
Abstract
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells (IMCs) with immunosuppressive functions, whereas IMCs originally differentiate into granulocytes, macrophages, and dendritic cells (DCs) to participate in innate immunity under steady-state conditions. At present, difficulties remain in identifying MDSCs due to lacking of specific biomarkers. To make identification of MDSCs accurately, it also needs to be determined whether having immunosuppressive functions. MDSCs play crucial roles in anti-tumor, angiogenesis, and metastasis. Meanwhile, MDSCs could make close interaction with osteoclasts, osteoblasts, chondrocytes, and other stromal cells within microenvironment of bone and joint, and thereby contributing to poor prognosis of bone-related diseases such as cancer-related bone metastasis, osteosarcoma (OS), rheumatoid arthritis (RA), osteoarthritis (OA), and orthopedic trauma. In addition, MDSCs have been shown to participate in the procedure of bone repair. In this review, we have summarized the function of MDSCs in cancer-related bone metastasis, the interaction with stromal cells within the bone microenvironment as well as joint microenvironment, and the critical role of MDSCs in bone repair. Besides, the promising value of MDSCs in the treatment for bone-related diseases is also well discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer-related bone metastasis, rheumatoid arthritis (RA), osteoarthritis (OA) and other bone-related diseases have threatened people's health seriously and bring economic burden to the society. Bone-related diseases, although less incidence than cardiovascular diseases as well as cancer, are generally difficult to cure and have poor prognosis. To develop therapeutic efficacy, several studies are dedicated towards finding effective and novel treatment strategies such as gene therapy and osteoimmunological treatment. The latter is based on the osteoimmune system created by the close interaction between skeletal system and immune system, and osteoimmune system contains all cells in the bone marrow; the novel field of osteoimmunology is developed to investigate the effect of the cross-talk among bone cells and immune cells on inflammation-related bone erosion, hematopoietic stem cell regulation, and tumor progression [1]. As bone marrow-derived cells, MDSCs are a heterogeneous population of immature myeloid cells (IMCs) [2]. Recent studies have focused on MDSCs as an important component of osteoimmunology and make the further investigation about the critical role of MDSCs in bone-related diseases [3].
MDSCs are bone marrow-derived suppressor cells that were first identified and noted in a lung cancer model in 1987 [4]. Previous studies have shown the essential role of MDSCs in malignant tumors. Meanwhile, several studies have focused on the precise mechanisms underlying the development, accumulation, and function of MDSCs. Accumulating evidence has demonstrated that MDSCs also play a significant role in autoimmune diseases, trauma, infectious diseases, etc. [5].
All in all, based on previous studies on the roles and functions of MDSCs in cancer, autoimmune diseases, and trauma, we make the hypothesis that the immunosuppressive function of MDSCs may be used as therapeutic target for bone-related diseases. In addition, MDSCs have other functions besides immunosuppression such as angiogenesis and transplantation immune tolerance induction, which will greatly expand the application prospect of MDSCs [6, 7]. We will summarize the functional role of MDSCs in bone-related diseases and develop novel therapeutic strategies in clinical treatment.
Main phenotypic and functional characteristics of MDSCs
MDSCs are a group of immature immune cells, derived from myeloid lineage, which are heterogeneous in morphology, phenotype, and function, and characterized by suppression of immune response. MDSCs are subdivided into two main groups of granulocytic/polymorphonuclear (G-MDSCs/PMN-MDSCs) and monocytic (M-MDSCs). PMN-MDSCs are alike neutrophils in morphology and phenotype, while M-MDSCs are similar to monocytes in morphology and phenotype. Previous studies have shown that MDSCs are found in both mice and humans, but with different phenotypic markers. CD11b+Gr1+ is the phenotypic marker of MDSCs in mice. According to the different expression of Ly6G and Ly6C on their surface, the phenotype of M-MDSCs in mice is CD11b+Ly6G−Ly6Chigh, while that of G-MDSCs is CD11b+Ly6G+Ly6Clow [8,9,10]. However, the phenotype of human MDSCs is more complex and diverse. MDSCs express CD33 and CD11b but not maturation markers such as HLA-DR in human [11]. M-MDSCs have the phenotype HLA-DR–/lowCD11b+CD33+CD14+CD15–, whereas PMN-MDSCs express HLA-DR–CD11b+CD33midCD14–CD15+ [12]. Interestingly, recent studies identified a new population of MDSCs, early-MDSCs (e-MDSCs), playing the role of precursors for both M-MDSCs and PMN- MDSCs, which are phenotypically HLA-DR−CD33+Lin−(CD3−CD14−CD15−CD19−CD56−) [13,14,15]. Obviously, the focus of current work is to find highly specific markers to accurately identify MDSCs. Fortunately, recent studies have shown that lectin-type oxidized LDL receptor 1 (LOX-1) can be used as a specific marker to accurately identify PMN-MDSCs [16]. However, specific markers to accurately identify M-MDSCs are still insufficient. In summary, accurate identification of MDSCs by cellular phenotype alone is insufficient and still requires the judgment of whether acting as immunosuppressive cells.
It is evident from the name itself that MDSCs are characterized by immunosuppressive function. In the complex immune network, MDSCs exert immunosuppressive functions through multiple pathways involving both soluble mediators and the interaction among different cells. Notwithstanding MDSCs interact with multiple immune cells, the most important target cells are T cells. MDSCs can competitively consume cysteine in the microenvironment, upregulate the activity of inducible nitric oxide synthase (iNOS), arginase 1 (Arg-1) and thus consume l-arginine to inhibit T cells generation. MDSCs can also inhibit T cells immune response by producing reactive oxygen species (ROS) [17]. By note, the mechanisms by which M-MDSCs and PMN-MDSCs exert immunosuppressive function are different. M-MDSCs significantly upregulate iNOS expression but not ROS mainly by activating STAT1, and iNOS generates large amounts of nitric oxide (NO). Many studies have demonstrated NO not only suppresses T cell proliferation, but also induces apoptosis of T cells [18]. Moreover, ROS likewise reacts with NO, leading to peroxynitrite production, and the latter nitrify TCR to induce T cells apoptosis [19]. However, PMN-MDSCs produce high levels of ROS and less NO by activating STAT3 as well as upregulating NADPH oxidase (NOX2) [20, 21]. Both types of MDSCs can upregulate the activity of Arg-1 [22]. In a word, M-MDSCs and PMN-MDSCs both have immunosuppressive function, but the mechanisms are not identical. Besides, MDSCs can also exert immunosuppression through multiple substances, including TGFβ, IL-10, COX2, indoleamine 2,3-dioxygenase (IDO), etc. [5]. And MDSCs highly express osteopontin (OPN) to inhibit T cells producing interferon-γ (IFN-γ) in the tumor microenvironment, thereby attenuating the cytotoxicity of CD8+T cells [23]. Respectively, OPN promotes the recruitment and immunosuppressive activity of MDSCs, and participates in proliferation, invasion, angiogenesis, and metastasis of tumor cells [24]. MDSCs not only inhibit acquired anti-tumor immunity, but also innate anti-tumor immunity. As the important effector of innate immunity, it is not surprising that natural killer (NK) cells are suppressed by MDSCs. Li et al. have found that MDSCs, through membrane-bound TGF-β1, not only inhibit NK cells cytotoxicity, but also suppress the expression of NKG2D and IFN-β in NK cells [25]. Moreover, the production of IL-10 by MDSCs also affects the function of NK cells [26]. DCs are another major target of MDSCs, which play an important role in the immune defense due to their superior antigen presentation and T-cell activation properties. Substantial results showed that the increase of MDSCs may be related to the inhibition of DCs differentiation, function, and accumulation [26, 27]. Unfortunately, it is still fully unclear the mechanism underlying the direct impact of MDSCs on DCs. Hu et al. found that MDSCs inhibited TLR-ligand-induced IL-12 production of DCs by IL-10 production and suppressed T cells stimulatory activity of DCs in a murine model of hepatocellular carcinoma [26, 28]. Furthermore, MDSCs express vascular endothelial growth factor (VEGF) under the stimulation of activated hypoxia-inducible factor 1α (HIF-1α) in tumor microenvironment, which inhibits the differentiation of DCs [29]. Besides, Wang et al. found Notch and STAT3 signaling were required for MDSCs exerting the function of suppressing the differentiation, maturation, and antigen presentation ability of DCs in vitro and in vivo, while STAT3 signaling promoted the expression of NOX2 and then produced a large amount of ROS to achieve the function of MDSCs [14, 30]. Beyond that, MDSCs can differentiate into tumor-associated macrophages (TAMs), tumor-associated dendritic cells (TADCs), and tumor-associated neutrophils (TANs), and these cells directly suppress NK cells and CD8+T cells through the expression of signal mediators such as Arg-1, iNOS, TGF-β, IL-10, and cysteine [31].
In addition to inhibiting anti-tumor immune responses, MDSCs also activate tumor growth by promoting angiogenesis and metastasis. There are several modulators such as VEGF, basic fibroblast growth factor (bFGF), VEGF analogue Bv8, and matrix metalloprotease 9 (MMP-9) produced by MDSCs that can induce angiogenesis and invasion at the tumor site [32]. Besides, MDSCS not only induce angiogenesis and protect tumor cells from immune detection, but also establish a pre-metastatic niche (PMN) to promote tumor cells metastasis [33]. Therefore, substantial reports have shown that MDSCs an play important role in inhibiting anti-tumor immunity as well as promoting angiogenesis and metastasis in the tumor microenvironment (Table 1).
Besides focusing on the function of MDSCs in cancer, on no account should we ignore the role of MDSCs in other non-cancer inflammatory conditions. The latter is currently mainly widely involved in sepsis, trauma, pregnancy, infectious diseases, autoimmune diseases, etc. [5]. Autoimmune diseases result primarily from the accumulation of immune cells and immunoglobulins generated in response to excessive inflammation. The excessive inflammatory response damages normal tissues and impairing functions, and when it occurs in joint tissues manifests as arthritis. Therefore, downregulating excessive inflammatory responses to return to a normal state is an effective therapeutic strategy against autoimmune diseases. This suggests that MDSCs act as a "nice man" in these diseases, since they are immunosuppressive cells. However, many studies have also shown that MDSCs could perform pro-inflammatory immune responses in autoimmune disorders [34]. In summary, MDSCs play double-edged sword role in autoimmune diseases, such as inflammatory arthritis.
Based on the origin of MDSCs, it is natural to speculate that MDSCs are abundant in bone marrow. Indeed, MDSCs are found at very high levels in bone marrow under pathological conditions according to several experimental results [12]. Therefore, MDSCs and bone cells share a common microenvironment. In addition, MDSCs are also found to exhibit high levels at inflammatory reaction sites, such as synovial fluid, which means it is reasonable to infer that MDSCs are widely present in the joint microenvironment under the pathological conditions of joint inflammation. In conclusion, we speculate that MDSCs may have potential interactions with osteoblasts, osteoclasts, chondrocytes, and other stromal cells in the bone microenvironment and joint microenvironment, indicating the crucial role of MDSCs in bone-related diseases.
The function of MDSCs in bone metastasis, bone microenvironment, joint microenvironment, and bone repair
MDSCs not only promote the growth of tumor cells, but also participate in tumor metastasis. Because the bone microenvironment provides suitable survival conditions for metastatic tumor cells, bone often serves as a preferred reservoir for tumor cells, facilitating further metastatic dissemination [35]. According to meaningful studies, bone metastasis in breast and prostate cancer may accounts for more than 80% of cases of metastatic bone disease [36]. Moreover, MDSCs are also involved in bone destruction, including osteolytic and osteoblastic lesions [37]. In addition, MDSCs show high levels in bone marrow, peripheral blood, and lesion location under pathological conditions, the latter including tumor sites and joint inflammation sites [12]. In this section, we will make the detailed illustration about the critical role of MDSCs in the bone microenvironment and joint microenvironment so as to perform the involvement of MDSCs in the progression of bone-related diseases. Besides, we will also summarize the multifaceted role of MDSCs in bone repair.
Regulatory role of MDSCs in bone metastasis
Distant metastasis is the major cause of failure in clinical treatment and mortality in cancer patients. Bone is the third most common site of metastasis, and bone metastasis is a multi-step process [38]. Initially, after epithelial-to-mesenchymal transition (EMT) as well as local invasion, few disseminated tumor cells are released and sowed into the PMN through peripheral blood circulation, laying basis for tumor cells colonization [39,40,41]. MDSCs regulate the PMN formation and evolution before the appearance of colonization [41]. Then the surviving tumor cells, adapting to the niches, grow immediately or enter a dormancy state upon interaction with the local environment, which may last several years [42]. Finally, when inhibited to a certain extent, dormant tumor cells can reactivate and form micro-metastasis, ultimately entering a growth phase [43]. And then the tumor cells gradually become independent of the microenvironment and modify bone with the development of metastasis [44].
The occurrence of such complex distant metastasis is inseparable from the contribution of MDSCs, which play pleiotropic roles throughout the process. MDSCs have been shown to secrete cytokines and chemokines that promote EMT [45]. The term "EMT" is used to describe malignant tumor cells change their phenotype from epithelial cells with polarity into mesenchymal cells with migratory ability [46]. The process promote tumor cells infiltration and metastasis, and may also allow tumor cells to escape apoptosis induced by certain factors [47]. Zhu et al. found that Ly6GmiLy6CloCD11b+CXCR2+ subpopulation (named CXCR2+ MDSCs) were predominately expanded and recruited in tumor microenvironment during breast cancer progression and metastasis, inducing EMT of breast cancer via IL-6 [48]. IL-6 has also been shown to initiate EMT via STAT3 signaling, which directly leads to the inhibition of E-cadherin expression and loss of cell–cell adhesion [49]. MDSCs can also secrete TGF-β1, another important inflammatory cytokine, to induce EMT, invasion, and metastasis via TGF-β1/Smad as well as nuclear factor kappa B (NF-κB) signaling [50]. Moreover, MDSCs trigger the expression of miRNA101 in tumor cells, which subsequently inhibits the co-repressor gene C-terminal binding protein-2 (CtBP2), and CtBP2 directly targets the stem cell core gene, resulting in increased proliferation of cancer stem cells as well as increasing metastatic and tumorigenic potential [51]. As mentioned above, MDSCs are the key criminals of tumor PMN formation. Liu et al. defined the PMN by six characteristics, including immunosuppression, inflammation, angiogenesis/vascular permeability, lymphangiogenesis, organotropism, and reprogramming [52]. The results of studies on various cancers have shown that chemokines, cytokines, and growth factors from MDSC, including TGF-β, Arg-1, ROS, COX-2, Bv8, MMP-9, S100A8, and S100A9, are involved in the formation and evolution of multiple features of PMN [53]. Unfortunately, the specific mechanism of MDSCs in PMN formation during bone metastasis remains largely unknown. In addition, MDSCs can also make effects on the growth phase of tumors via the release of VEGF, bFGF, VEGF analogues Bv8, and MMP-9, which regulates the tumor microenvironment at the metastatic site and thereby causing tumor growth as well as local angiogenesis [32].
With the exception of directly participating in the process of bone metastasis, MDSCs can also indirectly promote bone metastasis by inhibiting anti-tumor immune response. Besides the functions of MDSCs in the anti-tumor immune response described above, MDSCs can also suppress NK cells and CD8+T cells by differentiating into TAMs, TADCs, and TANs. TAMs, TADCs, and TANs achieve anti-tumor immune response by expressing iNOS, arginase, TGF-β, IL-10, and cysteine [31].
In the microenvironment, bone and tumor cells gradually form a vicious cycle, namely, tumor cells modify bone and then promote their own flourish [54]. Bone metastasis can break the balance of dynamic remodeling of bones, resulting in excessive osteolysis and/or osteogenesis, forming osteolytic or osteoblastic lesions [55]. Breast cancer metastasis are predominantly osteolytic, whereas metastasis from prostate cancer is predominantly osteoblastic. Nevertheless, some patients with bone metastasis have mixed osteolytic and osteoblastic lesions [56]. The main mechanisms of osteolytic and osteoblastic lesions are distinct from each other [57]. Essentially, both lesions result from breaking the dynamic cycle of normal bone remodeling by osteolytic and osteoblastic factors [58]. Osteoclasts dominate the formation of osteolytic lesions via the release of several osteoclastogenic factors from tumor cells [59]. Tumor-derived parathyroid hormone-related protein (PTHrP), FGF, platelet-derived growth factor (PDGF), insulin-like growth factor I(IGF-I), bone morphogenetic proteins (BMP), prostaglandin E2 (PGE2), epithelial growth factor (EGF), IL-1, and IL-6 can promote the growth of tumor cells themselves in the form of "autocrine", induce the production and release of receptor activator of nuclear factor kappa B ligand (RANKL) by osteoblasts as well as stromal cells, and decrease osteoprotegerin (OPG) levels [60, 61]. RANK is expressed on the surface of osteoclast precursors as well as osteoclasts, and binds RANKL to promote the fusion, differentiation as well as maturation of osteoclast precursors through transcription factors like mitogen activated protein kinase (MAPK), NF-κB signaling, and activator protein 1 (AP1); the binding of RANK to RANKL also enhances osteoclasts function through c-Src signaling [62]. OPG, as a "bait" receptor of RANKL, binds to RANK, thereby blocking the RANKL signaling. Therefore, upregulated RANKL level and downregulated OPG level break the dynamic balance of bone remodeling in the bone tumor microenvironment. After osteolysis, TGF-β, PDGF, IGF-1, Ca2+, and FGF are released from bone matrix, which can promote tumor cells growth and further osteolysis [63]. In osteoblastic lesions, osteoblasts play a dominant role by interacting with tumor cells. Osteoblasts-derived TGF-β, IGF, and FGF attract tumor cells and stimulate tumor cells proliferation and growth; tumor cells secret TGF-β, BMP, IGF, FGF, Wnts, endothelin-1 (ET-1), and PTHrP, which induce osteoblast osteogenesis and form a vicious cycle [64]. ET-1, as a mitogenic factor of osteoblasts, inhibits the expression of dick-kopf-1 (DKK-1) that is Wnts signaling antagonist, and thus induces osteoblasts differentiation [65]. Lipoprotein-related receptors 5 is the receptor of Wnts and can increase the level of β-catenin protein after binding; β-catenin protein as a transcription factor promotes osteoblast differentiation and synthesis of collagen precursor of bone matrix [66]. Bedsides, ET-1 can also bind to endothelin A receptor (ETAR) of osteoblasts to induce osteoblast proliferation and osteogenesis through β-catenin and MAPK [67]. Additionally, prostate specific antigen (PSA), a tumor marker, plays a momentous role in the balance of dynamic remodeling of bones; PSA inhibits RANKL expression but promotes OPG expression by osteoblasts to inhibit osteolytic activity, presenting osteoblastic activity [68]. PSA also sections PTHrP and inhibits the activity of osteoclasts [69]. On balance, the vicious cycle delineates how tumor cells manipulate osteoblasts as well as osteoclasts to generate required factors for promoting their own growth and establishment. Compelling evidence has emerged in recent years indicating that MDSCs are also a significant part of the cycle. The specific mechanisms how MDSCs contribute to vicious cycle will be described below.
Cross-talk among MDSCs, osteoclasts, and osteoblasts contributes to bone loss
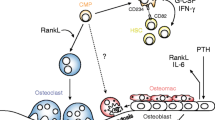
It has been shown that MDSCs are highly concentrated in bone marrow. Meanwhile, it may be performed the promising close relationship among osteoclasts, osteoblasts, and MDSCs in the bone marrow microenvironment [12, 70]. Note that in bone metastasis, the cross-talk among MDSCs, tumor cells, osteoclasts, and osteoblasts "portrays" a more complex vicious cycle. As progenitors of osteoclasts, macrophages are differentiated from MDSCs. Therefore, it is reasonable to speculate MDSCs within the bone microenvironment could also differentiate into osteoclasts, contributing to bone erosion and disease progression. Sawant et al. revealed a NO-dependent mechanism that could drive MDSCs to differentiate into osteoclasts through the HIF-1α signaling in the bone metastasis microenvironment of breast cancer [71]. As described previously, HIF-1α is elevated in MDSCs in the tumor microenvironment, and HIF-1α also induces NO production via iNOS [72]. Studies have shown that MDSCs not only differentiate into osteoclasts directly, but also may interact with osteoclasts through other ways. Osteoclasts that are derived from MDSCs also express MMP-9, which is critical for osteoclasts migrate continuously to enlarge the area of bone loss [73]. In addition, MDSCs may also stimulate endogenous macrophages to differentiate into osteoclasts by secreting NO, IL-1, IL-6, and macrophage colony-stimulating factor (M-CSF) [71, 74]. Noting that only MDSCs isolated from the bone microenvironment with bone metastasis could differentiate into mature and active osteoclasts; MDSCs isolated from a tumor-bearing mouse without bone metastasis are not able to differentiate into osteoclasts [75]. This indicates that bone microenvironment is essential for MDSCs to differentiate into osteoclasts. The tumor cells in breast cancer metastasized to the bone secrete monocyte chemoattractant protein 1 (MCP-1, also known as CCL-2), which can promote NO secretion that is a necessary molecular for inducing MDSCs to differentiate into osteoclasts [76]. In addition, MCP-1 can bind to CCR-2 (CCL-2 receptor) expressed by MDSCs, promoting the differentiation of MDSCs into osteoclasts [77]. Meanwhile, MDSCs release TGF-β to induce tumor cells to secret PTHrP, which stimulates osteoclast-mediated bone destruction [78]. Interestingly, the immunosuppressive function of MDSCs can also have influence on osteoclastogenesis to some extent. MDSCs dampen T cells to secret osteoclastogenesis inhibitors, including IFN-γ, IL-4, and IL-10, thereby indirectly boosting the process of bone resorption; IFN-γ interfere the RANKL-RANK signaling to suppress osteoclastogenesis [79]. In summary, MDSCs differentiate into osteoclasts directly or regulate osteoclastogenesis indirectly in cancer. As another important part of the bone microenvironment, osteoblasts also promote the release and activation of MDSCs by phosphorylating the Src family in MDSCs as well as upregulating VEGF-A and IL-6 to disrupt VCAM-1/integrin β1 axis [80]. Besides, multiple myeloma suppresses runt-related transcription factor 2 (Runx2) expression in osteoblasts, which in turn increase the number of MDSCs in bone marrow [81]. Moreover, in different mouse models of cancer, extra domain A (EDA)-FN produced by osteoblasts upregulates Arg-1 expression in MDSCs, inducing tumor growth [82]. In addition to OPN expressed by MDSCs (mentioned above), osteoblasts and osteoclasts also produce OPN to act on MDSCs as well as tumor cells [83]. In conclusion, osteoblasts promote the generation and activation of MDSCs in the bone microenvironment, and then MDSCs induce osteoclastogenesis through direct differentiation into osteoclasts or other indirect pathways in cancer, ultimately resulting in bone loss (Fig. 1).

The role of MDSCs in bone metastasis. Initially, MDSCs promote EMT of primary tumor cells via IL-6-STAT3, TGF-β1/Smad as well as NF-κB signaling. Besides, MDSCs trigger the expression of miRNA101 in tumor cells, which subsequently inhibits the CtBP2. Then, MDSCs secrete TGF-β, Arg-1, ROS, COX-2, Bv8, MMP-9, S100A8, S100A9, and OPN, which are involved in the formation and evolution of multiple features of PMN, and PMN provide a favorable survival environment for disseminated tumor cells. In addition, MDSCs can also lay an important role in the growth phase of tumors via the release of VEGF, bFGF, VEGF analogues Bv8, MMP-9, and OPN to regulate the tumor microenvironment at the metastatic site. Reciprocally, tumor cells secrete MCP-1, IL-1, IL-6, IL-10, TNF-α, and OPN to promote MDSCs. MDSCs differentiate into osteoclasts through HIF-1α signaling or express MMP-9 to induce osteoclasts migration. In addition, MDSCs may also stimulate endogenous macrophages to differentiate into osteoclasts by secreting NO, IL-1, IL-6, M-CSF. Osteoblasts also promote the release and activation of MDSCs through upregulating VEGF-A, IL-6, and EDA-FN. In addition, osteoblasts and osteoclasts also produce OPN to act on MDSCs as well as tumor cells. Moreover, MDSCs dampen T cells to secret osteoclastogenesis inhibitors, including IFN-γ, IL-4, and IL-10, thereby indirectly boosting the process of bone resorption
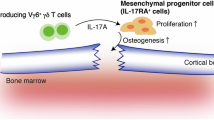
Moreover, recent studies elucidate a novel role of MDSCs as osteoclast progenitors not only in cancer, but also in inflammatory diseases (Fig. 2). Zhang et al. found a significant increase in MDSCs in bone marrow of collagen-induced arthritis (CIA) mice and that the number of circulating MDSCs correlates with disease activity. They also found MDSCs from CIA mice could differentiate into functional osteoclasts when cocultured with M-CSF and RANKL in vitro and in vivo, and IL-1α could promote the differentiation by activating the NF-κB signaling [84]. Additionally, many studies have shown that the proportion of circulating MDSCs positively correlates with the proportion of circulating Th17 cells [34]. As for the mechanisms of the simultaneous expansion, accumulating evidence lends support to the viewpoint that MDSCs secrete IL-1β and TGF-β to mediate CD4+T cell differentiation into Th17 cells, and the latter stimulate bone loss mainly through two ways [85]. On one hand, IL-17 employed by Th17 cells not only directly enhances the expression of RANKL in osteoblasts and fibroblast-like synoviocytes (FLS), but also induces macrophages to produce pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-6 [86]. These cytokines also activate osteoclastogenesis by inducing RANKL expression and inhibit osteoblasts differentiation by inducing DKK-1 expression [87]. On the other hand, Th17 cells are also found in the bone marrow and express high surface levels of RANKL [88]. In summary, MDSCs could upregulate the number and function of Th17 cells, which in turn induces the differentiation and activation of osteoclasts by various mechanisms. However, it is also shown that the proportion of circulating MDSCs are negatively correlated with the proportion of circulating Th17 cells in RA patients [89]. Therefore, whether Th17 cells and MDSCs are partners or rivals remains to be further studied. Moreover, many studies demonstrated that the levels of Treg cells were decreased in the peripheral blood, while the levels of MDSCs were increased [90]. However, the specific mechanism of the phenomenon is still unclear. Treg cells can inhibit bone loss by affecting the proliferation and differentiation of osteoclasts and osteoblasts. The mechanisms of Treg cells repressing osteoclasts mainly include cell contact-dependent mechanisms and inhibitory cytokine-dependent mechanisms [91]. Treg cells express high surface levels of cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), which is a vital molecule to mediate cell contact-dependent inhibition of osteoclasts generation [92]. Besides, many studies have demonstrated that Treg cells secrete inhibitory cytokines including granulocyte–macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-5, and IL-10 to inhibit osteoclasts generation [93]. And IL-10 can downregulate the expression of RANKL and M-CSF but upregulate OPG, which in turn inhibits osteoclasts maturation and differentiation [94]. In terms of acting on osteoblasts, Treg cells express TGF-β and activate intracellular effectors including MAPK and Smad-related proteins to promote the proliferation and differentiation of osteoblasts [95]. Besides, Treg cells could increase the level of Wnt10b, an osteoblastic Wnt ligand, thereby activating Wnt signaling in osteoblasts to induce osteoblasts differentiation [96]. Collectively, Treg cells could inhibit the differentiation of osteoclasts but promote the differentiation of osteoblasts, which suggests MDSCs could act on osteoblasts and osteoclasts indirectly by repressing Treg cells. However, the underlying mechanism of MDSCs suppressing Treg cells requires further investigation.

The role of MDSCs in the bone microenvironment and joint microenvironment when rheumatoid arthritis. In the bone microenvironment, MDSCs, with the help of other immune cells and cytokines, directly or indirectly interact with osteoclasts and osteoblasts to induce bone loss. The immune cells include Th17 cells, Treg cells, macrophages, and the cytokines include pro-osteoclastogenesis factors (TGF-β, IL-1β, M-CSF, RANKL, IL-1α, NF-κB, IL-6, TNF-α) and anti-osteoclastogenesis factors (CTLA-4, GM-CSF, IFN-γ, IL-5, IL-10, TGF-β, MAPK, Smad, Wnt10b). In the joint microenvironment, on one hand, MDSCs play a pro-inflammatory role by promoting Th17 cells. And various pro-inflammatory cytokines are involved, including IL-1β, IL-6, IL-26, IL-17A, IL-17F, IL-22, IL-23, TNF-α, and GM-CSF. On the other hand, MDSCs paly an anti-inflammatory role through the interaction with Th17 cells, Treg cells, macrophages, chondrocytes. Besides, anti-inflammatory factors such as TNF-α, IL-1β, IL-6, and IL-10 are involved in the interaction
Double-edged sword role of MDSCs in joint microenvironment
Arthritis is mainly caused by the infiltration of various immune cells, including T cells, B cells, neutrophils, etc. However, in the joint microenvironment, in addition to the above typical immune cells, compelling clinical and experimental studies have emerged in recent years indicating that MDSCs in synovial fluids, spleen, and peripheral blood from patients with arthritis are increased [34]. There have been many reports showing that MDSCs are involved in the poor progression of several diseases. However, opinions are divided into whether MDSCs play an anti-inflammatory or proinflammatory role in arthritis (Fig. 2).
MDSCs are well known for immunosuppressive function. By the same token, MDSCs inhibit inflammatory response through various mechanisms in arthritis. Jiao et al. found that increased circulating MDSCs were negatively correlated with Th17 cells in RA patients [89]. Furthermore, Zhang et al. found that adoptive transfer of MDSCs reduced the level of TNF-α, IL-6, IL-17, and IL-10 in joint microenvironment, while the number of Th17 cells were decreased [97]. However, the mechanism responsible for this phenomenon is unknown. Th17 cells are widely involved in the processes of arthritis diseases, including RA, psoriatic arthritis, systemic lupus erythematosus, and OA [98, 99]. Th17 cells could secret IL-17A, IL-17F, IL-22, and TNF-α or cell-contact with synovial fibroblasts to activate synovial fibroblasts [100,101,102]. The activated synovial fibroblasts produce MMPs, which are involved in cartilage destruction and matrix turnover [103]. Besides, chondrocytes are impaired by IL-17A and TNF-α secreted by Th17 cells, leading to cartilage damage [104]. The cross-talk between Th17 cells and immune cells, including macrophages and neutrophils, also can exacerbate the severity of RA. Honorati et al. found that IL-17 could promote the expression of VEGF by both chondrocytes and FLS in the development of OA, which led to the excessive proliferation of blood vessel network and synovial hypertrophy [105, 106]. Moreover, IL-17 could inhibit chondrocytes from producing proteoglycans and promote the synthesis of enzymes of the MMPs group. And IL-17 affects the expression of other cytokines that damage cartilage in the course of OA, such as IL-1β, TNF-α, IL-6, NO, and PGE2 [107]. Collectively, MDSCs protect stromal cells in the joint microenvironment from impeding intra-articular inflammation by suppressing Th17 cells. Moreover, Park et al. found that MDSCs could attenuate joint inflammation by promoting the proliferation of Treg cells in CIA mice. IL-10 may be a key cytokine for MDSCs to enhance Treg cells proliferation [108]. Treg cells could also exert their immune suppressive function like MDSCs. The positive effect of MDSCs on Th17 cells may counteract the aggravating arthritis process that FLS induce the conversion of Treg cells into Th17 cells or inhibit Treg cells via secreting IL-6 [109]. Additionally, Treg cells from OA patients have been found to secrete IL-10 and TGF-β at the synovium [110]. It has been proved that IL-10 could promote the transformation of the mesenchymal cells into chondrocytes by stimulating the expression of BMP-2 and BMP-6 [111]. And TGF-β could stimulate the production of proteoglycans, type II collagen, and chondrogenesis [112]. MDSCs affect not only adaptive immunity, but also innate immunity. Zhang et al. found that the amount of CD11b+CD68+ macrophages were reduced at the joint tissues in the CIA mice after treating with MDSCs [97]. However, the mechanism by which MDSCs suppress macrophages is unknown. Macrophages are activated in response to pro-inflammatory factors and in turn the activated macrophages produce pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-23, and GM-CSF. The cytokines then act on osteoblasts, chondrocytes, synovial fibroblasts, and neutrophils, inducing inflammation in the synovium and eventually leading to joint damage [113]. Furthermore, macrophages play important roles in the pathogenesis of OA. There are two main pathways of macrophages activation, among which the classical activation pathway is macrophages differentiate into M1 subset to initiate the inflammatory process [114]. Currently, the underlying mechanism of how M1 macrophages induce or aggravate OA remains unclear. One possible mechanism is that M1 macrophages could induce FLS to release MMPs as well as growth factors [115]. Another possible mechanism is that M1 macrophages alter chondrocyte metabolism, such that the R-spondin-2 protein secreted by the M1 subset activates β-catenin signaling on chondrocytes, leading to high expression of MMP1, MMP-3, MMP-9, MMP-13, IL-1β, TNF-α, IL-6, IL-8 [116]. Collectively, MDSCs may suppress macrophages by some unknown mechanism to alleviate intra-articular inflammation.
In addition to anti-inflammatory effects, MDSCs also play a proinflammatory role in joint microenvironment, which is incompatible with the expression of their name. Therefore, Mahmoud et al. named these cells as "myeloid-derived proinflammatory cells"(MDPCs) [12]. MDSCs exert proinflammatory function by interacting with other immune cells or stromal cells in the joint microenvironment. Guo et al. found that MDSCs and Th17 cells were simultaneously increased in synovial fluid of RA patients [34]. And the MDSCs of RA patients could promote both the differentiation of Th17 cells and Th17 responses in vitro. When MDSCs were depleted in CIA mice, the differentiation of Th17 cells as well as IL-17A and IL-1β production were also inhibited. After adoptive transferred MDSCs, the severity of the arthritis was increased along with the rise of both Th17 cells differentiation and the level of IL-17A as well as IL-1β [117]. Besides, studies have found that MDSCs produce high levels of IL-1β [34]. Combined with the role of Th17 cells in arthritis, MDSCs promote the differentiation of Th17 cells as well as the Th17 responses by secreting IL-1β in RA patients and CIA mice, thereby aggravating inflammatory reaction, destruction of cartilage, and synovial hypertrophy. Moreover, it has been reported that the OPN level is highly elevated in the synovial fluid of patients with RA and OA, and the increased OPN was suggested to participate in the pathogenesis and progression of RA and OA by promoting inflammation [118, 119]. And OPN has been demonstrated to induce MDSCs recruitment. However, whether MDSCs produce OPN within the joint microenvironment as they do in the tumor microenvironment remains unclear [23]. Taken together, MDSCs influence joint stromal cells either indirectly by interacting with important immune cells or directly by acting on them within the joint microenvironment. The cross-talk among them determines the double-edged sword effect of MDSCs in the joint microenvironment.
MDSCs are widely involved in bone repair
The efficacy of bone repair determines the prognosis of trauma, arthritis, tumors, and other orthopedic diseases, so it is necessary to clarify the mechanism of bone repair and the influencing factors on bone repair [120]. And the multifaceted roles of MDSCs in bone repair will be discussed as follows.
The first phase of bone repair is acute inflammation, so bone repair is not solely the involvement of osteoblasts, osteoclasts as well as other stromal cells, but also inflammatory cells [121]. Neutrophils, macrophages, and other cells infiltrate the injury sites and promote the release of a variety of cytokines, chemokines, and growth factors, forming the early acute inflammation of bone repair. The early acute inflammatory response could create a favorable microenvironment for bone repair through clearing immune complexes, nascent malignant cells, and necrotic tissue debris. However, dysregulated inflammation suppresses bone formation, therefore it is necessary to prevent excessive inflammatory response [122]. Fortunately, MDSCs could exert their immunosuppressive effect to restrain excessive inflammatory response and maintain immune homeostasis [22]. When trauma occurs, IMCs are recruited to the sites of injury under the stimulation of cytokines as well as chemokines and behave as MDSCs under the stimulation of the injury environment [123]. Taken together, MDSCs can dampen excessive acute inflammatory responses and maintain homeostasis through immunosuppressive characteristics, for which MDSCs play an important role in bone repair. Moreover, Kawai et al. found that MDSCs may be involved in bone formation by expressing Runx2 and osteocalcin, which both promote osteoblasts differentiation when bone healing [124]. However, when large segment of bone defects is inflicted by malignant tumors, chronic inflammation or extensive trauma, the immunosuppressive function of MDSCs is instead detrimental to bone repair. Because of the overexpression of Arg-1, MDSCs inhibit body's defense, leading to difficulty in bone healing and even infections [125]. Moreover, the promotion of osteoclastogenesis by MDSCs is also reversed to bone repair.
In the process of bone repair, when angiogenesis is poor, it leads to failure of bone healing, termed a nonunion. As mentioned above, MDSCs could promote tumor angiogenesis via the production of VEGF-A, bFGF, Bv8, and MMP9. However, whether MDSCs could enhance angiogenesis in bone repair is controversial. Levy et al. demonstrated that a large number of IMCs infiltrated into the injury site from the initial stages of fracture healing to complete bone healing. And they also found that removal of MDSCs led to poor fracture healing but accelerated fracture healing with adoptive transfer of IMCs. The functional assays on IMCs revealed that IMCs were not as functional as MDSCs [126]. Conversely, Wang et al. found that MDSCs could promote angiogenesis in the polymethyl methacrylate (PMMA)-induced membrane via expression of VEGF-A, angiopoietin 2 (Ang2), and HIF-1α [127]. And the PMMA-induced membrane plays a key role in bone repair. In summary, MDSCs are widely involved in the bone repair, but the mechanisms involved remain unclear, and further studies will lead to new therapeutic strategies for bone repair.
Therapeutic strategies for bone-related diseases by targeting MDSCs
Cancer-related bone metastasis
Metastasis is a critical stage of cancer progression, remaining to be a major challenge in treatment of cancer and a leading cause of cancer-related mortality [128]. Bone is one of the most common sites for certain metastatic cancer, and the processes mainly include the growth of the primary tumor, EMT, the PMN formation and evolution, colonization, survival or dormancy, reactivation, the growth of disseminated tumor cells, and bone modification [40, 44, 53]. MDSCs are widely involved in these processes [53]. Therefore, in recent years, a variety of MDSCs-targeted immune therapies have been developed to prevent the processes of bone metastasis [3].
The strategies of MDSCs-targeted immune therapies for cancer-related bone metastasis rely on four main principles: (1) Blocking the positive effect of MDSCs on primary tumor. Because the primary tumor is the initial factor causing bone metastasis, this principle is indispensable. (2) Inhibiting the role of MDSCs in the process of bone metastasis, such as EMT, the formation of PMN and so on. (3) Targeting MDSCs in the bone microenvironment to reduce vicious cycle. (4) Increasing the cell death rate of MDSCs to disrupt homeostasis of MDSCs. Combined with these principles, MDSCs-targeted immune therapeutic strategies include limiting the generation, recruitment, and localization of MDSCs, impeding the immunosuppressive function of MDSCs, blocking the effect of MDSCs on PMN formation, targeting MDSCs death pathway, and suppressing the osteoclastogenesis of MDSCs (Table 2, Fig. 3).

The therapeutic strategies for cancer-related bone metastasis by targeting MDSCs. Based on the principles for MDSCs-targeted immune therapies that we have discussed, therapeutic strategies for cancer-related bone metastasis by targeting MDSCs fall into five main groups. Starting from the section with blue as the background color, five therapeutic strategies are shown clockwise. The strategies include: a limiting the generation, recruitment, and localization of MDSCs, b impeding the immunosuppressive function of MDSCs, c blocking the effect of MDSCs on PMN formation, d targeting MDSCs death pathway, e suppressing the osteoclastogenesis of MDSCs
Firstly, limiting the generation, recruitment, and localization of MDSCs mainly includes two aspects. On the one hand, prevention of MDSCs generation and recruitment could be workable. Obviously, the generation of MDSCs can be inhibited if the MDSCs-prone cytokines and chemokines are blocked. Current therapeutics that exploit this strategy include curcumin (an IL-6 inhibitor), BMP-4 (reducing the expression of G-CSF), r84 (an antibody against VEGF) and sulforaphane (SFN, an inhibitor of MIF) [129,130,131,132]. At the transcriptional level, the generation of MDSCs is inhibited by interferon regulatory factor-8 (IRF-8) that is a master regulator of normal myelopoiesis, but IRF-8 is often silenced in MDSCs because of tumor-derived factors [133]. Therefore, elevating IRF-8 expression is a potential approach to inhibit MDSCs generation, and upregulating IRF-8 conversely promotes normal myelopoiesis to produce monocytes, neutrophils, and DCs to boost anti-tumor innate immunity. Interestingly, MDSCs highly expressed OPN by silencing IRF-8 [134]. Therefore, elevating the level of IRF-8 could not only inhibit the enhanced recruitment and function of OPN on MDSCs, but also suppress the promoting role of OPN in cancer. Drugs proven effective to hinder MDSCs from the marrow to the tumor microenvironment include chemokine (C-X-C motif) receptor 2 (CXCR2) inhibitor, chemokine (C-X-C motif) receptor 4 (CXCR4) antagonists, and colony stimulating factor 1 receptor (CSF-1R) inhibitors [135]. On the other hand, considering that MDSCs could differentiate into mature myeloid cells without immunosuppressive function, thus the treatment that promote MDSCs differentiation is also feasible. It has been reported that all-trans retinoic acid (ATRA), Vitamin D3, Vitamin A, STAT3 inhibitors, and DNA demethylating agent 5-aza-2'-deoxycytidine could induce MDSCs differentiation [136,137,138]. Similarly, promoting the polarization of MDSCs into macrophages is also a practicable method, in which representative drugs are docetaxel and paclitaxel [139].
Secondly, impeding the immunosuppressive function of MDSCs is a viable therapeutic strategy. As mentioned above, MDSCs upregulate iNOS expression by activating STAT1, ROS expression by activating STAT3 or NOX2, and Arg-1 expression to exert immunosuppressive effects. More recent approaches using STAT3 inhibitors aim to block the activation of STAT3, such as sunitinib, AZD9150, and BBI608, or a conjugate of the STAT3 antisense oligonucleotide (ASO) tethered to immunostimulatory toll-like receptor 9 (TLR9) agonist (CpG-STAT3ASO) conjugates. And all of these STAT3 inhibitors could reduce the immunosuppressive function of MDSCs [140]. Moreover, the suppressive effect of MDSCs could also be eliminated using p-STAT3 or STAT3-targeted siRNA to interfere with STAT3 mRNA [141]. Nuclear factor E2-related factor 2 (Nrf2), a transcription factor, attenuates the ROS accumulation in MDSCs [142]. Synthetic triterpenoid, such as omaveloxolone (RTA-408), CDDO-Me (RTA-402), and CDDO-Im (RTA-403), could upregulate the level of Nrf2 to reduce the production of ROS and prevent the immune suppressive effect of MDSCs [143,144,145]. N-Hydroxy-nor-L-arginine (nor-NOHA), an Arg-1 inhibitor, could reduce the level of Arg-1 to block the immunosuppressive activity of MDSCs [146]. Targeting cyclooxygenase-2 (COX-2) could reduce the synthesis of PGE2, which induces MDSCs to produce Arg-1 and iNOS [147]. And the synthesis of PGE2 is regulated by COX-2, RIPK3 as well as fatty acid transport protein 2(FATP2), which means these molecules could be potential target for treatment [140]. COX-2 inhibitors include acetylsalicylic acid, NS-398, aspirin, and celecoxib. Besides, lipofermata is a selective inhibitor of FATP2. Similarly, the application of phosphodiesterase 5 (PDE5) inhibitors, including sildenafil, tadalafil, and vardenafil, could also be workable for reducing the production of Arg-1 and iNOS in MDSCs [148]. Moreover, it has been reported that 1-methyl-L-tryptophan (1-MT) or STAT3 antagonist JSI-124 could reduce the level of MDSC-produced IDO; STAT3-dependent IDO expression is required for exerting immunosuppressive effects of MDSCs in breast cancer [149].
Thirdly, blocking the effect of MDSCs on PMN formation. S1PR1-STAT3 signaling is key for MDSCs colonization at future metastatic sites, therefore targeting S1PR1-STAT3 signaling in MDSCs by CpG-STAT3 siRNA or CpG-S1PR1 siRNA would effectively reduce the formation of PMN and thereby preventing metastasis [150]. Besides, reducing the levels of PMN formation factors produced by MDSCs might also be workable, therefore the inhibitor of TGF-β, Arg-1, ROS, COX-2, Bv8, MMP-9, S100A8, and S100A9 might be potential therapeutic approaches [24].
Fourthly, targeting the cell death pathways of MDSCs may be an effective therapeutic strategy to inhibit MDSCs. The homeostasis of MDSCs is not only regulated by differentiation or generation pathways, but also by cell death pathways. Sinha et al. found that FasL( +) T cells counter kill MDSCs via the Fas-FasL apoptosis pathway [151]. However, MDSCs increase the expression of Bcl-xL and decrease the expression of Bax by down-regulating IRF-8, thereby negatively modulating the Fas-FasL apoptosis signaling pathway [152]. Guha et al. found that STAT3 inhibitors (STATTIC or BBI608) precisely induce cell death through the Bax-dependent apoptosis pathway, and the finding established the link between STAT3 inhibition and Fas-FasL apoptosis of MDSCs [153]. Therefore, Bcl-xL is a potential target to induce MDSCs death. Furthermore, Condamine et al. described that the reason for higher cell death rate of MDSCs compared to neutrophils and monocytes is that ER stress regulates MDSCs through TNF-related apoptosis-induced ligand receptors (TRAIL-Rs)-mediated apoptosis, which suggests that TRAIL-Rs may serve as potential targets for inhibiting MDSCs [154]. Moreover, as tumor cells apoptosis inducer, ceramide also activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress MDSCs, which has performed the potential clinical value of ceramidase inhibitor [155]. Besides, Smith et al. found that decitabine (DAC) promoted cell death through the disrupting DNA methylation of RIP1-dependent targets of necroptosis, and further studies revealed that MDSCs autocrine IL-6 to regulate STAT3-DNMT epigenetic axis, thereby silencing the TNFα-RIP1 necroptosis pathway [156]. In addition to DAC, there are other chemotherapeutic agents that can induce the death of MDSCs. It has been reported that MDSCs can be eliminated by chemotherapy with gemcitabine, 5-fluorouracil, CD33 monoclonal antibody (mAb), liver-X receptors (LXRs) agonists (including GW3965 and RGX-104), and peptibodies consisting of S100A9-derived peptides, which all could impair the survival of MDSCs [157,158,159,160,161]. But the mechanism of their impairment is not the same.
Finally, the approaches to suppress the osteoclastogenesis of MDSCs are also effective. Many studies have shown that cysteine protease (including legumain) and cysteine cathepsin could suppress osteoclastogenesis of MDSCs in breast cancer [162]. Intriguingly, the Nrf2 activator RTA-408 not only blocks the immunosuppressive effects of MDSCs, but also attenuates osteoclastogenesis by inhibiting STING dependent NF-κB signaling [163]. Besides, NG-monomethyl-l-arginine acetate (l-NMMA), an iNOS inhibitor, prevents MDSCs from differentiating into osteoclasts by inhibiting NO signaling [164]. In fact, there are many other modalities of MDSCs-targeted immune therapies beyond the above-mentioned chemotherapy. These treatments include radiotherapy using radiation to kill MDSCs, combination immunotherapy synergistically with other classical immunotherapies, and epigenetic therapy.
Osteosarcoma (OS)
OS is the most common bone malignancy in children and teenagers [165]. However, the treatment of OS has not progressed over the years and is still mainly based on surgical resection and chemotherapy, resulting in no improvement in overall survival. Fortunately, several evidence demonstrated that MDSCs extensively infiltrated OS tissues and suppressed immune responses, therefore MDSCs have gained increasing attention as the new target of OS immunotherapy (Table 2).
It has been shown that metformin (Met) inhibits the growth of K7M2neo OS by regulating MDSCs, and the inhibition is independent of T cells. The mechanism behind the above phenomenon is that Met can regulate the metabolism of MDSCs to decrease oxidative phosphorylation (OXPHOS) while increasing glycolysis. Besides, ROS production in MDSCs could also be inhibited by Met [166]. Recently, Jiang et al. shed light on the OS-infiltrating MDSCs were CXCR4 positive, and the binding of CXCR4 to SDF-1 could reduce the apoptosis of MDSCs by activating downstream AKT pathway. They also found that AMD3100, a CXCR4 antagonist, could synergize with anti-PD-1 antibody immunotherapy to treat OS in a murine [167]. Also augmenting anti-PD-1 antibody immunotherapy is (S)-(−)-N-[2-(3-Hydroxy-1H-indol-3-yl)-methyl]-acetamide (SNA), which is specific inhibitor of phosphatidylinositide 3-kinases δ/γ (PI3Kδ/γ). Shi et al. reveled that SNA treatment could decrease the expression of arginase and iNOS, as well as the phosphorylation of AKT and S6 downstream of PI3K on OS-infiltrating MDSCs [168]. In addition, SNA treatment promoted the polarization of MDSCs towards the TAM-M1 population in OS. IL-18 could induce MDSCs to migrate into the OS tissue, which means that IL-18 inhibitor is likewise potential MDSCs-targeted therapy [169]. As standard treatment for OS, neoadjuvant chemotherapy, including the use of doxorubicin, cisplatin, and ifosfamide (IFO), has similarly been shown to eliminate MDSCs on patients with OS [170]. Similarly, ATRA treatment reduces the quantity of M-MDSCs and diminishes the suppressive potency of PMN-MDSCs in mice models of sarcoma, thereby treating OS [171].
The cure rate for patients with localized disease approaches 70%, whereas the 5-year overall survival rate for patients with metastatic disease is less than 25% [165]. Therefore, therapies that prevent OS metastasis are critical for patients with OS. Since MDSCs can hinder T-cell infiltration into the PMN, especially pulmonary metastasis, MDSCs targeted therapy may also be a potential therapeutic approach to block OS metastasis [172].
Rheumatoid arthritis (RA)
RA is an autoimmune disease that causes polyarticular, symmetrical, aggressive joint erosion by inducing synovitis and subsequent cartilage as well as bone destruction. In the joint cavity of RA patients, leukocytes, especially Th17 cells, Th1 cells, Treg cells, monocytes, and macrophages, infiltrate into it and cause synovial hyperplasia. The infiltration of leukocytes and the secretion of proinflammatory cytokines drive the maturation of osteoclast precursors into osteoclasts, thereby eroding the bone. Meanwhile, synovial cells proliferate excessively and secrete a large number of MMPs as well as collagenases, which degrade proteoglycans and type II collagen, the main components of extracellular matrix (ECM), and ultimately destroy articular cartilage and subchondral bone [173]. Targeting pro-inflammatory cytokines, several biological agents, including tozumab, infliximab, etanercept, adalimumab, and rituximab, have been approved for marketing in the current treatment of RA and achieved some efficacy [174, 175]. However, biological agents are not effective for all patients with RA, so there is a need to develop new treatment.
MDSCs play a double-edged role in RA and CIA. MDSCs exert proinflammatory effects mainly through the following aspects: (1) MDSCs could directly differentiate into osteoclasts through NF-κB signaling or regulate other immune cells, including Th17 cells, Treg cells and so on, to promote osteoclastogenesis; (2) MDSCs enhance Th17 cells function by IL-1β, thereby aggravating cartilage damage and synovitis. The anti-inflammatory effects of MDSCs include: (1) MDSCs could inhibit Th17 cells responses; (2) MDSCs could promote Treg cells proliferation and enhance the anti-inflammatory effect of Treg cells; (3) MDSCs could reduce the number of macrophages, thereby reducing the pro-inflammatory factors secreted by macrophages, such as TNF-α, IL-1b, IL-6, IL-23, and GM-CSF. The greatest feature of MDSCs over single target biological agents is their role as double-edged sword, which also dictates that MDSCs will be diverse for use as therapeutics. Zhang et al. found that the adoptive transfer of MDSCs ameliorated arthritis by reducing the amount of Th17 cells and macrophages in the joint tissue of CIA and the level of inflammatory cytokines [97]. And the adoptive transfer of MDSCs therapy exerts an anti-inflammatory effect by the Arg-1 pathway [176]. Besides, Sun et al. found that piperlonguminine (PL) could enhance the expansion of MDSCs to alleviate RA [177]. Hence, PL may be a candidate therapeutic agent for RA based on its effect on MDSCs. Intriguingly, MDSCs-derived exosomes also have potential value for the treatment of RA. Zhu et al. found that G-MDSC-derived exosomes attenuated CIA by decreasing the number of Th17 cells and Th1 cells in vivo and in vitro [178]. As critical for MDSCs generation, IRF-8 deficiency similarly promotes RANKL-mediated osteoclastogenesis in RA [179]. Thus, promoting the expression of IRF-8 may not only inhibit osteoclastogenesis, but also hinder the generation of MDSCs and subsequently inhibit the destructive role of MDSCs in RA. All in all, further understanding the role of MDSCs in RA is required, which will facilitate the application of MDSCs-based therapies to RA (Table 2).
Osteoarthritis (OA)
OA is a common joint disease characterized by articular cartilage degeneration, mainly involving the knee joint, hip joint, and distal fingertip joint. The traditional view is that OA is a non-inflammatory disease, but an increasing number of studies have shown that OA pathogenesis is closely related to synovial inflammation. Currently, studies show that T cells, B cells, macrophages, and NK cells infiltrate the joint synovium of OA patients and contribute to the disease process [180]. In addition, MDSCs infiltration is also present in synovial fluid of OA patients, although the degree of infiltration is not as high as in RA [34]. Therefore, MDSCs may play an immunoregulatory role in the pathogenesis of OA, but the mechanism responsible for how MDSCs regulate the disease progression of OA is unknown. Here, we summarize the potential role of MDSCs in OA pathogenesis and the related research progress, aiming to provide direction for the in-depth study of OA pathogenesis as well as the development of intervention drugs targeting MDSCs (Table 2).
Synovitis is presented in both OA and RA patients, and MDSCs have been shown to be elevated within the joint microenvironment. Therefore, based on the method of analogy, we speculate that MDSCs might also be making interaction with immune or stromal cells in the joint microenvironment of OA patients. Our first conjecture is that MDSCs are equally likely to reduce the number of macrophages. Macrophages play a vital role in inducing or aggravating OA by altering the metabolism of chondrocyte or promoting the release of MMPs and growth factors [115, 116]. In summary, MDSCs may alleviate OA via inhibitory effects on macrophages. The second conjecture is that MDSCs may also increase the number of Treg cells and in turn promote the chondrogenesis and the synthesis of proteoglycan and type II collagen. However, it has been reported that there is a large reduction in the number of Treg cells in OA [110]. Therefore, the exact interaction between Treg cells and MDSCs requires further study. Finally, we speculate that MDSCs may also play a pro-inflammatory role in OA, because of the potential relationship between MDSCs and Th17 cells. This potential relationship is that MDSCs may similarly promote the differentiation of human Th17 cells and Th17 responses, as both Th17 cells and MDSCs are found to be elevated in the synovial fluid of OA patients like RA patients [34]. Th17 cells could inhibit chondrogenesis proteoglycans, secret inflammatory factor, and promote MMP synthesis, thereby contributing the progression of OA [107]. Therefore, MDSCs may damage cartilage by regulating Th17 cells. According to the above hypothesis, it is not difficult to conclude that MDSCs may also play a double-edged sword role in OA disease progression.
In addition, another major pathogenesis of OA is that the abnormal bone resorption in subchondral bone is able to change the normal mechanical environment of articular cartilage, thereby inducing articular cartilage to undergo degenerative pathologies, ultimately leading to OA [181]. And whether MDSCs participate in this process is not known. No studies have reported MDSCs levels in the subchondral bone or peripheral blood of OA patients, and if further studies show an increase, the role played by MDSCs in subchondral bone destruction would be well worth exploring and may provide a novel therapeutic modality for clinical treatment of OA. Overall, the above speculation requires further study and may provide feasible ideas for clinical treatment.
Orthopedic trauma
Orthopedic trauma is manifested by varying degrees of trauma, to a lesser extent by minor fractures and to a greater extent by extensive bone loss. Regardless of the degree of trauma, MDSCs have influence on orthopedic trauma, which also determines their potentially great value for orthopedic trauma treatment (Table 2).
Since tissue repair after minor trauma is often limited, preventing the excessive inflammatory response is critical for minor trauma. The immunosuppressive effect of MDSCs is just to restrain excessive inflammatory response and maintain immune homeostasis [22]. Besides, Arg-1-derived MDSCs could also metabolize arginine to ornithine, which stimulates the proliferation of fibroblasts and the production of collagen to heal wound effectively [182]. Taken together, MDSCs contribute to minor trauma healing. However, the role of MDSCs is quite opposite upon extensive orthopedic trauma. Post traumatic infections such as extensive bone fractures are a significant cause of delayed wound healing and even death. And the occurrence of infection may be associated with MDSCs. Ochoa et al. have found that Arg-1 expression is increased in MDSCs after trauma [125]. The mechanism of Arg-1 overexpression by MDSCs is the secretion of anti-inflammatory factors by increased Th12 cells after trauma [183]. As mentioned earlier, Arg-1 will cause an arginine deficiency state, which in turn will inhibit the number and function of T cells. Therefore, inhibiting MDSCs to express Arg-1 or applicating Arg-1 inhibitors appropriately after trauma may become novel therapies to avoid the occurrence of infections. Extensive orthopedic trauma is often accompanied by the absence of the periosteum, which plays an important role in bone formation and defect reconstruction [184]. Clinically, the PMMA cement is often used to induce the organism to form periosteum, and the PMMA-induced membrane establishes a blood supply for the large bone defect repair [185]. Wang et al. demonstrated that MDSCs played an important role in the formation and angiogenesis processes of the PMMA-induced membrane. Interestingly, MDSCs express VEGF-A, Ang2, and HIF-1α to promote the angiogenesis of endothelial cells via STAT3 signaling, which is different from the mechanism of enhancing tumor angiogenesis [127]. It suggests that MDSCs derived from PMMA-induced membrane may serve as a stimulating angiogenesis target in bone repair. Allogeneic bone grafts are often required for patients with large bone defects after injection of cement to form periosteum, and the role of MDSCs in allogeneic bone grafts is currently unknown. But based on the functional studies of MDSCs in other organs or tissues allografts, we could roughly speculate about the function of MDSCs in bone grafts. The common complication of allografts is immune rejection, and bone grafts are no exception. Fortunately, numerous studies have shown that MDSCs participate in maintaining immune tolerance in allograft transplantation [186]. And MDSCs could inhibit allograft rejection of various organ by secreting iNOS, IDO, Arg-1 as well as interacting with other immune cells, including Treg cells, CD8+T cells, and NKT cells [187]. Therefore, we speculate that MDSCs may also maintain bone transplantation immune tolerance. If further studies validate this speculation, using drugs that induce MDSCs generation or adoptive transfer MDSCs is maybe a potential therapeutic strategy to reduce the immune rejection of bone grafts. Taken together, MDSCs play an important role in orthopedic trauma, indicating their potential clinical therapeutic value. But considering that MDSCs act variously on different degrees of trauma and different timing of trauma healing process, thus we should pay attention to the application of MDSCs timing when using MDSCs as therapeutic strategy.
Conclusion and future prospective
MDSCs are heterogeneous in morphology, phenotype, and function, therefore, targeting MDSCs has high specificity and efficacy, which may have great prospect in the treatment of bone-related diseases. Meanwhile, MDSCs are mainly produced in large quantities under pathological conditions in peripheral blood, tumor sites, inflammatory sites, and bone marrow, while they are less under physiological conditions. Targeting MDSCs in many ways mimics classical immunotherapy methods (such as immune checkpoint, cellular immunotherapy, etc.) and may cause fewer side effects. However, due to technology limitation and lack of highly specific markers, it is still difficult to identify complex phenotypes of MDSCs accurately and reliably, which limits the clinical application of MDSCs. The regulation of MDSCs in bone-related diseases is complex and serves double-edged sword role. MDSCs function in the bone and joint microenvironment, where MDSCs make interaction with osteoclasts, osteoblasts, chondrocytes, and other stromal cells to build a complex network. Therefore, drugs without precise function and targets may cause unwanted side effects such as aggravating disease progression, which demonstrates the need to find methods to identify the specificity of MDSCs. In addition, MDSCs may also serve as disease marker to predict prognosis of bone-related diseases.
The mechanisms of the immunosuppressive action of MDSCs still need more studies; the process of MDSCs accumulation and interaction with other immune cells are unclear. Meanwhile, current studies on MDSCs are limited to immune aspect and ignore interaction between MDSCs and bone cells including osteoclasts and osteoblasts. MDSCs, other immune cells, osteoclasts, osteoblasts, and chondrocytes exist in the same microenvironment, therefore paying more attention to the association between MDSCs and other stromal cells may provide a new intervention target for MDSCs-based therapy. In addition, studies related to the other functions of MDSCs such as angiogenesis and transplantation immune tolerance induction also perform significant value.
Taken together, the clinical significance of MDSCs in bone-related diseases has been established. The further key research is to determine whether targeting MDSCs could provide tangible clinical benefits and how to identify complex phenotypes of MDSCs accurately and reliably. Meanwhile, cross-talk among MDSCs, osteoclasts, and osteoblasts make the association between skeletal system and immune system. It is strongly believed that more mechanisms of osteoimmunology will be clarified and more drug targeting MDSCs in bone-related disease will be detected in further studies.
Availability of data and material
Not applicable.
References
Tsukasaki M, Takayanagi H (2019) Osteoimmunology: evolving concepts in bone-immune interactions in health and disease. Nat Rev Immunol 19:626–642. https://doi.org/10.1038/s41577-019-0178-8
Goh C, Narayanan S, Hahn YS (2013) Myeloid-derived suppressor cells: the dark knight or the joker in viral infections? Immunol Rev 255:210–221. https://doi.org/10.1111/imr.12084
Xiang L, Gilkes DM (2019) The contribution of the immune system in bone metastasis pathogenesis. Int J Mol Sci. https://doi.org/10.3390/ijms20040999
Young MR, Newby M, Wepsic HT (1987) Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res 47:100–105
Gabrilovich DI (2017) Myeloid-Derived Suppressor Cells. Cancer. Immunol Res 5:3–8. https://doi.org/10.1158/2326-6066.Cir-16-0297
Binsfeld M, Muller J, Lamour V, De Veirman K, De Raeve H, Bellahcène A, Van Valckenborgh E, Baron F, Beguin Y, Caers J, Heusschen R (2016) Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 7:37931–43. https://doi.org/10.18632/oncotarget.9270
Yang F, Li Y, Wu T, Na N, Zhao Y, Li W, Han C, Zhang L, Lu J, Zhao Y (2016) TNFα-induced M-MDSCs promote transplant immune tolerance via nitric oxide. J Mol Med (Berl) 94:911–920. https://doi.org/10.1007/s00109-016-1398-z
Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, Khoury SJ (2007) CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol 179:5228–5237. https://doi.org/10.4049/jimmunol.179.8.5228
Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111:4233–4244. https://doi.org/10.1182/blood-2007-07-099226
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181:5791–5802. https://doi.org/10.4049/jimmunol.181.8.5791
Talmadge JE, Gabrilovich DI (2013) History of myeloid-derived suppressor cells. Nat Rev Cancer 13:739–752. https://doi.org/10.1038/nrc3581
Yaseen MM, Abuharfeil NM, Darmani H, Daoud A (2021) Recent advances in myeloid-derived suppressor cell biology. Front Med 15:232–251. https://doi.org/10.1007/s11684-020-0797-2
Veglia F, Perego M, Gabrilovich D (2018) Myeloid-derived suppressor cells coming of age. Nat Immunol 19:108–119. https://doi.org/10.1038/s41590-017-0022-x
Kumar V, Patel S, Tcyganov E, Gabrilovich DI (2016) The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 37:208–220. https://doi.org/10.1016/j.it.2016.01.004
Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S (2012) Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother 61:1155–1167. https://doi.org/10.1007/s00262-012-1294-5
Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, Partlova S, Garfall A, Vogl DT, Xu X, Knight SC, Malietzis G, Lee GH, Eruslanov E, Albelda SM, Wang X, Mehta JL, Bewtra M, Rustgi A, Hockstein N, Witt R, Masters G, Nam B, Smirnov D, Sepulveda MA, Gabrilovich DI (2016) Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. https://doi.org/10.1126/sciimmunol.aaf8943
Rodríguez PC, Ochoa AC (2008) Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 222:180–191. https://doi.org/10.1111/j.1600-065X.2008.00608.x
Yang Y, Li C, Liu T, Dai X, Bazhin AV (2020) Myeloid-derived suppressor cells in tumors: from mechanisms to antigen specificity and microenvironmental regulation. Front Immunol 11:1371. https://doi.org/10.3389/fimmu.2020.01371
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2009) Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 182:5693–5701. https://doi.org/10.4049/jimmunol.0900092
Su YL, Banerjee S, White SV, Kortylewski M (2018) STAT3 in tumor-associated myeloid cells: multitasking to disrupt immunity. Int J Mol Sci. https://doi.org/10.3390/ijms19061803
Wang Z, Jiang J, Li Z, Zhang J, Wang H, Qin Z (2010) A myeloid cell population induced by Freund adjuvant suppresses T-cell-mediated antitumor immunity. J Immunother 33:167–177. https://doi.org/10.1097/CJI.0b013e3181bed2ba
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. https://doi.org/10.1038/nri2506
Klement JD, Paschall AV, Redd PS, Ibrahim ML, Lu C, Yang D, Celis E, Abrams SI, Ozato K, Liu K (2018) An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J Clin Invest 128:5549–5560. https://doi.org/10.1172/jci123360
Chiodoni C, Sangaletti S, Colombo MP (2017) Matricellular proteins tune myeloid-derived suppressor cell recruitment and function in breast cancer. J Leukoc Biol 102:287–292. https://doi.org/10.1189/jlb.3MR1016-447R
Li H, Han Y, Guo Q, Zhang M, Cao X (2009) Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol 182:240–249. https://doi.org/10.4049/jimmunol.182.1.240
Hu CE, Gan J, Zhang RD, Cheng YR, Huang GJ (2011) Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol 46:156–164. https://doi.org/10.3109/00365521.2010.516450
Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, Svane IM (2010) Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR–/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol 72:540–547. https://doi.org/10.1111/j.1365-3083.2010.02463.x
Won WJ, Deshane JS, Leavenworth JW, Oliva CR, Griguer CE (2019) Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 3:47–65. https://doi.org/10.15698/cst2019.02.176
Li YL, Zhao H, Ren XB (2016) Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med 13:206–214. https://doi.org/10.20892/j.issn.2095-3941.2015.0070
Wang SH, Lu QY, Guo YH, Song YY, Liu PJ, Wang YC (2016) The blockage of Notch signalling promoted the generation of polymorphonuclear myeloid-derived suppressor cells with lower immunosuppression. Eur J Cancer 68:90–105. https://doi.org/10.1016/j.ejca.2016.08.019
Wu MY, Li CJ, Yiang GT, Cheng YL, Tsai AP, Hou YT, Ho YC, Hou MF, Chu PY (2018) Molecular regulation of bone metastasis pathogenesis. Cell Physiol Biochem 46:1423–1438. https://doi.org/10.1159/000489184
Marvel D, Gabrilovich DI (2015) 32. J Clin Invest 125:3356–64. https://doi.org/10.1172/jci80005
Chan CY, Yuen VW, Wong CC (2019) Hypoxia and the Metastatic Niche. Adv Exp Med Biol 1136:97–112. https://doi.org/10.1007/978-3-030-12734-3_7
Guo C, Hu F, Yi H, Feng Z, Li C, Shi L, Li Y, Liu H, Yu X, Wang H, Li J, Li Z, Wang XY (2016) Myeloid-derived suppressor cells have a proinflammatory role in the pathogenesis of autoimmune arthritis. Ann Rheum Dis 75:278–285. https://doi.org/10.1136/annrheumdis-2014-205508
Pantel K, Alix-Panabières C (2014) Bone marrow as a reservoir for disseminated tumor cells: a special source for liquid biopsy in cancer patients. Bonekey Rep 3:584. https://doi.org/10.1038/bonekey.2014.79
Coleman RE (2001) Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev 27:165–176. https://doi.org/10.1053/ctrv.2000.0210
Park SI, Soki FN, McCauley LK (2011) Roles of bone marrow cells in skeletal metastases: no longer bystanders. Cancer Microenviron 4:237–246. https://doi.org/10.1007/s12307-011-0081-8
Fornetti J, Welm AL, Stewart SA (2018) Understanding the bone in cancer metastasis. J Bone Miner Res 33:2099–2113. https://doi.org/10.1002/jbmr.3618
Demirkan B (2013) The roles of epithelial-to-mesenchymal transition (EMT) and mesenchymal-to-epithelial transition (MET) in breast cancer bone metastasis: potential targets for prevention and treatment. J Clin Med 2:264–282. https://doi.org/10.3390/jcm2040264
Todd VM, Johnson RW (2020) Hypoxia in bone metastasis and osteolysis. Cancer Lett 489:144–154. https://doi.org/10.1016/j.canlet.2020.06.004
Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, Bissell MJ, Cox TR, Giaccia AJ, Erler JT, Hiratsuka S, Ghajar CM, Lyden D (2017) Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17:302–317. https://doi.org/10.1038/nrc.2017.6
Sosnoski DM, Norgard RJ, Grove CD, Foster SJ, Mastro AM (2015) Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin Exp Metastasis 32:335–344. https://doi.org/10.1007/s10585-015-9710-9
Kan C, Vargas G, Pape FL, Clézardin P (2016) Cancer cell colonisation in the bone microenvironment. Int J Mol Sci. https://doi.org/10.3390/ijms17101674
Croucher PI, McDonald MM, Martin TJ (2016) Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer 16:373–386. https://doi.org/10.1038/nrc.2016.44
Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, Kato M, Prevost-Blondel A, Thiery JP, Abastado JP (2011) Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol 9:e1001162. https://doi.org/10.1371/journal.pbio.1001162
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265–273. https://doi.org/10.1038/nrc2620
Heerboth S, Housman G, Leary M, Longacre M, Byler S, Lapinska K, Willbanks A, Sarkar S (2015) EMT and tumor metastasis. Clin Transl Med 4:6. https://doi.org/10.1186/s40169-015-0048-3
Zhu H, Gu Y, Xue Y, Yuan M, Cao X, Liu Q (2017) CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 8:114554–67. https://doi.org/10.18632/oncotarget.23020
Xiao J, Gong Y, Chen Y, Yu D, Wang X, Zhang X, Dou Y, Liu D, Cheng G, Lu S, Yuan W, Li Y, Zhao Z (2017) IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am J Physiol Renal Physiol 313:F310–F318. https://doi.org/10.1152/ajprenal.00428.2016
Massagué J (2008) TGFbeta in cancer. Cell 134:215–230. https://doi.org/10.1016/j.cell.2008.07.001
Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG, Kotarski J, Tarkowski R, Wicha M, Cho K, Giordano T, Liu R, Zou W (2013) Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 39:611–621. https://doi.org/10.1016/j.immuni.2013.08.025
Liu Y, Cao X (2016) Characteristics and significance of the pre-metastatic niche. Cancer Cell 30:668–681. https://doi.org/10.1016/j.ccell.2016.09.011
Wang Y, Ding Y, Guo N, Wang S (2019) MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol 10:172. https://doi.org/10.3389/fimmu.2019.00172
Sterling JA, Edwards JR, Martin TJ, Mundy GR (2011) Advances in the biology of bone metastasis: how the skeleton affects tumor behavior. Bone 48:6–15. https://doi.org/10.1016/j.bone.2010.07.015
Clines GA, Guise TA (2005) Hypercalcaemia of malignancy and basic research on mechanisms responsible for osteolytic and osteoblastic metastasis to bone. Endocr Relat Cancer 12:549–583. https://doi.org/10.1677/erc.1.00543
Brook N, Brook E, Dharmarajan A, Dass CR, Chan A (2018) Breast cancer bone metastases: pathogenesis and therapeutic targets. Int J Biochem Cell Biol 96:63–78. https://doi.org/10.1016/j.biocel.2018.01.003
Keller ET, Brown J (2004) Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem 91:718–729. https://doi.org/10.1002/jcb.10662
Guise TA, Mohammad KS, Clines G, Stebbins EG, Wong DH, Higgins LS, Vessella R, Corey E, Padalecki S, Suva L, Chirgwin JM (2006) Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res 12:6213s-s6216. https://doi.org/10.1158/1078-0432.Ccr-06-1007
Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, Amadori D, Kang Y (2013) Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell 24:542–556. https://doi.org/10.1016/j.ccr.2013.09.008
Roodman GD (2004) Mechanisms of bone metastasis. N Engl J Med 350:1655–1664. https://doi.org/10.1056/NEJMra030831
Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massagué J, Mundy GR, Guise TA (1999) TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 103:197–206. https://doi.org/10.1172/jci3523
Yogo K, Ishida-Kitagawa N, Takeya T (2007) Negative autoregulation of RANKL and c-Src signaling in osteoclasts. J Bone Miner Metab 25:205–210. https://doi.org/10.1007/s00774-007-0751-2
Shi Y, Massagué J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700. https://doi.org/10.1016/s0092-8674(03)00432-x
Ibrahim T, Flamini E, Mercatali L, Sacanna E, Serra P, Amadori D (2010) Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 116:1406–1418. https://doi.org/10.1002/cncr.24896
Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD Jr (2003) The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 349:2483–2494. https://doi.org/10.1056/NEJMoa030847
Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, Gerald WL, Massagué J (2009) WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138:51–62. https://doi.org/10.1016/j.cell.2009.04.030
Bagnato A, Loizidou M, Pflug BR, Curwen J, Growcott J (2011) Role of the endothelin axis and its antagonists in the treatment of cancer. Br J Pharmacol 163:220–233. https://doi.org/10.1111/j.1476-5381.2011.01217.x
Yonou H, Horiguchi Y, Ohno Y, Namiki K, Yoshioka K, Ohori M, Hatano T, Tachibana M (2007) Prostate-specific antigen stimulates osteoprotegerin production and inhibits receptor activator of nuclear factor-kappaB ligand expression by human osteoblasts. Prostate 67:840–848. https://doi.org/10.1002/pros.20574
Cramer SD, Chen Z, Peehl DM (1996) Prostate specific antigen cleaves parathyroid hormone-related protein in the PTH-like domain: inactivation of PTHrP-stimulated cAMP accumulation in mouse osteoblasts. J Urol 156:526–531. https://doi.org/10.1097/00005392-199608000-00076
Bussard KM, Gay CV, Mastro AM (2008) The bone microenvironment in metastasis; what is special about bone? Cancer Metast Rev 27:41–55. https://doi.org/10.1007/s10555-007-9109-4
Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X, Ponnazhagan S (2013) Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 73:672–682. https://doi.org/10.1158/0008-5472.Can-12-2202
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2010) HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207:2439–2453. https://doi.org/10.1084/jem.20100587
Blavier L, Delaissé JM (1995) Matrix metalloproteinases are obligatory for the migration of preosteoclasts to the developing marrow cavity of primitive long bones. J Cell Sci 108(Pt 12):3649–3659
Nilforoushan D, Gramoun A, Glogauer M, Manolson MF (2009) Nitric oxide enhances osteoclastogenesis possibly by mediating cell fusion. Nitric Oxide 21:27–36. https://doi.org/10.1016/j.niox.2009.04.002
Sawant A, Ponnazhagan S (2013) Myeloid-derived suppressor cells as osteoclast progenitors: a novel target for controlling osteolytic bone metastasis. Cancer Res 73:4606–4610. https://doi.org/10.1158/0008-5472.Can-13-0305
Soria G, Ben-Baruch A (2008) The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett 267:271–285. https://doi.org/10.1016/j.canlet.2008.03.018
Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, Liu Y, Li D, Yuan Y, Zhang GM, Feng ZH (2007) CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett 252:86–92. https://doi.org/10.1016/j.canlet.2006.12.012
Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling JA (2012) Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology 1:1484–1494. https://doi.org/10.4161/onci.21990
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T (2000) T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408:600–605. https://doi.org/10.1038/35046102
Park S, Lee KJ, Lee EJ, Whang YM, Cho SW (2020) Abstract P5–04–29: Osteoblasts regulate mobilization of the myeloid-derived suppressor cells from the bone marrow of breast cancer patients. In: AACR. https://cancerres.aacrjournals.org/content/80/4_Supplement/P5-04-29
Xu X, Zhang C, Trotter TN, Gowda PS, Lu Y, Ponnazhagan S, Javed A, Li J, Yang Y (2020) Runx2 deficiency in osteoblasts promotes myeloma progression by altering the bone microenvironment at new bone sites. Cancer Res 80:1036–1048. https://doi.org/10.1158/0008-5472.Can-19-0284
Rossnagl S, Altrock E, Sens C, Kraft S, Rau K, Milsom MD, Giese T, Samstag Y, Nakchbandi IA (2016) EDA-fibronectin originating from osteoblasts inhibits the immune response against cancer. PLoS Biol 14:e1002562. https://doi.org/10.1371/journal.pbio.1002562
Fisher LW, Fedarko NS (2003) Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res 44(Suppl 1):33–40
Zhang H, Huang Y, Wang S, Fu R, Guo C, Wang H, Zhao J, Gaskin F, Chen J, Yang N, Fu SM (2015) Myeloid-derived suppressor cells contribute to bone erosion in collagen-induced arthritis by differentiating to osteoclasts. J Autoimmun 65:82–89. https://doi.org/10.1016/j.jaut.2015.08.010
Yi H, Guo C, Yu X, Zuo D, Wang XY (2012) Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol 189:4295–4304. https://doi.org/10.4049/jimmunol.1200086
Raphael I, Nalawade S, Eagar TN, Forsthuber TG (2015) T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 74:5–17. https://doi.org/10.1016/j.cyto.2014.09.011
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13:156–163. https://doi.org/10.1038/nm1538
Kim KW, Kim HR, Kim BM, Cho ML, Lee SH (2015) Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am J Pathol 185:3011–3024. https://doi.org/10.1016/j.ajpath.2015.07.017
Jiao Z, Hua S, Wang W, Wang H, Gao J, Wang X (2013) Increased circulating myeloid-derived suppressor cells correlated negatively with Th17 cells in patients with rheumatoid arthritis. Scand J Rheumatol 42:85–90. https://doi.org/10.3109/03009742.2012.716450
Jiao Z, Wang W, Jia R, Li J, You H, Chen L, Wang Y (2007) Accumulation of FoxP3-expressing CD4+CD25+ T cells with distinct chemokine receptors in synovial fluid of patients with active rheumatoid arthritis. Scand J Rheumatol 36:428–433. https://doi.org/10.1080/03009740701482800
Zaiss MM, Axmann R, Zwerina J, Polzer K, Gückel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G (2007) Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum 56:4104–4112. https://doi.org/10.1002/art.23138
Yuan FL, Li X, Lu WG, Xu RS, Zhao YQ, Li CW, Li JP, Chen FH (2010) Regulatory T cells as a potent target for controlling bone loss. Biochem Biophys Res Commun 402:173–176. https://doi.org/10.1016/j.bbrc.2010.09.120
Kelchtermans H, Geboes L, Mitera T, Huskens D, Leclercq G, Matthys P (2009) Activated CD4+CD25+ regulatory T cells inhibit osteoclastogenesis and collagen-induced arthritis. Ann Rheum Dis 68:744–750. https://doi.org/10.1136/ard.2007.086066
Bozec A, Zaiss MM (2017) T regulatory cells in bone remodelling. Curr Osteoporos Rep 15:121–125. https://doi.org/10.1007/s11914-017-0356-1
Zhu L, Hua F, Ding W, Ding K, Zhang Y, Xu C (2020) The correlation between the Th17/Treg cell balance and bone health. Immun Ageing 17:30. https://doi.org/10.1186/s12979-020-00202-z
Tyagi AM, Yu M, Darby TM, Vaccaro C, Li JY, Owens JA, Hsu E, Adams J, Weitzmann MN, Jones RM, Pacifici R (2018) The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of WNT10B expression. Immunity 49:1116–31.e7. https://doi.org/10.1016/j.immuni.2018.10.013
Zhang L, Zhang Z, Zhang H, Wu M, Wang Y (2014) Myeloid-derived suppressor cells protect mouse models from autoimmune arthritis via controlling inflammatory response. Inflammation 37:670–677. https://doi.org/10.1007/s10753-013-9783-z
Tesmer LA, Lundy SK, Sarkar S, Fox DA (2008) Th17 cells in human disease. Immunol Rev 223:87–113. https://doi.org/10.1111/j.1600-065X.2008.00628.x
Hussein MR, Fathi NA, El-Din AM, Hassan HI, Abdullah F, Al-Hakeem E, Backer EA (2008) Alterations of the CD4(+), CD8 (+) T cell subsets, interleukins-1beta, IL-10, IL-17, tumor necrosis factor-alpha and soluble intercellular adhesion molecule-1 in rheumatoid arthritis and osteoarthritis: preliminary observations. Pathol Oncol Res 14:321–328. https://doi.org/10.1007/s12253-008-9016-1
Hot A, Zrioual S, Toh ML, Lenief V, Miossec P (2011) IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis 70:341–348. https://doi.org/10.1136/ard.2010.132233
Tran CN, Lundy SK, White PT, Endres JL, Motyl CD, Gupta R, Wilke CM, Shelden EA, Chung KC, Urquhart AG, Fox DA (2007) Molecular interactions between T cells and fibroblast-like synoviocytes: role of membrane tumor necrosis factor-alpha on cytokine-activated T cells. Am J Pathol 171:1588–1598. https://doi.org/10.2353/ajpath.2007.070004
van Hamburg JP, Corneth OB, Paulissen SM, Davelaar N, Asmawidjaja PS, Mus AM, Lubberts E (2013) IL-17/Th17 mediated synovial inflammation is IL-22 independent. Ann Rheum Dis 72:1700–1707. https://doi.org/10.1136/annrheumdis-2012-202373
van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, Dolhain RJ, Lubberts E (2011) Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum 63:73–83. https://doi.org/10.1002/art.30093
Brennan FM, Hayes AL, Ciesielski CJ, Green P, Foxwell BM, Feldmann M (2002) Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum 46:31–41. https://doi.org/10.1002/1529-0131(200201)46:1%3c31::Aid-art10029%3e3.0.Co;2-5
Honorati MC, Cattini L, Facchini A (2007) VEGF production by osteoarthritic chondrocytes cultured in micromass and stimulated by IL-17 and TNF-alpha. Connect Tissue Res 48:239–245. https://doi.org/10.1080/03008200701541767
Honorati MC, Neri S, Cattini L, Facchini A (2006) Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthr Cartil 14:345–352. https://doi.org/10.1016/j.joca.2005.10.004
Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D (2014) The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm 2014:561459. https://doi.org/10.1155/2014/561459
Park MJ, Lee SH, Kim EK, Lee EJ, Baek JA, Park SH, Kwok SK, Cho ML (2018) Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep 8:3753. https://doi.org/10.1038/s41598-018-21856-2
Cooles FA, Isaacs JD, Anderson AE (2013) Treg cells in rheumatoid arthritis: an update. Curr Rheumatol Rep 15:352. https://doi.org/10.1007/s11926-013-0352-0
Yudoh K, Matsuno H, Nakazawa F, Yonezawa T, Kimura T (2000) Reduced expression of the regulatory CD4+ T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis Rheum 43:617–627. https://doi.org/10.1002/1529-0131(200003)43:3%3c617::Aid-anr19%3e3.0.Co;2-b
Umulis D, O’Connor MB, Blair SS (2009) The extracellular regulation of bone morphogenetic protein signaling. Development 136:3715–3728. https://doi.org/10.1242/dev.031534
Blaney Davidson EN, van der Kraan PM, van den Berg WB (2007) TGF-beta and osteoarthritis. Osteoarthr Cartil 15:597–604. https://doi.org/10.1016/j.joca.2007.02.005
Paulissen SM, van Hamburg JP, Dankers W, Lubberts E (2015) The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine 74:43–53. https://doi.org/10.1016/j.cyto.2015.02.002
Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM (2000) M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 164:6166–6173. https://doi.org/10.4049/jimmunol.164.12.6166
Blom AB, van Lent PL, Libregts S, Holthuysen AE, van der Kraan PM, van Rooijen N, van den Berg WB (2007) Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum 56:147–157. https://doi.org/10.1002/art.22337
Zhang H, Lin C, Zeng C, Wang Z, Wang H, Lu J, Liu X, Shao Y, Zhao C, Pan J, Xu S, Zhang Y, Xie D, Cai D, Bai X (2018) Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann Rheum Dis 77:1524–1534. https://doi.org/10.1136/annrheumdis-2018-213450
Zhang H, Wang S, Huang Y, Wang H, Zhao J, Gaskin F, Yang N, Fu SM (2015) Myeloid-derived suppressor cells are proinflammatory and regulate collagen-induced arthritis through manipulating Th17 cell differentiation. Clin Immunol 157:175–186. https://doi.org/10.1016/j.clim.2015.02.001
Qian J, Xu L, Sun X, Wang Y, Xuan W, Zhang Q, Zhao P, Wu Q, Liu R, Che N, Wang F, Tan W, Zhang M (2018) Adiponectin aggravates bone erosion by promoting osteopontin production in synovial tissue of rheumatoid arthritis. Arthritis Res Ther 20:26. https://doi.org/10.1186/s13075-018-1526-y
Gao SG, Li KH, Zeng KB, Tu M, Xu M, Lei GH (2010) Elevated osteopontin level of synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis patients. Osteoarthr Cartil 18:82–87. https://doi.org/10.1016/j.joca.2009.07.009
Betz VM, Betz OB, Harris MB, Vrahas MS, Evans CH (2008) Bone tissue engineering and repair by gene therapy. Front Biosci 13:833–841. https://doi.org/10.2741/2724
Marsell R, Einhorn TA (2011) The biology of fracture healing. Injury 42:551–555. https://doi.org/10.1016/j.injury.2011.03.031
Mountziaris PM, Spicer PP, Kasper FK, Mikos AG (2011) Harnessing and modulating inflammation in strategies for bone regeneration. Tissue Eng Part B Rev 17:393–402. https://doi.org/10.1089/ten.TEB.2011.0182
Zhang X, Sarkar K, Rey S, Sebastian R, Andrikopoulou E, Marti GP, Fox-Talbot K, Semenza GL, Harmon JW (2011) Aging impairs the mobilization and homing of bone marrow-derived angiogenic cells to burn wounds. J Mol Med (Berl) 89:985–995. https://doi.org/10.1007/s00109-011-0754-2
Kawai H, Oo MW, Tsujigiwa H, Nakano K, Takabatake K, Sukegawa S, Nagatsuka H (2021) Potential role of myeloid-derived suppressor cells in transition from reaction to repair phase of bone healing process. Int J Med Sci 18:1824–1830. https://doi.org/10.7150/ijms.51946
Ochoa JB, Bernard AC, O’Brien WE, Griffen MM, Maley ME, Rockich AK, Tsuei BJ, Boulanger BR, Kearney PA, Morris SM Jr (2001) Arginase I expression and activity in human mononuclear cells after injury. Ann Surg 233:393–399. https://doi.org/10.1097/00000658-200103000-00014
Levy S, Feduska JM, Sawant A, Gilbert SR, Hensel JA, Ponnazhagan S (2016) Immature myeloid cells are critical for enhancing bone fracture healing through angiogenic cascade. Bone 93:113–124. https://doi.org/10.1016/j.bone.2016.09.018
Wang W, Zuo R, Long H, Wang Y, Zhang Y, Sun C, Luo G, Zhang Y, Li C, Zhou Y, Li J (2020) Advances in the Masquelet technique: Myeloid-derived suppressor cells promote angiogenesis in PMMA-induced membranes. Acta Biomater 108:223–236. https://doi.org/10.1016/j.actbio.2020.03.010
Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12:895–904. https://doi.org/10.1038/nm1469
Singh M, Ramos I, Asafu-Adjei D, Quispe-Tintaya W, Chandra D, Jahangir A, Zang X, Aggarwal BB, Gravekamp C (2013) Curcumin improves the therapeutic efficacy of Listeria(at)-Mage-b vaccine in correlation with improved T-cell responses in blood of a triple-negative breast cancer model 4T1. Cancer Med 2:571–582. https://doi.org/10.1002/cam4.94
Cao Y, Slaney CY, Bidwell BN, Parker BS, Johnstone CN, Rautela J, Eckhardt BL, Anderson RL (2014) BMP4 inhibits breast cancer metastasis by blocking myeloid-derived suppressor cell activity. Cancer Res 74:5091–5102. https://doi.org/10.1158/0008-5472.Can-13-3171
Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA (2009) Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS ONE 4:e7669. https://doi.org/10.1371/journal.pone.0007669
Simpson KD, Templeton DJ, Cross JV (2012) Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol 189:5533–5540. https://doi.org/10.4049/jimmunol.1201161
Netherby CS, Abrams SI (2017) Mechanisms overseeing myeloid-derived suppressor cell production in neoplastic disease. Cancer Immunol Immunother 66:989–996. https://doi.org/10.1007/s00262-017-1963-5
Moorman HR, Poschel D, Klement JD, Lu C, Redd PS, Liu K (2020) Osteopontin: a key regulator of tumor progression and immunomodulation. Cancers (Basel). https://doi.org/10.3390/cancers12113379
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268. https://doi.org/10.1038/nri3175
Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI (2006) All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res 66:9299–9307. https://doi.org/10.1158/0008-5472.Can-06-1690
Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI (2005) Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res 65:9525–9535. https://doi.org/10.1158/0008-5472.Can-05-0529
Daurkin I, Eruslanov E, Vieweg J, Kusmartsev S (2010) Generation of antigen-presenting cells from tumor-infiltrated CD11b myeloid cells with DNA demethylating agent 5-aza-2’-deoxycytidine. Cancer Immunol Immunother 59:697–706. https://doi.org/10.1007/s00262-009-0786-4
Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY (2010) A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res 16:4583–4594. https://doi.org/10.1158/1078-0432.Ccr-10-0733
Wang Y, Jia A, Bi Y, Wang Y, Yang Q, Cao Y, Li Y, Liu G (2020) Targeting myeloid-derived suppressor cells in cancer immunotherapy. Cancers (Basel). https://doi.org/10.3390/cancers12092626
Trovato R, Fiore A, Sartori S, Canè S, Giugno R, Cascione L, Paiella S, Salvia R, De Sanctis F, Poffe O, Anselmi C, Hofer F, Sartoris S, Piro G, Carbone C, Corbo V, Lawlor R, Solito S, Pinton L, Mandruzzato S, Bassi C, Scarpa A, Bronte V, Ugel S (2019) Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer 7:255. https://doi.org/10.1186/s40425-019-0734-6
Zhang D, Rennhack J, Andrechek ER, Rockwell CE, Liby KT (2018) Identification of an unfavorable immune signature in advanced lung tumors from Nrf2-deficient mice. Antioxid Redox Signal 29:1535–1552. https://doi.org/10.1089/ars.2017.7201
Hiramoto K, Satoh H, Suzuki T, Moriguchi T, Pi J, Shimosegawa T, Yamamoto M (2014) Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev Res (Phila) 7:835–844. https://doi.org/10.1158/1940-6207.Capr-14-0094
Creelan BC, Gabrilovich DI, Gray JE, Williams CC, Tanvetyanon T, Haura EB, Weber JS, Gibney GT, Markowitz J, Proksch JW, Reisman SA, McKee MD, Chin MP, Meyer CJ, Antonia SJ (2017) Safety, pharmacokinetics, and pharmacodynamics of oral omaveloxolone (RTA 408), a synthetic triterpenoid, in a first-in-human trial of patients with advanced solid tumors. Onco Targets Ther 10:4239–4250. https://doi.org/10.2147/ott.S136992
Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, Sporn MB, Liby KT, Rawal B, Lee JH, Gabrilovich DI (2010) Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res 16:1812–1823. https://doi.org/10.1158/1078-0432.Ccr-09-3272
Romano A, Parrinello NL, La Cava P, Tibullo D, Giallongo C, Camiolo G, Puglisi F, Parisi M, Pirosa MC, Martino E, Conticello C, Palumbo GA, Di Raimondo F (2018) PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev Mol Diagn 18:675–683. https://doi.org/10.1080/14737159.2018.1470929
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC (2005) Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 202:931–939. https://doi.org/10.1084/jem.20050715
Peak TC, Richman A, Gur S, Yafi FA, Hellstrom WJ (2016) The role of PDE5 inhibitors and the NO/cGMP pathway in cancer. Sex Med Rev 4:74–84. https://doi.org/10.1016/j.sxmr.2015.10.004
Yu J, Du W, Yan F, Wang Y, Li H, Cao S, Yu W, Shen C, Liu J, Ren X (2013) Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 190:3783–3797. https://doi.org/10.4049/jimmunol.1201449
Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, Raubitschek A, Hoon DSB, Forman S, Figlin RA, Liu J, Jove R, Yu H (2012) S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 21:642–654. https://doi.org/10.1016/j.ccr.2012.03.039
Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S (2011) Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 117:5381–5390. https://doi.org/10.1182/blood-2010-11-321752
Hu X, Bardhan K, Paschall AV, Yang D, Waller JL, Park MA, Nayak-Kapoor A, Samuel TA, Abrams SI, Liu K (2013) Deregulation of apoptotic factors Bcl-xL and Bax confers apoptotic resistance to myeloid-derived suppressor cells and contributes to their persistence in cancer. J Biol Chem 288:19103–19115. https://doi.org/10.1074/jbc.M112.434530
Guha P, Gardell J, Darpolor J, Cunetta M, Lima M, Miller G, Espat NJ, Junghans RP, Katz SC (2019) STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene 38:533–548. https://doi.org/10.1038/s41388-018-0449-z
Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, El-Deiry WS, Winograd R, Vonderheide RH, English NR, Knight SC, Yagita H, McCaffrey JC, Antonia S, Hockstein N, Witt R, Masters G, Bauer T, Gabrilovich DI (2014) ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest 124:2626–2639. https://doi.org/10.1172/jci74056
Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, Marshall B, Schoenlein PV, Lebedyeva IO, Liu K (2016) Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget 7:83907–25. https://doi.org/10.18632/oncotarget.13438
Smith AD, Lu C, Payne D, Paschall AV, Klement JD, Redd PS, Ibrahim ML, Yang D, Han Q, Liu Z, Shi H, Hartney TJ, Nayak-Kapoor A, Liu K (2020) Autocrine IL6-mediated activation of the STAT3-DNMT axis silences the TNFα-RIP1 necroptosis pathway to sustain survival and accumulation of myeloid-derived suppressor cells. Cancer Res 80:3145–3156. https://doi.org/10.1158/0008-5472.Can-19-3670
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM (2005) Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 11:6713–6721. https://doi.org/10.1158/1078-0432.Ccr-05-0883
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F (2010) 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 70:3052–3061. https://doi.org/10.1158/0008-5472.Can-09-3690
Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC, Ries RE, Aplenc R, Hirsch BA, Raimondi SC, Walter RB, Bernstein ID, Gamis AS, Alonzo TA, Meshinchi S (2017) CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from Randomized Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol 35:2674–2682. https://doi.org/10.1200/jco.2016.71.2513
Liang H, Shen X (2020) LXR activation radiosensitizes non-small cell lung cancer by restricting myeloid-derived suppressor cells. Biochem Biophys Res Commun 528:330–335. https://doi.org/10.1016/j.bbrc.2020.04.137
Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, Qian J, Hailemichael Y, Nurieva R, Dwyer KC, Roth J, Yi Q, Overwijk WW, Kwak LW (2014) Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med 20:676–681. https://doi.org/10.1038/nm.3560
Edgington-Mitchell LE, Rautela J, Duivenvoorden HM, Jayatilleke KM, van der Linden WA, Verdoes M, Bogyo M, Parker BS (2015) Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid-derived suppressor cells in breast cancer. Oncotarget 6:27008–22. https://doi.org/10.18632/oncotarget.4714
Sun X, Xie Z, Hu B, Zhang B, Ma Y, Pan X, Huang H, Wang J, Zhao X, Jie Z, Shi P, Chen Z (2020) The Nrf2 activator RTA-408 attenuates osteoclastogenesis by inhibiting STING dependent NF-κb signaling. Redox Biol 28:101309. https://doi.org/10.1016/j.redox.2019.101309
Sawant A, Ponnazhagan S (2013) Myeloid-derived suppressor cells as a novel target for the control of osteolytic bone disease. Oncoimmunology 2:e24064. https://doi.org/10.4161/onci.24064
Mirabello L, Troisi RJ, Savage SA (2009) Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 115:1531–1543. https://doi.org/10.1002/cncr.24121
Uehara T, Eikawa S, Nishida M, Kunisada Y, Yoshida A, Fujiwara T, Kunisada T, Ozaki T, Udono H (2019) Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int Immunol 31:187–198. https://doi.org/10.1093/intimm/dxy079
Jiang K, Li J, Zhang J, Wang L, Zhang Q, Ge J, Guo Y, Wang B, Huang Y, Yang T, Hao D, Shan L (2019) SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int Immunopharmacol 75:105818. https://doi.org/10.1016/j.intimp.2019.105818
Shi X, Li X, Wang H, Yu Z, Zhu Y, Gao Y (2019) Specific inhibition of PI3Kδ/γ enhances the efficacy of anti-PD1 against osteosarcoma cancer. J Bone Oncol 16:100206. https://doi.org/10.1016/j.jbo.2018.11.001
Guan Y, Zhang R, Peng Z, Dong D, Wei G, Wang Y (2017) Inhibition of IL-18-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy against osteosarcoma cancer. J Bone Oncol 9:59–64. https://doi.org/10.1016/j.jbo.2017.10.002
Deng C, Xu Y, Fu J, Zhu X, Chen H, Xu H, Wang G, Song Y, Song G, Lu J, Liu R, Tang Q, Huang W, Wang J (2020) Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci 111:1899–1909. https://doi.org/10.1111/cas.14398
Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, El-Etriby R, Galli S, Tsokos MG, Orentas RJ, Mackall CL (2016) Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res 4:869–880. https://doi.org/10.1158/2326-6066.Cir-15-0230
Ligon JA, Choi W, Cojocaru G, Fu W, Hsiue EH, Oke TF, Siegel N, Fong MH, Ladle B, Pratilas CA, Morris CD, Levin A, Rhee DS, Meyer CF, Tam AJ, Blosser R, Thompson ED, Suru A, McConkey D, Housseau F, Anders R, Pardoll DM, Llosa N (2021) Pathways of immune exclusion in metastatic osteosarcoma are associated with inferior patient outcomes. J Immunother Cancer. https://doi.org/10.1136/jitc-2020-001772
Kraan MC, Reece RJ, Smeets TJ, Veale DJ, Emery P, Tak PP (2002) Comparison of synovial tissues from the knee joints and the small joints of rheumatoid arthritis patients: Implications for pathogenesis and evaluation of treatment. Arthritis Rheum 46:2034–2038. https://doi.org/10.1002/art.10556
Abbasi M, Mousavi MJ, Jamalzehi S, Alimohammadi R, Bezvan MH, Mohammadi H, Aslani S (2019) Strategies toward rheumatoid arthritis therapy; the old and the new. J Cell Physiol 234:10018–10031. https://doi.org/10.1002/jcp.27860
Smolen JS, Aletaha D, McInnes IB (2016) Rheumatoid arthritis. Lancet 388:2023–2038. https://doi.org/10.1016/s0140-6736(16)30173-8
Kurkó J, Vida A, Ocskó T, Tryniszewska B, Rauch TA, Glant TT, Szekanecz Z, Mikecz K (2014) Suppression of proteoglycan-induced autoimmune arthritis by myeloid-derived suppressor cells generated in vitro from murine bone marrow. PLoS ONE 9:e111815. https://doi.org/10.1371/journal.pone.0111815
Sun J, Xu P, Du X, Zhang Q, Zhu Y (2015) Piperlongumine attenuates collagen-induced arthritis via expansion of myeloid-derived suppressor cells and inhibition of the activation of fibroblast-like synoviocytes. Mol Med Rep 11:2689–2694. https://doi.org/10.3892/mmr.2014.3001
Zhu D, Tian J, Wu X, Li M, Tang X, Rui K, Guo H, Ma J, Xu H, Wang S (2019) G-MDSC-derived exosomes attenuate collagen-induced arthritis by impairing Th1 and Th17 cell responses. Biochim Biophys Acta Mol Basis Dis 1865:165540. https://doi.org/10.1016/j.bbadis.2019.165540
Zhao B, Takami M, Yamada A, Wang X, Koga T, Hu X, Tamura T, Ozato K, Choi Y, Ivashkiv LB, Takayanagi H, Kamijo R (2009) Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med 15:1066–1071. https://doi.org/10.1038/nm.2007
Miller RJ, Malfait AM, Miller RE (2020) The innate immune response as a mediator of osteoarthritis pain. Osteoarthr Cartil 28:562–571. https://doi.org/10.1016/j.joca.2019.11.006
Castañeda S, Roman-Blas JA, Largo R, Herrero-Beaumont G (2012) Subchondral bone as a key target for osteoarthritis treatment. Biochem Pharmacol 83:315–323. https://doi.org/10.1016/j.bcp.2011.09.018
Hoemann CD, Chen G, Marchand C, Tran-Khanh N, Thibault M, Chevrier A, Sun J, Shive MS, Fernandes MJ, Poubelle PE, Centola M, El-Gabalawy H (2010) Scaffold-guided subchondral bone repair: implication of neutrophils and alternatively activated arginase-1+ macrophages. Am J Sports Med 38:1845–1856. https://doi.org/10.1177/0363546510369547
Marik PE, Flemmer M (2012) The immune response to surgery and trauma: Implications for treatment. J Trauma Acute Care Surg 73:801–808. https://doi.org/10.1097/TA.0b013e318265cf87
Allen MR, Hock JM, Burr DB (2004) Periosteum: biology, regulation, and response to osteoporosis therapies. Bone 35:1003–1012. https://doi.org/10.1016/j.bone.2004.07.014
Cuthbert RJ, Churchman SM, Tan HB, McGonagle D, Jones E, Giannoudis PV (2013) Induced periosteum a complex cellular scaffold for the treatment of large bone defects. Bone 57:484–492. https://doi.org/10.1016/j.bone.2013.08.009
Boros P, Ochando JC, Chen SH, Bromberg JS (2010) Myeloid-derived suppressor cells: natural regulators for transplant tolerance. Hum Immunol 71:1061–1066. https://doi.org/10.1016/j.humimm.2010.08.001
Zhang J, Hodges A, Chen SH, Pan PY (2021) Myeloid-derived suppressor cells as cellular immunotherapy in transplantation and autoimmune diseases. Cell Immunol 362:104300. https://doi.org/10.1016/j.cellimm.2021.104300
Funding
This work was supported by the Medical Innovation of Graduate Students in Chongqing (Grant No. CYS19360).
Author information
Authors and Affiliations
Contributions
ZGL and CY were major contributors in writing the manuscript. ZGL, CD and CY created all the figures and tables. ZGL, JLT and CY performed literature search. ZGL and YQC made substantial contributions to the design of the manuscript and revised it critically for important intellectual content. All authors have read and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ling, Z., Yang, C., Tan, J. et al. Beyond immunosuppressive effects: dual roles of myeloid-derived suppressor cells in bone-related diseases. Cell. Mol. Life Sci. 78, 7161–7183 (2021). https://doi.org/10.1007/s00018-021-03966-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03966-9