted to determine the level of polyandry present in the South African population.
Knowledge of genetic patterns in biological invasions is an important step before utilizing biological control strategies, as a deeper understanding of the genetics in the invasive region is required. Future research could incorporate the data obtained here to design appropriate invasion control methods suitable to South Africa. Background knowledge of the origin of the invasion could reveal where to find natural enemies or pathogens that could be used as potential biological control agents (Lester et al. 2014). Genetically mediated control is another method that is becoming of more interest to biologists and which requires an understanding of the target pest genetics (Dearden et al. 2018).
Overall, the South African wasps represent one genetic population with no sub-structuring. Potential causes leading to lack of genetic differentiation may include high dispersal rates as determined for the ant Formica fusca (Johansson et al. 2018) and/or genetic drift reducing variability following bottleneck events (Sakai et al. 2001; Azzurro et al. 2006). We also found that the South African German wasp population originated from multiple introductions. These introductions could have occurred independent of one another, or as single events involving multiple queens. It is important that future research try to shed more light on this because if multiple independent introduction events occurred in the past, future introductions may be likely.
References
Abdelkrim J, Pascal M, Calmet C, Samadi S (2005) Importance of assessing population genetic structure before eradication of invasive species: examples from insular Norway rat populations. Conserv Biol 19:1509–1518
Arca M, Capdevielle-Dulac C, Villemant C et al (2012) Development of microsatellite markers for the yellow-legged Asian hornet, Vespa velutina, a major threat for European bees. Conserv Genet Resour 4:283. https://doi.org/10.1007/s12686-011-9525-1
Archer ME (1998) The world distribution of the Euro-Asian species of Paravespula (Hym., Vespinae). Entomol Mon Mag 134:279–284
Azzurro E, Golani D, Bucciarelli G, Bernardi G (2006) Genetics of the early stages of invasion of the Lessepsian rabbitfish Siganus luridus. J Exp Mar Biol Ecol 333:190–201
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Beggs JR, Brockerhoff EG, Corley JC et al (2011) Ecological effects and management of invasive alien Vespidae. Biocontrol 56:505–526
Belkhir K, Borsa P, Chikhi L, et al (1998) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier, France
Beye M, Hasselmann M, Fondrk MK, Page RE, Omholt SW (2003) The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 114:419–429
Bonckaert W, Vuerinckx K, Billen J, Hammond RL, Keller L, Wenseleers T (2008) Worker policing in the German wasp Vespula germanica. Behav Ecol 19:272–278
Brenton-Rule EC, Dobelmann J, Baty JW et al (2018) The origins of global invasions of the German wasp (Vespula germanica) and its infection with four honey bee viruses. Biol Invasions 20:3445–3460
Chapman RE, Bourke AFG (2001) The influence of sociality on the conservation biology of social insects. Ecol Lett 4:650–662
Crozier RH, Page RE (1985) On being the right size: male contributions and multiple mating in social Hymenoptera. Behav Ecol Sociobiol 18:105–115
D’adamo P, Sackmann P, Corley JC, Rabinovich M (2002) The potential distribution of German wasps (Vespula germanica) in Argentina. N Z J Zool 29:79–85
Daly D, Archer ME, Watts PC et al (2002) Polymorphic microsatellite loci for eusocial wasps (Hymenoptera: Vespidae). Mol Ecol Notes 2:273–275
de Villiers M, Kriticos DJ, Veldtman R (2017) Including irrigation in niche modelling of the invasive wasp Vespula germanica (Fabricius) improves model fit to predict potential for further spread. PLoS ONE 12:e0181397. https://doi.org/10.1371/journal.pone.0181397
Dearden PK, Gemmell NJ, Mercier OR et al (2018) The potential for the use of gene drives for pest control in New Zealand: a perspective. J R Soc N Z 48:225–244
Ding G, Xu H, Oldroyd BP, Gloag RS (2017) Extreme polyandry aids the establishment of invasive populations of a social insect. Heredity 119:381–387
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Dobelmann J, Loope KJ, Wilson-Rankin E et al (2017) Fitness in invasive social wasps: the role of variation in viral load, immune response and paternity in predicting nest size and reproductive output. Oikos 126:1208–1218
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Ann Rev Ecol Syst 24:217–242
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19:4113–4130
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Foster KR, Ratnieks FLW, Gyllenstrand N, Thorén PA (2001) Colony kin structure and male production in Dolichovespula wasps. Mol Ecol 10:1003–1010
Giliomee JH (2011) Recent establishment of many alien insects in South Africa: a cause for concern. Afr Entomol 19:151–155
Gloag R, Ding G, Christie JR et al (2017) An invasive social insect overcomes genetic load at the sex locus. Nat Ecol Evol 1:0011. https://doi.org/10.1038/s41559-016-0011
Goodisman MAD, Matthews RW, Crozier RH (2001) Hierarchical genetic structure of the introduced wasp Vespula germanica in Australia. Mol Ecol 10:1423–1432
Goodisman MA, Matthews RW, Crozier RH (2002) Mating and reproduction in the wasp Vespula germanica. Behav Ecol Sociobiol 51:497–502
Hasegawa E, Takahashi J (2002) Microsatellite loci for genetic research in the hornet Vespa mandarinia and related species. Mol Ecol Notes 2:306–308
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Haupt K (2015) Assessment of the invasive wasp Vespula germanica in South Africa. M.Sc. Thesis. University of Stellenbosch
Huelsenbeck JP, Ronquist F (2001) MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Johansson H, Seppa P et al (2018) Weak population structure in the ant Formica fusca. PeerJ 6:e5024. https://doi.org/10.7717/peerj.5024
Kearse M, Moir R, Wilson A et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Keller L, Reeve HK (1994) Genetic variability, queen number, and polyandry in social Hymenoptera. Evol (N Y) 48:694–704
Kenis M, Auger-Rozenberg M-A, Roques A et al (2009) Ecological effects of invasive alien insects. Biol Invasions 11:21–45
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lester PJ, Beggs JR (2019) Invasion success and management strategies for social Vespula wasps. Annu Rev Entomol 64:51–71
Lester PJ, Gruber MAM, Brenton-Rule EC et al (2014) Determining the origin of invasions and demonstrating a lack of enemy release from microsporidian pathogens in common wasps (Vespula vulgaris). Divers Distrib 20:964–974
Loope KJ, Chien C, Juhl M (2014) Colony size is linked to paternity frequency and paternity skew in yellowjacket wasps and hornets. BMC Evol Biol 14:277. https://doi.org/10.1186/s12862-014-0277-x
Lowe S, Browne M, Boudjelas S, De Poorter M (2004) 100 of the World’s Worst Invasive Alien Species: a selection from the Global Invasive Species Database. Invasive Species. Published by The Invasive Species Specialist Group (ISSG) a specialist group of the Species Survival Commission (SSC) of the World Conservation Union (IUCN)
Markert JA, Champlin DM, Gutjahr-Gobell R et al (2010) Population genetic diversity and fitness in multiple environments. BMC Evol Biol 10:205. https://doi.org/10.1186/1471-2148-10-205
Marsden CD, Ortega-Del Vecchyo D, O’Brien DP et al (2016) Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc Natl Acad Sci USA 113:152–157
Masciocchi M, Corley J (2013) Distribution, dispersal and spread of the invasive social wasp (Vespula germanica) in Argentina. Austral Ecol 38:162–168
Moller H (1996) Lessons for invasion theory from social insects. Biol Conserv 78:125–142
Myers N, Mittermeier RA, Mittermeier CG et al (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Nieminen M, Singer MC, Fortelius W et al (2001) Experimental confirmation that inbreeding depression increases extinction risk in butterfly populations. Am Nat 157:237–244
Oksanen J, Blanchet G, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos PM, Stevens HH, Wagner H (2019) vegan: Community Ecology Package. R package version 2.3–5. http://CRAN.R-project.org/package=vegan
Passera L (1994) Characteristics of tramp species. In: Williams DF (ed) Exotic ants: biology, impact and control of introduced species. Westview Press, Oxford, pp 23–43
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pimentel D, Zuniga R, Morrison D (2005) Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol Econ 52:273–288
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pyšek P, Richardson DM (2010) Invasive species, environmental change and management, and health. Annu Rev Environ Resour 35:25–55
Richardson DM, Macdonald IAW, Forsyth GG (1989) Reductions in plant species richness under stands of alien trees and shrubs in the Fynbos biome. South Afr For J 149:1–8
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Sackmann P, Rabinovich M, Corley JC (2001) Successful removal of German yellowjackets (Hymenoptera: Vespidae) by toxic baiting. J Econ Entomol 94:811–816
Sakai AK, Allendorf FW, Holt JS et al (2001) The population biology of invasive species. Annu Rev Ecol Syst 32:305–332
Schmack JM, Brenton-Rule EC, Veldtman R et al (2019) Lack of genetic structuring, low effective population sizes and major bottlenecks characterise common and German wasps in New Zealand. Biol Invasions 21:3185–3201
Spradbery JP, Maywald GF (1992) The distribution of the European or German wasp, Vespula germanica (F.) (Hymenoptera: Vespidae), in Australia: past, present and future. Aust J Zool 40:495–510
Sutton JT, Robertson BC, Jamieson IG (2011) Dye shift: a neglected source of genotyping error in molecular ecology. Mol Ecol Resour 11:514–520
Thomas CR (1960) The European wasp (Vespula germanica Fab.) in New Zealand. DSIR Information Series No. 27
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Thorén PA, Paxton RJ, Estoup A (1995) Unusually high frequency of (CT)n and (GT)n microsatellite loci in a yellowjacket wasp, Vespula rufa (L.) (Hymenoptera: Vespidae). Insect Mol Biol 4:141–148
Tsutsui ND, Suarez AV, Holway DA, Case TJ (2000) Reduced genetic variation and the success of an invasive species. Proc Natl Acad Sci USA 97:5948–5953
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
van Wilgen BW, De Lange WJ (2011) The costs and benefits of biological control of invasive alien plants in South Africa. Afr Entomol 19:504–514
Vandewoestijne S, Schtickzelle N, Baguette M (2008) Positive correlation between genetic diversity and fitness in a large, well-connected metapopulation. BMC Biol 6:46. https://doi.org/10.1186/1741-7007-6-46
Veldtman R, Addison P, Tribe GD (2012) Current status and potential future impact of invasive vespid wasps (Vespula germanica and Polistes dominulus) in South Africa. IOBC/WPRS Bull 75:217–227
Whitehead VB, Prins AJ (1975) The European wasp, Vespula germanica (F.), in the Cape Peninsula. J Entomol Soc South Afr 38:39–42
Wilson EO, Hölldobler B (2005) Eusociality: origin and consequences. Proc Natl Acad Sci USA 102:13367–13371
Acknowledgements
This work was funded by Victoria University of Wellington. We thank Julia Schmack for sharing her DNA microsatellite data with us and two anonymous reviewers for their useful comments that improved our original manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Eloff, J., Veldtman, R., Bulgarella, M. et al. Population genetics of the invasive wasp Vespula germanica in South Africa. Insect. Soc. 67, 229–238 (2020). https://doi.org/10.1007/s00040-020-00752-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00040-020-00752-x