
Abstract
Invasive species cause severe ecological and economic impacts in their introduced ranges. Vespula wasps, native to Eurasia, are a major threat to New Zealand native ecosystems. Understanding factors that influence the success of wasp invasion is pivotal for the development of control strategies. Here, we compare genetic diversity and structure of Vespula germanica and Vespula vulgaris between regions of their native and introduced ranges using microsatellite markers. Our study found lower diversity and lack of genetic structure for both invasive Vespula species within New Zealand. The significant reduction in allelic richness, gene diversity and effective population size illustrate a major bottleneck in New Zealand V. germanica and V. vulgaris populations. Strong signatures of population structure were found for both Vespula species with two clusters being identified as optimal k, approximately corresponding to the native and the invaded ranges. Our results highlight the fact that the lack of genetic diversity does not impede successful invasions in V. germanica and V. vulgaris and encourage further research into mechanisms that promote the success of invasive social insects. Overall, this study provides insights into the genetics of invasive Vespula wasps that can be useful for the development of efficient management strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biological invasions result in severe impacts on ecosystems, the economy and human health (Clavero and García-Berthou 2005; Pimentel et al. 2005; Pyšek and Richardson 2010). Social insects like wasps, ants and termites are a particularly harmful group of invaders; their social lifestyle, high dispersal and reproduction rates as well as strong competitive abilities and the potential of rapid spread throughout the invaded range facilitate the invasion process (Moller 1996; Chapman and Bourke 2001; Suarez et al. 2002; Kenis et al. 2009).
Reduced genetic diversity through founder events, measured through reduced heterozygosity and number of alleles, is expected to result in decreased fitness in the invaded range (Nei et al. 1975; Allendorf and Lundquist 2003; Reed and Frankham 2003). Yet, the majority of studies demonstrates high invasion success despite severe bottlenecks (Golani et al. 2007; Dlugosch and Parker 2008; Puillandre et al. 2008; Burne et al. 2017; Zhang and Evans 2017). In contrast, some studies found high levels of genetic diversity in invasive populations, suggesting that a large founding population and/or multiple introductions lead to invasion success (Kolbe et al. 2004). Examples for elevated genetic diversity within the invaded range include the invasive brown anole, Anolis sagrei, and the European paper wasp, Polistes dominula, both introduced to the United States (Johnson and Starks 2004; Kolbe et al. 2004) and the mosquitofish, Gambusia affinis, invasive in New Zealand (Purcell et al. 2012). A recent study on the mosquito Aedes albopictus, invasive in several islands in the Indian Ocean, indicates that genetic diversity is likely to increase with increasing time since establishment (Sherpa et al. 2018).
The effective population size (Ne) is a powerful parameter to determine the rate of change in the composition of a population caused by genetic drift, the level of variability in a population, and the effectiveness of selection relative to drift (Wright 1931; Charlesworth 2009). Ne describes the amount of the gene pool that is passed on to the next generation; it determines within-species diversity and potential degree of inbreeding (Charlesworth 2009; Wang et al. 2016) and thus, is key for assessing the viability of invasive populations (Zayed 2004; Zayed et al. 2007; Luikart et al. 2010; Laugier et al. 2016). Reduced genetic diversity through founder effects during the invasion process can result in low Ne which leads to high levels of genetic drift and reduces the population’s ability to adapt to changing environments (Reed and Frankham 2003; Zayed et al. 2007). Departures from the ideal of random mating alter Ne (Sugg and Chesser 1994). For example, multiple mating or polyandry increases Ne in reptiles (Davis et al. 2001; Pearse and Avise 2001; Moore et al. 2008), mammals (Shurtliff et al. 2005) and land snails (Murray 1964; but see also Karl 2008), while complementary sex determination leads to the production of infertile diploid males (Whiting 1933; Beye et al. 2003), which is expected to result in even lower Ne than predicted under haplodiploidy only (Zayed 2004). Exceptions to this theory are the invasive Asian honey bee, Apis cerana, which overcame the genetic depletion during the invasion of Australia due to natural selection of rare csd alleles (Gloag et al. 2017) and the solitary bee, Lasioglossum leucozonium, which successfully invaded North America despite extremely reduced levels of genetic variation, a significant bottleneck and lack of population structure (Zayed and Packer 2007). Thus, genetic variability may be conserved through mechanisms of natural selection, which compensates even for severe bottlenecks in hymenopterans (Gloag et al. 2017).
Vespula wasps, native to Eurasia, are highly efficient invaders and the economic and ecological impacts in their invaded ranges are numerous. Social behaviour, a polyphagous diet and the initiation of colonies by a single mated queen are considered the reasons for their exceptional global invasion success (Moller 1996; Hanna et al. 2014; Lester and Beggs 2019). Vespula wasps have become invasive in Argentina, Oceania, South Africa and the United States including all major islands of Hawaii (Visscher and Vetter 2003; Beggs et al. 2011) and recent studies show that climate change is likely to increase the invasion pressure by Vespula wasps on a global scale (Parmesan 2006; Hulme 2009).
The highest densities of Vespula wasps in the world are found in New Zealand South Island’s honeydew-beech forests (Nothofagus spp.) (Moller et al. 1991; Beggs et al. 1998). Invaded ecosystems undergo numerous negative shifts including the decline of native taxa (Beggs et al. 2011; Gardner-Gee and Beggs 2013; Lester et al. 2013; Burne et al. 2017). As social wasps use invertebrate prey to rear their larvae, high densities of Vespula wasps restructure insect communities (Toft and Rees 1998; Beggs and Rees 1999).
Two Vespula species invaded New Zealand. The German wasp (Vespula germanica) established on the North Island of New Zealand around 1945 (Clapperton et al. 1989). The common wasp (Vespula vulgaris) was first recorded in New Zealand in 1921 (Donovan 1984) becoming widespread and very abundant since the 1970s. Mitochondrial DNA (mtDNA) suggests that England and Scotland were the most likely sources of V. germanica wasps into New Zealand (Brenton-Rule et al. 2018) whereas the New Zealand population of V. vulgaris seem to have originated from England and Ireland (Lester et al. 2014). An assessment of the effective population size of V. germanica and V. vulgaris within New Zealand and a comparison with those in their native ranges are lacking.
We hypothesised that genetic bottlenecks during the introduction of V. germanica and V. vulgaris into New Zealand reduced the effective population size, heterozygosity and number of alleles considerably when compared to those in the native range (Dlugosch and Parker 2008). If we find evidence of a bottleneck, then higher levels of overall genetic diversity in the native range are expected when compared to the New Zealand introduced range of both Vespula species. We genotyped 10 and 14 microsatellite loci for V. germanica and V. vulgaris, respectively, to determine whether: (1) New Zealand populations were founded by a small number of individuals; (2) genetic diversity reflects a population bottleneck, and (3) these wasp populations are genetically structured within their invaded and native ranges.
Materials and methods
Wasp collection
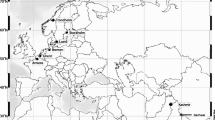
Foraging wasps, V. germanica (n = 44) and V. vulgaris (n = 40) were collected throughout their native range in Europe and invaded ranges. Samples for V. germanica from the native range included specimens from Austria (n = 2), Belgium (n = 2), England (n = 2), France (n = 3), Germany (n = 1), Italy (n = 1), Portugal (n = 2), Scotland (n = 3), Spain (n = 2), Sweden (n = 1) and Switzerland (n = 1). The invasive range was represented by V. germanica individuals from Australia (n = 1), New Zealand (n = 20) and South Africa (n = 3). Vespula vulgaris samples from the native range included specimens from Belgium (n = 10) and Germany (n = 10) and from the invasive range in New Zealand (n = 20). Sampling in New Zealand for both species includes six offshore islands on the northern east coast of the North Island (Fig. 1). Specimen collection information is presented in Supplemental Table S1. Individuals were collected and immediately placed in 99% ethanol, and frozen upon arrival to the laboratory until DNA extraction. Following the criteria of Brenton-Rule et al. (2018), we considered the United Kingdom (UK) samples (England and Scotland) separated from mainland Europe. First, because the UK is geographically separated from the mainland. Secondly, it is the known source of the Vespula spp. invasions into New Zealand (Lester et al. 2014; Brenton-Rule et al. 2018).

Sampling locations of Vespula germanica and V. vulgaris in the native European range (left) and the introduced range in New Zealand (right). The V. germanica samples collected from Australia and South Africa are not shown
Genetics
We extracted DNA from whole wasps using a modified chloroform protocol (GENEzol reagent, Geneaid Biotech, Taiwan was used in combination with β-mercaptoethanol, Sigma Aldrich, St Louis, MI, USA). We tested microsatellite primers previously developed for other vespine wasps in V. germanica and V. vulgaris (Thorén et al. 1995; Foster et al. 2001; Daly et al. 2002; Hasegawa and Takahashi 2002; Arca et al. 2012). Primer pairs were initially tried on six V. germanica individuals (30 loci assayed of which 24 amplified) and seven V. vulgaris individuals (31 loci assayed of which 27 amplified) from New Zealand. We finally assayed the multilocus genotype of 44 V. germanica individuals for 10 variable loci (of the 24 loci that amplified, 6 were non-variable and 8 presented excessive stuttering therefore these 14 loci were discarded) and of 40 V. vulgaris individuals at 14 variable loci (of the 27 loci that amplified successfully, 8 were non-variable and 5 loci presented excessive stuttering and were discarded; Supplemental Table S2). We screened one worker per nest as nestmates are related and not independent from one another (Goodisman et al. 2001). A M13 tag (TGTAAAACGACGGCCAGT) was added to the 5′-end of the forward primer of each locus. Each locus was amplified in 10 μL PCRs that contained 1 μL of template DNA, 0.2 μM forward primer, 0.8 μM reverse primer, 0.8 μM M13 primer (labelled with FAM, NED, PET or HEX), 0.8 uL Bovine Serum Albumin (Sigma Aldrich), ultra-pure water and 1 × MyTaq Mix (Bioline, London, UK). Multiplex PCR thermocycling conditions and primer annealing temperatures are reported in Supplemental Tables S2 and S3. Genotyping was performed on an ABI 3130x1 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) at Massey Genome Service (Massey University, Palmerston North, New Zealand). Alleles were sized using the internal size standard GeneScan 500 LIZ (Applied Biosystems) and scored by hand using Geneious v.10.2.3 (Kearse et al. 2012). To avoid dye shifts (Sutton et al. 2011) we assigned one dye per locus (FAM, NED, PET or HEX) and whenever possible genotyped all individuals for one locus in the same run. Possible scoring errors caused by null alleles, stutter and allelic dropout were assessed with Microchecker v2.2.3 (van Oosterhout et al. 2004). To estimate genotyping errors, 10% of the samples were re-amplified and genotyped at least once for quality control. We found no inconsistencies between replicates. Allelic scores for V. germanica are presented in Supplemental Table S4 and for V. vulgaris in Supplemental Table S5.
Analysis of genetic diversity
Allele frequencies, observed (Ho) and expected (He) heterozygosities at the assayed microsatellite loci within the native or introduced V. germanica and V. vulgaris populations were estimated using GenAlEx v.6.5 (Peakall and Smouse 2012). Allelic richness per locus and population as an unbiased measure of the number of alleles adjusted by sample size was estimated using FSTAT v.2.9.3 (Goudet 1995). Gene diversity was also calculated with FSTAT for both species. We used Wilcoxon signed-rank test to compare allelic richness and gene diversity between the invaded and native ranges for both species. Possible deviation from Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) between all locus pairs and by population (1000 dememorisations, 1000 batches, and 10,000 iterations per batch) were analysed using Genepop v.4.2 (Rousset 2008). Significance levels (p = 0.05) for departure from HWE and LD were corrected for multiple comparisons.
Analysis of genetic structure
We estimated the degree of population differentiation within V. germanica and V. vulgaris by calculating FST and RST in Arlequin v.3.5.2.2 (Excoffier and Lischer 2010). A Bayesian clustering approach was used to assign individuals to admixture proportions based solely on allele frequencies without including a priori information as implemented in the program STRUCTURE v.2.3.4 (Pritchard et al. 2000; Hubisz et al. 2009). Data were analysed using the admixture model assuming correlated frequencies with a 100,000 burn-in period and a million Markov Chain Monte Carlo iterations, the possible number of populations (k) ranged from 1 to 10 for V. germanica and from 1 to 12 for V. vulgaris; analyses were repeated 10 times to ensure consistency across runs. We used the Evanno et al. (2005) method to determine the optimal number of genetic clusters (k) given the data as implemented in STRUCTURE HARVESTER web v0.6.94 (Earl and vonHoldt 2012).
We performed a factorial correspondence analysis (FCA) of multilocus genotypes to visualise the genetic variation at the individual level as implemented in GENETIX v.4.05 (Belkhir et al. 1998).
We estimated contemporary effective population sizes (Ne) for V. germanica and V. vulgaris populations with the software NeESTIMATOR v.2 (Do et al. 2014) using the linkage disequilibrium method. This method evaluates non-random associations formed among alleles at different loci that occur when Ne is low and thus genetic drift influences allelic frequencies (Luikart et al. 2010). We used the random mating model and 95% confidence limits were obtained by jackknifing over loci. To test the effects that low-frequency alleles have on effective population size estimates, rare alleles (alleles with frequency < Pcrit) were excluded (Sonsthagen et al. 2017). We estimated Ne with Pcrit values from the lowest frequency of 0.01–0.09 and without frequency restriction. If Ne estimates vary across a range of Pcrit values, this suggests a history of gene flow and/or the presence of first-generation dispersers. If Ne remains stable across a range of Pcrit values, this indicates isolated populations (Waples and England 2011; Sonsthagen et al. 2017).
Results
Genetic diversity
Vespula germanica
All loci in the native population were in Hardy–Weinberg equilibrium (HWE) although five loci in the introduced population deviated significantly from HWE after sequential Bonferroni correction (Holm 1979). All loci pairs were in linkage disequilibrium (LD) for V. germanica in the native and the invasive populations following Bonferroni correction. Microchecker found no evidence of scoring errors or large alleles dropout. However, there is evidence of null alleles for the native population at locus List-2007 (estimated frequency of 0.1388). For the introduced population, null alleles were detected for six loci (Rufa-5 with an estimated frequency of 0.2307, List-2007: 0.1204, List-2011: 0.1841, List-2019: 0.1325, VMA-6: 0.15 and R4-114: 0.159); which might explain the deviation from HWE. The null allele frequencies at these loci was estimated to be low (< 0.25) and previous research demonstrated that low frequency null alleles have little influence on the detection of genetic differentiation (Carlsson 2008; Rico et al. 2017), therefore, we decided to retain these seven loci.
Observed (Ho) and expected (He) heterozygosities ranged from 0.108 to 0.536 in the native range and 0.071–0.347 in the invaded range, respectively. Alleles per locus ranged from 1 to 12 in V. germanica (mean = 4.1, Table 1). The native population presented a larger number of alleles (mean = 5.1) when compared to the introduced population (mean = 3.2, Table 2). The number of private alleles was also larger for the native population with a total of 24 private alleles from 7 loci (mean = 2.4) while for the introduced V. germanica population, only 5 private alleles were found each corresponding to a different locus (mean = 0.5, Table 1).
Introduced populations of V. germanica show extremely low levels of genetic diversity. Allelic richness was significantly reduced in the introduced population when compared to the native population (Table 2; Wilcoxon sign-rank test, n = 10, p = 0.028). The same pattern was observed for gene diversity (Table 2; Wilcoxon sign-rank test, n = 10, p = 0.037).
Vespula vulgaris
No significant LD was detected among paired loci comparisons by population and no deviations from HWE were observed after Bonferroni correction. The V. vulgaris data shows no evidence of scoring errors, large allele dropout or null alleles for either population. Ho and He ranged from 0.184 to 1.000 in the European native range. In the New Zealand invaded range, Ho and He varied from 0.000 to 0.895. Alleles per locus ranged from 2 to 10 in the native population (mean = 5.7) and from 1 to 7 in the invaded population (mean = 3.5, Tables 1, 2). As expected, the number of alleles was larger in the native than in the invasive population of V. vulgaris with the native population having a larger number of private alleles (mean = 2.786) than the invasive population (mean = 0.571, Table 1).
As found for the congener species, V. vulgaris showed significantly reduced allelic richness in the introduced range when compared to the native range wasps (Table 2; Wilcoxon sign-rank test, n = 14, p = 0.001). Gene diversity was also significantly lower in the invaded range than in the native range (Table 2; Wilcoxon sign-rank test, n = 14, p = 0.006).
Genetic structure
Vespula germanica
A strong signature of population structure was found for V. germanica with frequency differences detected among microsatellite loci (FST = 0.166, p < 0.0001; RST = 0.101, p = 0.017). The STRUCTURE HARVESTER analysis of Δk indicated that the optimal k was 2, Δk = 371.678 (Fig. 2; Suppl. Fig. S1) corresponding roughly to the native European population and the invasive localities. Secondary optima were detected at k = 5, Δk = 9.074 and at k = 7, Δk = 2.255.

STRUCTURE outputs for Vespula germanica inferred from 10 microsatellite loci. k = 2 was selected as the most likely k value by Structure Harvester but higher levels of k revealed further population subdivision. Secondary optima identified at k = 5 and k = 7 are also shown
At k = 2, samples collected in the introduced ranges of New Zealand and Australia clustered with the five samples collected from the United Kingdom (3 from Scotland, 2 from England) confirming that the United Kingdom is the origin of the New Zealand and Australian introductions. However, the samples collected in the introduced range in South Africa grouped with samples from European countries (Austria, Belgium, France, Germany, Italy, Spain, Portugal, and Sweden) suggesting that continental Europe is the most likely origin for the South African V. germanica introduction (Fig. 2).
At k = 5, wasps were assigned to five admixture proportions. Samples from mainland Europe were mostly assigned with equal probability to two proportions. The five samples from the United Kingdom grouped into a third proportion with high probability as did four of the New Zealand wasps and the only Australian wasp. The remaining 16 New Zealand wasps grouped on a fourth proportion and the three South African samples clustered together in the fifth admixture proportion (Fig. 2).
At k = 7, additional population structuring was evident. Samples from mainland Europe were assigned with equal probability to four admixture proportions. The five samples from the United Kingdom grouped into a fifth proportion together with one Australian and four New Zealand wasps. Sixteen New Zealand wasps grouped on a sixth proportion and the three South African samples clustered together in one of the four admixture proportions identified for the European samples (Fig. 2).
The first two axes of FCA for V. germanica explained 16.56% and 10.70% of the total variance observed (Fig. 4a) with the three South African samples grouping apart from the rest of the individuals. When excluding the South African samples, axis 1 explained 13.09% and axis 2 9.22% of the variance, with the introduced population clustering tightly together (Fig. 4b).
Vespula vulgaris
Population differentiation was high among microsatellite loci for this species as well (FST = 0.108, p < 0.0001; RST = 0.109, p = 0.002). STRUCTURE HARVESTER uncovered an optimal k at 2, Δk = 198.902. These two admixture proportions represented the native population (proportion 1: Belgium and Germany, Fig. 3) and the New Zealand invasion (proportion 2, Fig. 3) with the exception of one sample collected in the Karori suburb of Wellington that seemed to have originated from either Belgium or Germany. These results suggest that the main V. vulgaris invasion into New Zealand has not occurred from Belgium or Germany but from a population not sampled for this study. Suboptimal k were detected at k = 3, Δk = 21.153 and k = 5, Δk = 4.444.

STRUCTURE outputs for Vespula vulgaris inferred from 14 microsatellite loci. k = 2 was selected as the most likely k value by Structure Harvester. Secondary optima identified at k = 3 and k = 5 are also shown
At k = 3, the samples from Belgium and Germany and one sample from the Karori suburb of Wellington grouped with approximately equal probability into two admixture proportions whereas the New Zealand samples clustered in a third independent group (Fig. 3).
At k = 5, the European samples, plus the Karori sample, were assigned equally to four admixture proportions. Fifteen New Zealand wasps collected from offshore islands and other northern localities of the North Island grouped together in a fifth proportion while four other New Zealand samples: three collected from Nelson Lakes in the South Island and one collected in Rotokakahi Lake, Rotorua, North Island were assigned to the five proportions previously identified (Fig. 3).
The FCA shows higher genetic diversity in samples from the native range than in the introduced populations, with the first two axes explaining 8.96% and 7.16% of the observed variance, respectively (Fig. 4c).

Factorial correspondence analysis of individual multilocus genotypes. Multilocus scores are computed in the bivariate space defined by the first two factorial components. a FCA performed on all 44 V. germanica individuals based on 10 microsatellite loci, b same analyses for V. germanica excluding the 3 South African samples, and c results for all Vespula vulgaris individuals (n = 40) based on 14 microsatellite loci
Effective population sizes (Ne)
A signature of a reduced effective population size for the invasive population of V. germanica was observed based on the linkage disequilibrium method (Fig. 5a). Both native and introduced populations presented relatively stable values of Ne across Pcrit values, with the introduced population presenting narrow confidence limits. The native population upper bound confidence interval was infinity.

Changes in contemporary effective population sizes (Ne) estimates for V. germanica (top) and V. vulgaris (bottom) as a function of excluding rare alleles (Pcrit). The solid lines represent the point estimate of Ne, and dashed lines are the associated 5% and 95% confidence limits. Upper 95% confidence limits for the native population of V. germanica was ∞ for all estimates. The native population of V. vulgaris presented values of Ne = ∞ for all values of Pcrit except 0.07, with upper 95% confidence limits = ∞ for all estimates
Native V. vulgaris represent a large and stable population across Pcrit, suggestive of a panmictic population with substantial levels of gene flow across its natural range. The introduced V. vulgaris population however, presents small estimates of effective population size with very narrow confidence intervals (Fig. 5b).
Discussion
We compared levels of genetic diversity within the native and the invaded range of V. germanica and V. vulgaris and found a strong reduction in genetic diversity in the invaded range of New Zealand for both species, as predicted. Vespula germanica in the invaded range showed 70% of the expected heterozygosity and 63% of the allelic richness found in the native range. Vespula vulgaris in the invaded range had 68% of the expected heterozygosity and 61% of the allelic richness found in the European samples. For both species, only 20–21% of private alleles present in the native range were found in the invaded range, a reduction of 80%. Our findings indicate a strong genetic bottleneck for V. germanica and V. vulgaris in their invaded range of New Zealand and suggest that New Zealand populations were founded by a small number of individuals. The reduction of genetic diversity through bottleneck effects in invasive Vespula wasps is in line with studies on other invasive social wasps (Goodisman et al. 2001; Husseneder et al. 2012; Tsuchida et al. 2014; Arca et al. 2015; Chau et al. 2015; Cheng et al. 2016; Takeuchi et al. 2017).
A similar relation between reduced genetic diversity, particularly allelic richness, and invasion success has been observed in other invasive social insects, such as ants and termites. There are well-known examples of bottleneck effects in populations of the Formosan subterranean termite, Coptotermes formosanus, which is invasive in the United States (Vargo and Husseneder 2009; Husseneder et al. 2012). The Argentine ant, Linepithema humile, successfully invaded parts of Europe, South Africa, the United States, and New Zealand despite displaying low levels of genetic diversity (Tsutsui et al. 2000; Giraud et al. 2002; Suarez et al. 2008; Cheng et al. 2016). Allelic richness in Argentine ant colonies was reduced by 55.5% in Hawaii (Tsutsui and Case 2001) and 40% in New Zealand (Corin et al. 2007).
There is a possible link between low genetic diversity and changes in social phenotypes which leads to invasion success in invasive social hymenopteran (Chapman and Bourke 2001). Some invasive Vespula species develop perennial, polygynous nests in their invaded ranges (Spradbery 1973; Akre and Reed 1981; Plunkett et al. 1989; Donovan et al. 1992; Leathwick and Godfrey 1996; Visscher and Vetter 2003). The mechanisms underlying the occurrence of multi-year Vespula colonies with multiple queens are poorly understood, yet the lack of thermal constraints has been considered to be a major factor (Visscher and Vetter 2003). Additionally, recent studies on Vespula pensylvanica suggest that ancestral weak nestmate discrimination may facilitate the adaptation of polygyny in the invaded range (Loope et al. 2018). Another change in social phenotype that might facilitate the invasion success of Vespula wasps is gene flow induced by queen movement and resulting in an increased exchange of genetic information between meta-populations. Colonies in the introduced ranges have been found to contain more workers that have been produced by multiple, foreign queens (Goodisman et al. 2001; Hanna et al. 2014). In addition, polyandry is likely to have helped Vespula wasp invasion in New Zealand; only a single multi-mated queen might have successfully established a whole new population (Goodisman et al. 2007; Schmid-Hempel et al. 2007; Arca et al. 2015; Dobelmann et al. 2017). Further, V. germanica and V. vulgaris might be successful invaders despite drastic genetic bottlenecks due to their haplodiploid sex determination system, which exposes recessive, deleterious mutations to selection (Schmid-Hempel et al. 2007). Thus, offspring of such purged lines might be able to tolerate high levels of genetic load and inbreeding (Schmid-Hempel et al. 2007; Zayed et al. 2007; Gloag et al. 2017).
While genetic structure of V. germanica and V. vulgaris populations differed between the native and the invaded ranges, genetic differentiation of New Zealand wasps was low. In both species, the genetic variation within New Zealand was not sufficient to distinguish between meta-populations. Low levels of genetic structure found in our study contrast with studies on invasive Vespula wasp populations in Australia (Goodisman et al. 2001) and Hawaii (Chau et al. 2015) but are in line with work on invasive paper wasp populations, Polistes spp., in New Zealand (Tsuchida et al. 2014). The difference between our results and the findings from the Hawaiian Islands and Australia might be due to weaker dispersal barriers among sample sites in New Zealand. Low levels of genetic structure in New Zealand may indicate ongoing gene flow within New Zealand wasps or underline strong founder effects during the introduction of Vespula wasps.
Contrary to other studies on genetic structure in invasive social wasps (Hoffman et al. 2008; Chau et al. 2015), V. germanica showed some genetic structure within its native range, with populations from mainland Europe being genetically different from the United Kingdom wasps. However, because we could not secure V. vulgaris specimens from the United Kingdom, we were not able to detect if there is the same differentiation in its native range as observed for V. germanica. Previous studies found that in its native range, V. pensylvanica seem to be panmictic with large populations and unrestricted gene flow (Chau et al. 2015), in agreement with our estimates of effective population size for V. vulgaris in its native range.
Our study confirms that V. germanica populations in New Zealand originated from a source population in the United Kingdom (England and Scotland) in agreement with mitochondrial DNA data (Brenton-Rule et al. 2018). The South African V. germanica introduction, however, seems to have its origin somewhere in mainland Europe; suggesting an introduction that was independent from the New Zealand invasion and not introduced into South Africa from the New Zealand stock. Further research with increased samples from both, South Africa and Northern European countries is necessary to determine the country of origin of the South African introduction.
A study using mitochondrial DNA data showed that the V. vulgaris populations found in New Zealand likely originated in England and Ireland (Lester et al. 2014). We could not secure samples from these countries for our current study. We can confirm that neither Belgium nor Germany are the source populations. However, one sample collected in a suburb of Wellington seems to have originated from either Belgium or Germany, suggesting an independent invasion event in the Wellington region, likely through maritime traffic as the Wellington harbour is a busy transport hub.
We found a reduction of effective population sizes (Ne) for both, V. germanica and V. vulgaris, in their invaded ranges in agreement with allelic richness and other population differentiation estimates.
This is the first study to compare both Vespula wasp species from their native and introduced ranges as well as six offshore islands on the east coast of New Zealand. The population structure analyses did not identify these islands as discrete from mainland New Zealand wasp populations therefore we have little evidence for considering them as genetically isolated from the mainland. Further research to understand the colonisation patterns of Vespula wasps into offshore islands and to gain insights into the effect of geographic factors on dispersal patterns of invasive wasps would be beneficial. It is a key question for conservation management whether each island invasion is an independent colonisation process or if invasive species establish in an area by ‘hopping’ from island to island (Parkes et al. 2017; Russell et al. 2017). For example, a recent point of discussion for conservation management in New Zealand is if establishing marine reserves around islands could prevent introductions or reintroductions of invasive species into unique island ecosystems (Department of Conservation and Ministry of the Environment 2000; Secretariat of the Convention on Biological Diversity 2004; Edgar et al. 2017; Sala and Giakoumi 2018).
Studies on the population genetic patterns of invasive populations are key for developing a sound understanding of the evolutionary mechanisms underpinning invasion success and the development of control strategies including novel techniques such as gene editing. Overall, insights into the invasion of V. germanica and V. vulgaris across New Zealand’s mainland and offshore islands may provide essential knowledge on invasion processes and help manage invasions by social wasps.
References
Akre RD, Reed HC (1981) Population cycles of yellowjackets (Hymenoptera: Vespinae) in the Pacific Northwest. Environ Entomol 10(3):267–274
Allendorf FW, Lundquist LL (2003) Introduction: population biology, evolution, and control of invasive species. Conserv Biol 17:24–30
Arca M, Capdevielle-Dulac C, Villemant C et al (2012) Development of microsatellite markers for the yellow-legged Asian hornet, Vespa velutina, a major threat for European bees. Conserv Genet Resour 4:283. https://doi.org/10.1007/s12686-011-9525-1
Arca M, Mougel F, Guillemaud T, Dupas S, Rome Q, Perrard A, Muller F et al (2015) Reconstructing the invasion and the demographic history of the yellow-legged hornet, Vespa velutina, in Europe. Biol Invasions 17:2357–2371. https://doi.org/10.1007/s10530-015-0880-9
Beggs JR, Rees JS (1999) Restructuring of Lepidoptera communities by introduced Vespula wasps in a New Zealand beech forest. Oecologia 119:565–571
Beggs JR, Toft RJ, Malham JP et al (1998) The difficulty of reducing introduced wasp (Vespula vulgaris) populations for conservation gains. N Z J Ecol 22:55–63
Beggs JR, Brockerhoff EG, Corley JC, Kenis M, Masciocchi M, Muller F, Rome Q, Villemant C (2011) Ecological effects and management of invasive alien Vespidae. Biocontrol 56:505. https://doi.org/10.1007/s10526-011-9389-z
Belkhir K, Borsa P, Goudet J, Chikhi L, Bonhomme F (1998) GENETIX, Logiciel sous Windows pour la Génétique des Populations. Laboratoire Génome et Populations. CNRS UPR 9060. Université de Montpellier II, Montpellier
Beye M, Hasselmann M, Fondrk MK, Page RE, Omholt SW (2003) The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 114:419–429. https://doi.org/10.1016/S0092-8674(03)00606-8
Brenton-Rule EC, Dobelmann J, Baty JW, Brown RL, Dvorak L et al (2018) The origins of global invasions of the German wasp (Vespula germanica) and its infection with four honey bee viruses. Biol Invasions 20:3445–3460
Burne AR, Ritchie PA, Gruber M, Lester PJ (2017) A genetic bottleneck in populations of a New Zealand endemic ant associated with density of an invasive predatory wasp. Insectes Soc 64:65–74
Carlsson J (2008) Effects of microsatellite null allele on assignment testing. J Hered 99:616–623
Chapman RE, Bourke AF (2001) The influence of sociality on the conservation biology of social insects. Ecol Lett 4:650–662
Charlesworth B (2009) Effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195
Chau LM, Hanna C, Jenkins LT, Kutner RE, Burns EA, Kremen C, Goodisman MAD (2015) Population genetic structure of the predatory, social wasp Vespula pensylvanica in its native and invasive range. Ecol Evol 5:5573–5587. https://doi.org/10.1002/ece3.1757
Cheng D, Liang G, Xu Y (2016) Meta-analysis reveals asymmetric reduction in the genetic diversity of introduced populations of exotic insects. Biol Invasions 18:1163–1175. https://doi.org/10.1007/s10530-016-1058-9
Clapperton BK, Moller H, Sandlant GR (1989) Distribution of social wasps (Hymenoptera: Vespidae) in New Zealand in 1987. N Z J Zool 16:315–323. https://doi.org/10.1080/03014223.1989.10422896
Clavero M, García-Berthou E (2005) Invasive species are a leading cause of animal extinctions. Trends Ecol Evol 20:110
Corin S, Abbott K, Ritchie P, Lester PJ (2007) Large scale unicoloniality: the population and colony structure of the invasive Argentine ant (Linepithema humile) in New Zealand. Insect Soc 54:275–282. https://doi.org/10.1007/s00040-007-0942-9
Daly D, Archer ME, Watts PC et al (2002) Polymorphic microsatellite loci for eusocial wasps (Hymenoptera: Vespidae). Mol Ecol Notes 2:273–275
Davis LM, Glenn TC, Elsey RM, Dessauer HC, Sawyer RH (2001) Multiple paternity and mating patterns in the American alligator, Alligator mississippiensis. Mol Ecol 10:1011–1024. https://doi.org/10.1046/j.1365-294X.2001.01241.x
Department of Conservation and Ministry of the Environment (2000) The New Zealand biodiversity strategy 2000–2020. Department of Conservation and Ministry of the Environment. ISBN: O-478-21919-9
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Dobelmann J, Loope KJ, Wilson-Rankin E, Quinn O, Baty JW, Gruber MAM, Lester PJ (2017) Fitness in invasive social wasps: the role of variation in viral load, immune response and paternity in predicting nest size and reproductive output. Oikos 126:1208–1218. https://doi.org/10.1111/oik.04117
Donovan BJ (1984) Occurrence of the common wasp, Vespula vulgaris (L.) (Hymenoptera: Vespidae) in New Zealand. N Z J Zool 11:417–427. https://doi.org/10.1080/03014223.1984.10428256
Donovan BJ, Howie A, Schroeder NC, Wallace AR, Read P (1992) Comparative characteristics of nests of Vespula germanica (F.) and Vespula vulgaris (L.) (Hymenoptera: Vespinae) from Christchurch City, New Zealand. N Z J Zool 19:61–71
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Edgar GJ, Stuart-Smith RD, Thomson RJ, Freeman DJ (2017) Consistent multi-level trophic effects of marine reserve protection across northern New Zealand. PLoS ONE 12:e0177216. https://doi.org/10.1371/journal.pone.0177216
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin Suite Ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Foster KR, Ratnieks FLW, Gyllenstrand N, Thorén PA (2001) Colony kin structure and male production in Dolichovespula wasps. Mol Ecol 10:1003–1010
Gardner-Gee R, Beggs JR (2013) Invasive wasps, not birds, dominate in a temperate honeydew system. Austral Ecol 38:346–354
Giraud T, Pedersen JS, Keller L (2002) Evolution of supercolonies: the Argentine ants of southern Europe. Proc Natl Acad Sci USA 99:6075–6079. https://doi.org/10.1073/pnas.092694199
Gloag R, Ding G, Christie JR, Buchmann G, Beekman M, Oldroyd BP (2017) An invasive social insect overcomes genetic load at the sex locus. Nat Ecol Evol 1:0011
Golani D, Azzurro E, Corsini-Foka M, Falautano M, Andaloro F, Bernardi G (2007) Genetic bottlenecks and successful biological invasions: the case of a recent Lessepsian migrant. Biol Lett 3:541–545. https://doi.org/10.1098/rsbl.2007.0308
Goodisman MA, Matthews RW, Crozier RH (2001) Hierarchical genetic structure of the introduced wasp Vespula germanica in Australia. Mol Ecol 10:1423–1432
Goodisman M, Sankovich K, Kovacs J (2007) Genetic and morphological variation over space and time in the invasive fire ant Solenopsis invicta. Biol Invasions 9:571–584. https://doi.org/10.1007/s10530-006-9059-8
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-Statistics. J Hered 86:485–486
Hanna C, Cook E, Thompson A, Dare L, Palaski A, Foote D, Goodisman M (2014) Colony social structure in native and invasive populations of the social wasp Vespula pensylvanica. Biol Invasions 16:283–294. https://doi.org/10.1007/s10530-013-0517-9
Hasegawa E, Takahashi J (2002) Microsatellite loci for genetic research in the hornet Vespa mandarinia and related species. Mol Ecol Notes 2:306–308
Hoffman EA, Kovacs JL, Goodisman MAD (2008) Genetic structure and breeding system in a social wasp and its social parasite. BMC Evol Biol 8:239. https://doi.org/10.1186/1471-2148-8-239
Holm S (1979) A simple sequential rejective method procedure. Scand J Stat 6:65–70
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Hulme PE (2009) Trade, transport and trouble: managing invasive species pathways in an era of globalization. J Appl Ecol 46:10–18. https://doi.org/10.1111/j.1365-2664.2008.01600.x
Husseneder C, Simms D, Delatte J, Wang C, Grace J, Vargo E (2012) Genetic diversity and colony breeding structure in native and introduced ranges of the Formosan subterranean termite, Coptotermes formosanus. Biol Invasions 14:419–437. https://doi.org/10.1007/s10530-011-0087-7
Johnson RN, Starks PT (2004) A surprising level of genetic diversity in an invasive wasp: Polistes dominulus in the northeastern United States. Ann Entomol Soc Am 97:732–737
Karl SA (2008) The effect of multiple paternity on the genetically effective size of a population. Mol Ecol 17:3973–3977. https://doi.org/10.1111/j.1365-294X.2008.03902.x
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kenis M, Auger-Rozenberg M, Roques A, Timms L, Péré C, Cock M, Settele J, Augustin S, Lopez-Vaamonde C (2009) Ecological effects of invasive alien insects. Biol Invasions 11:21–45. https://doi.org/10.1007/s10530-008-9318-y
Kolbe JJ, Glor RE, Schettino LR, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177
Laugier G, Le Moguédec G, Su W, Tayeh A, Soldati L, Serrate B, Estoup A, Facon B (2016) Reduced population size can induce quick evolution of inbreeding depression in the invasive ladybird Harmonia axyridis. Biol Invasions 18:2871–2881. https://doi.org/10.1007/s10530-016-1179-1
Leathwick DM, Godfrey PL (1996) Overwintering colonies of the common wasp (Vespula vulgaris) in Palmerston North, New Zealand. N Z J Zool 23:355–358
Lester PJ, Beggs JR (2019) Invasion success and management strategies for social Vespula wasps. Annu Rev Entomol 64:51–71. https://doi.org/10.1146/annurev-ento-011118-111812
Lester PJ, Beggs JR, Brown RL, Edwards ED, Groenteman R, Toft RJ, Twidle AM, Ward DF (2013) The outlook for control of New Zealand’s most abundant, widespread and damaging invertebrate pests: social wasps. N Z Sci Rev 70:56–62
Lester PJ, Gruber MAM, Brenton-Rule E, Archer M, Corley JC, Dvořák L, Masciocchi M, Oystaeyen A (2014) Determining the origin of invasions and demonstrating a lack of enemy release from microsporidian pathogens in the common wasps (Vespula vulgaris). Divers Distrib 20:964–974
Loope KJ, Millar JG, Wilson Rankin EE (2018) Weak nestmate discrimination behavior in native and invasive populations of a yellowjacket wasp (Vespula pensylvanica). Biol Invasions 20:3431–3444
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW (2010) Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11:355–373. https://doi.org/10.1007/s10592-010-0050-7
Moller H (1996) Lessons for invasion theory from social insects. Biol Conserv 78:125–142
Moller H, Clapperton BK, Alspach PA, Tilley J (1991) Comparative seasonality of Vespula germanica (F.) and Vespula vulgaris (L.) colonies (Hymenoptera: Vespidae) in urban Nelson, New Zealand. N Z J Zool 18:111–120
Moore J, Nelson N, Keall S, Daugherty C (2008) Implications of social dominance and multiple paternity for the genetic diversity of a captive-bred reptile population (tuatara). Conserv Genet 9:1243–1251. https://doi.org/10.1007/s10592-007-9452-6
Murray J (1964) Multiple mating and effective population size in Cepaea nemoralis. Evolution 18:283. https://doi.org/10.2307/2406402
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Parkes J, Byrom A, Edge K (2017) Eradicating mammals on New Zealand island reserves: what is left to do? N Z J Ecol 41:263–270. https://doi.org/10.20417/nzjecol.41.25
Parmesan C (2006) Ecological and evolutionary responses to recent climate change. Annu Rev Ecol Evol Syst 37:637–669. https://doi.org/10.1146/annurev.ecolsys.37.091305.110100
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pearse DE, Avise JC (2001) Turtle mating systems: behavior, sperm storage, and genetic paternity. J Hered 92:206
Pimentel D, Zuniga R, Morrison D (2005) Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol Econ 52:273–288
Plunkett GM, Moller H, Hamilton C, Clapperton BK, Thomas CD (1989) Overwintering colonies of German (Vespula germanica) and common wasps (Vespula vulgaris) (Hymenoptera: Vespidae) in New Zealand. N Z J Zool 16:345–353
Pritchard JC, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puillandre N, Dupas S, Dangles O, Zeddam J, Capdevielle-Dulac C, Barbin K, Torres-Leguizamon M, Silvain J (2008) Genetic bottleneck in invasive species: the potato tuber moth adds to the list. Biol Invasions 10:319–333
Purcell KM, Ling N, Stockwell CA (2012) Evaluation of the introduction history and genetic diversity of a serially introduced fish population in New Zealand. Biol Invasions 14:2057–2065
Pyšek P, Richardson DM (2010) Invasive species, environmental change and management, and health. Annu Rev Environ Resour 35:25–55
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Rico C, Cuesta JA, Drake P, Macpherson E, Bernatchez L, Marie AD (2017) Null alleles are ubiquitous at microsatellite loci in the Wedge Clam (Donax trunculus). PeerJ 5:e3188. https://doi.org/10.7717/peerj.3188
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Russell JC, Meyer J, Holmese ND, Pagad S (2017) Invasive alien species on islands: impacts, distribution, interactions and management. Environ Conserv 44:359–370. https://doi.org/10.1017/S0376892917000297
Sala E, Giakoumi S (2018) No-take marine reserves are the most effective protected areas in the ocean. ICES J Mar Sci 75:1166–1168. https://doi.org/10.1093/icesjms/fsx059
Schmid-Hempel P, Schmid-Hempel R, Brunner PC, Seeman OD, Allen GR (2007) Invasion success of the bumblebee, Bombus terrestris, despite a drastic genetic bottleneck. Heredity 99:414–422. https://doi.org/10.1038/sj.hdy.6801017
Secretariat of the Convention on Biological Diversity (2004) The ecosystem approach. Secretariat of the Convention on Biological Diversity. ISBN: 92-9225-023-x
Sherpa S, Rioux D, Pougnet-Lagarde C, Després L (2018) Genetic diversity and distribution differ between long-established and recently introduced populations in the invasive mosquito Aedes albopictus. Infect Genet Evol 58:145–156
Shurtliff QR, Pearse DE, Rogers DS (2005) Parentage analysis of the canyon mouse (Peromyscus crinitus): evidence for multiple paternity. J Mammal 86:531–540. https://doi.org/10.1644/1545-1542(2005)86[531:PAOTCM]2.0.CO;2
Sonsthagen SA, Wilson RE, Underwood JG (2017) Genetic implications of bottleneck effects of differing severities on genetic diversity in naturally recovering populations: an example from Hawaiian coot and Hawaiian gallinule. Ecol Evol 7:9925–9934. https://doi.org/10.1002/ece3.3530
Spradbery JP (1973) Wasps: an account of the biology and natural history of solitary and social wasps. University of Washington Press, Seattle
Suarez AV, Holway DA, Liang D, Tsutsui ND, Case TJ (2002) Spatiotemporal patterns of intraspecific aggression in the invasive Argentine ant. Anim Behav 64:697–708
Suarez AV, Robinson GE, Toth AL, Smith CR (2008) Genetic and genomic analyses of the division of labour in insect societies. Nat Rev Genet 9:735–748. https://doi.org/10.1038/nrg2429
Sugg DW, Chesser RK (1994) Effective population sizes with multiple paternity. Genetics 137:1147–1155
Sutton JT, Robertson BC, Jamieson IG (2011) Dye shift: a neglected source of genotyping error in molecular ecology. Mol Ecol Resour 11:514–520. https://doi.org/10.1111/j.1755-0998.2011.02981.x
Takeuchi T, Takahashi R, Kiyoshi T, Nakamura M, Minoshima Y, Takahashi J (2017) The origin and genetic diversity of the yellow-legged hornet, Vespa velutina introduced in Japan. Insect Soc 64:313–320. https://doi.org/10.1007/s00040-017-0545-z
Thorén PA, Paxton RJ, Estoup A (1995) Unusually high frequency of (CT)n and (GT)n microsatellite loci in a yellowjacket wasp, Vespula rufa (L.) (Hymenoptera: Vespidae). Insect Mol Biol 4:141–148
Toft RJ, Rees JS (1998) Reducing predation of orb-web spiders by controlling common wasps (Vespula vulgaris) in a New Zealand beech forest. Ecol Entomol 23:90–95. https://doi.org/10.1046/j.1365-2311.1998.00100.x
Tsuchida K, Kudô K, Ishiguro N (2014) Genetic structure of an introduced paper wasp, Polistes chinensis antennalis (Hymenoptera, Vespidae) in New Zealand. Mol Ecol 23:4018–4034. https://doi.org/10.1111/mec.12852
Tsutsui ND, Case TJ (2001) Population genetics and colony structure of the Argentine ant (Linepithema humile) in its native and introduced ranges. Evolution 55:976–985. https://doi.org/10.1111/j.0014-3820.2001.tb00614.x
Tsutsui ND, Suarez AV, Holway DA, Case TJ (2000) Reduced genetic variation and the success of an invasive species. Proc Natl Acad Sci USA 97:5948–5953
van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vargo EL, Husseneder C (2009) Biology of subterranean termites: insights from molecular studies of Reticulitermes and Coptotermes. Annu Rev Entomol 54:379–403. https://doi.org/10.1146/annurev.ento.54.110807.090443
Visscher PK, Vetter RS (2003) Annual and multi-year nests of the western yellowjacket, Vespula pensylvanica, in California. Insect Soc 50:160–166. https://doi.org/10.1007/s00040-003-0636-x
Wang J, Santiago E, Caballero A (2016) Prediction and estimation of effective population size. Heredity 117:193
Waples RS, England PR (2011) Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics 189:633–644
Whiting PW (1933) Selective fertilization and sex-determination in Hymenoptera. Science 78:537–538
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97
Zayed A (2004) Effective population size in Hymenoptera with complementary sex determination. Heredity 93:627
Zayed A, Packer L (2007) The population genetics of a solitary oligolectic sweat bee, Lasioglossum (Sphecodogastra) oenotherae (Hymenoptera: Halictidae). Heredity 99:397
Zayed A, Constantin ŞA, Packer L (2007) Successful biological invasion despite a severe genetic load. PLoS ONE 2:e868
Zhang M, Evans TA (2017) Determining urban exploiter status of a termite using genetic analysis. Urban Ecosyst 20:535–545
Acknowledgements
Thanks to the landowners and managers for access to the various sites for collecting wasps. We thank Jana Dobelmann who shared V. vulgaris DNA extractions, Carolien van Zyl for sharing South African V. germanica samples from her PhD project and Dr Lara Shepherd who helped with analyses. We also thank two anonymous referees and the Handling Editor for their useful comments that improved our manuscript.
Funding
This work was partially funded by the Ministry of Business, Innovation and Employment (New Zealand’s Biological Heritage NSC, C09X1501) and by Victoria University of Wellington.
Author information
Authors and Affiliations
Contributions
JMS collected the New Zealand samples and together with JPL and JRB conceived the project. ECBR, RV and TW collected or provided samples. MB and JMS led the genetic work, analysed and interpreted the data. JMS and MB led the writing with input from all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Schmack, J.M., Brenton-Rule, E.C., Veldtman, R. et al. Lack of genetic structuring, low effective population sizes and major bottlenecks characterise common and German wasps in New Zealand. Biol Invasions 21, 3185–3201 (2019). https://doi.org/10.1007/s10530-019-02039-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-019-02039-0