
Abstract
The skeleton is a common destination for many cancer metastases including breast and prostate cancer. There are many characteristics of bone that make it an ideal environment for cancer cell migration and colonization. Metaphyseal bone, found at the ends of long bone, in ribs, and in vertebrae, is comprised of trabecular bone interspersed with marrow and rich vasculature. The specialized microvasculature is adapted for the easy passage of cells in and out of the bone marrow. Moreover, the metasphyseal regions of bone are constantly undergoing remodeling, a process that releases growth factors from the matrix. Bone turnover also involves the production of numerous cytokines and chemokines that provide a means of communication between osteoblasts and osteoclasts, but co-incidentally can also attract and support metastatic cells. Once in the marrow, cancer cells can interact directly and indirectly with osteoblasts and osteclasts, as well as hematopoietic and stromal cells. Cancer cells secrete factors that affect the network of cells in the bone microenvironment as well as interact with other cytokines. Additionally, transient cells of the immune system may join the local mileau to ultimately support cancer cell growth. However, most metastasized cells that enter the bone marrow are transient; a few may remain in a dormant state for many years. Advances in understanding the bone cell-tumor cell interactions are key to controlling, if not preventing metastasis to bone.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Tumor cell metastasis
The skeleton is a favored site of metastasis for a number of common tumors. Bone metastases are by far more prevalent than primary tumors of the bone. Based on post-mortem examination, approximately 70% of patients who die from breast or prostate cancer have bone metastases [1]. The incidences from thyroid, kidney, and lung cancer also are high (about 40%). In contrast, it has been noted that bone metastases from cancers of the gastrointestinal tract are uncommon. In many cases, cancer cell metastases are diagnosed in patients before diagnosis of the primary disease. A better understanding of the specificity and the pathogenesis of metastasis will allow for better therapeutic treatments and quality of life for patients.
The metastasis of a primary tumor to distant organs requires a series of coordinated steps. Proliferation of the primary tumor is supported by tumor autocrine factors or local growth factors, such as vascular endothelial growth factor (VEGF), tumor growth factor-beta (TGF-β), and interleukin-6 (IL-6). For a tumor to reach a clinically detectable size, localized neovascularization or angiogenesis must occur. The development of new blood vessels provide an endless supply of nutrients as well as a route for tumor cell migration to secondary sites. Subsequently, local invasion takes place, which is accomplished by the destruction of the extracellular matrix, including the basement membrane and connective tissue. The process of invasion through a basement membrane is a hallmark characteristic of a metastatic cell. Additionally, tumor cells experience increased motility and reduced adherence allowing them to migrate into lymph or blood vessels. Intravasation into blood vessels at the primary site may occur as a result of excess force or response to a soluble chemotactic factor gradient. After circulating through the vasculature, tumor cells may adhere to vessel endothelium of the target organ and extravasate into the tissue. This movement is facilitated by cancer cell secretion of matrix metalloproteinases (MMPs) and cathepsin-K that destroy surrounding tissue. Finally, tumor cells thrive at the secondary site, a defining characteristic of metastatic tumor cells, only if there is an appropriate environment of paracrine or autocrine factors that aid in growth and vascularization [2–4]. The distribution pattern of cancer cells to the bone is believed to be due to the venous flow from breast and prostate towards the vena cava and into the vertebral venous plexus [5]. Once in the circulation, entry of the cancer cells into the venous circulation of the bone marrow may be facilitated by the slow blood flow and particular anatomy of the venous sinusoids. Nonetheless these steps alone do not explain survival and growth of the cancer cells in the bone.
2 Bone structure
In order to understand the bone-tumor microenvironment, one must consider bone structure and function. Bone is a specialized type of connective tissue, which provides structural support, protective functions, and plays a major role in the regulation of calcium levels in the body [6]. Type I collagen accounts for 95% of the organic bone matrix [7]. The remaining 5% includes proteoglycans and a variety of other non-collagenous proteins [6]. This largely collagenous matrix is hardened through the mineralization process, in which hydroxyapatite (3Ca[PO4]2[OH]2) crystals are deposited in the organic matrix [8]. Mineralization increases bone resistance to compression [9], and also contains numerous growth factors, including TGF-β, which are released upon bone resorption [10].
The bones of the body are classified as long bones (e.g. the tibia, femur, and humerus) and flat bones (e.g. the skull, ileum, and mandible). Both types contain cortical and trabecular bone, albeit in different concentrations. Cortical bone, the compact, dense outer protective layer of bone, is made up of tightly packed collagen fibrils [6]. This form of bone is vital for supporting the weight load of the body. On the other hand, trabecular bone, also known as cancellous bone, has a loosely organized, porous matrix and is located in the interior of bone, near the ends. Trabecular bone is metabolically active. All bone matrix undergoes remodeling, but trabecular bone has a greater turnover rate than cortical bone [6].
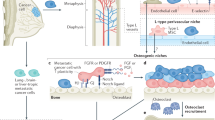
Long bones are divided into the diaphysis, metaphysis, and epiphysis in a growing individual [11] (Fig. 1). The long bone ends, or epiphyses, are located above the growth plate, where bone elongation occurs. The diaphysis, which is the long, narrow shaft of the bone, is primarily composed of cortical bone. The metaphysis, located near the ends of bones just below the growth plate, is predominantly composed of trabecular bone and is surrounded by hematopoietic marrow, fatty marrow, and blood vessels [11].

Diagram of a long bone indicating the major regions and the major structures within the metaphysis, i.e. trabecular bone and the sinusoidal endothelium
3 Cells in the bone microenvironment
Bone is a dynamic structure that undergoes constant remodeling in order to respond to mechanical strain and maintain calcium homeostasis. Bone resorption and deposition occur in a tightly regulated fashion that is orchestrated by three cell types: osteoblasts, osteocytes, and osteoclasts. Osteoblasts are derived from mesenchymal stem cells located in the bone marrow stroma. They synthesize osteoid (i.e. new bone matrix), comprised primarily of collagen and non-collagenous proteins, and also aid in mineralization of the bone matrix. Upon stimulation by bone morphogenetic proteins and local growth factors, the mesenchymal stem cells proliferate and form pre-osteoblasts, which subsequently differentiate into mature osteoblasts [12]. After synthesizing new bone matrix, the osteoblast either undergoes apoptosis or becomes embedded in the bone as an osteocyte [13]. These cells have long processes that allow them to remain in contact with other osteocytes and with osteoblasts that line the bone surface. The processes connect the entire matrix through a series of canaliculi [14, 15].
Osteoclasts, responsible for bone resorption, are derived from monocytes in the bone marrow [16]. Monocytes are activated to form osteoclasts through osteoblasts. Osteoblasts express the receptor-activator for NF-κB ligand (RANK-L) on their external surfaces; RANK-L binds to the receptor RANK found on the surface of monocytes. In the presence of macrophage colony stimulating factor (M-CSF), RANK-L promotes cellular fusion of several monocytes to form a multinucleated osteoclast [16]. Activated osteoclasts bind to the bone matrix through αvβ3, αvβ5, α2β1 integrins located on the membrane surface and also secrete acid and lysosomal enzymes which degrade bone [13, 16].
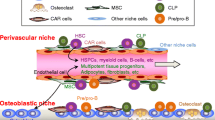
Other cell types located within the bone microenvironment may also contribute to the bone metastatic niche. These cells can generally be grouped into two categories: stromal and transient. Mesenchymal stem cells in the bone marrow give rise to stromal cells which can differentiate into adipocytes, fibroblasts, chrondrocytes, or osteoblasts. Stromal cells have been found to support the differentiation, proliferation, and survival of both hematopoetic and cancer cells. In particular, it has been found that stromal cells express vascular cell adhesion molecule (VCAM-1). Michigami and colleagues discovered that the presence of VCAM-1 on stromal cells increased the production of bone-resorbing cytokines by myeloma cells. Neutralizing antibody to VCAM-1 or to α4β1 integrin reduced osteolysis [17]. The adipocyte has been found to secrete tumor necrosis factor-alpha (TNF-α), IL-6, and leptin, which stimulate bone resorption and inhibit osteoblast proliferation [18, 19]. Factors secreted by adipocytes have also been implicated in breast cancer proliferation, invasiveness, survival, and angiogenesis [19]. Another type of mesenchymal cell, the fibroblast, has been shown to affect breast cancer cell invasion and contribute to metastatic bone disease. Fibroblast-secreted syndecan-1 was found to increase breast cancer cell proliferation in vivo [20]. In addition, fibroblasts secrete inactive MMP-2, which can be activated by breast cancer cells to subsequently increase their invasiveness and migration [21]. Fibroblasts have also been found to stimulate osteoclasts, and thus bone resorption, in a RANK-L dependent manner [22].
Vascular endothelial cells contribute to the formation of a favorable cancer cell microenvironment. New blood vessels, which arise from endothelial cells, are essential for the survival of cancer cells. Investigators have shown that the bone marrow, which has a high microvessel density, is associated with increased bone-tumor metastasis and survival of tumor cells [23]. It has also been found that many tumor-cell secreted factors stimulate endothelial cell proliferation, differentiation, and angiogenesis [23], thus producing a feedback loop that facilitates tumor cell survival in a secondary location.
Transient cells also contribute to the metastatic bone microenvironment. These cells include erythrocytes, T cells, and platelets, all derived from hematopoietic stem cells. In one study it was shown that platelets were directed by MDA-MB-231, a metastatic breast cancer cell line, to secrete LPA (Lysophosphatidic acid ([1-acyl-sn-glycero-3-phosphate]), a phospholipid with diverse biological activities [24]. Overexpression of this molecule and its receptor has been shown to increase tumor growth and metastasis [25]. Platelets may also adhere to cancer cells in the blood stream, allowing them to evade natural killer immune cell surveillance [26]. In addition, platelets may aid in cancer cell attachment to vascular endothelial cell walls [26]. T lymphocytes have been found to express RANKL, also known as TRANCE, and aid in osteoclast formation and activation [16, 27]. Peripheral T cells also secrete TNF-α, which is involved in osteoclastogenesis, inhibits osteoblast cell differentiation, and is a pro-apoptotic factor for osteoblasts [27, 28]. Besides enhancing osteoclastic bone resorption, T cells may be affected by bone metastatic cancer cells as well. As bone resorption is enhanced, TGF-β is released from the bone. This factor can inhibit both T cell proliferation and activity, and natural killer cell function [29]. Thus the immune response is suppressed and tumor cells may escape surveillance. In addition, tumor cell-secreted parathyroid hormone related peptide (PTHrP) and IL-8 may activate T cells, thus enhancing bone resorption and suppressing T cell function [29]. Plasma cells, or antibody-producing B cells, upregulate the receptor CXCR4 upon completion of differentiation [27]. Breast cancer cells, as well as stromal cells, express the ligand to this receptor CXCL12 [Stromal-derived factor-1 (SDF-1)] [27]. This ligand-receptor interaction may facilitate cancer cell migration into and within the bone microenvironment.
Tumor-associated macrophages (TAMs) are an important component of the inflammatory response in tissues [30]. These cells are derived from monocytes and are recruited by monocyte chemotactic peptide (MCP) chemokines [30]. Although activated TAMs kill many cancerous cells through secretion of IL-2, interferon, and IL-12, they also secrete a variety of potent angiogenic and lymphangiogenic growth factors, cytokines, and proteases [31, 32], all of which are involved in the promotion of tumor cell growth and survival. TAMs, as well as tumor cells, produce IL-10 which suppresses the anti-tumor response of cytotoxic T cells [33]. TAMs also induce the expression of VCAM-1 on mesothelial cells, which can aid in tumor cell invasion [34].
4 Bone remodeling
Mineralized bone has an abundance of growth factors, calcium ions, cell adhesion molecules, cytokines, and chemokines that, when released into the microenvironment during bone remodeling, make the skeleton an attractive site for metastatic cancer cells. This observation was first described in 1889 by Stephen Paget, who recognized the nonrandom movement of cancer cells within the body that was unexplained by blood flow. Paget stated “When a plant goes to seed, its seeds are carried in all directions; but they can only grow if they fall on congenial soil” [35]. This ‘seed and soil’ hypothesis helps to explain the preferential metastasis of certain types of cancers to the bone microenvironment, which provides a fertile soil where cancer cells can grow. Osteoclasts further contribute to this environment by acting as plows to break up the ‘soil’ and release its ‘nutrients’ for cancer cell growth and maintenance. Studies have shown that there is a close relationship between bone resorption and tumor cell growth [36].
The relative activities of osteoblasts and osteoclasts are normally tightly coupled in order to maintain a balance between bone formation and degradation. Bone remodeling is regulated both by systemic hormones and locally produced cytokines [37]. Cells in the bone marrow, especially stromal and immune cells, produce cytokines and growth factors that influence the activities of osteoblasts and osteoclasts [38]. However, this balance between bone synthesis and resorption is disturbed in several pathological conditions, including osteoporosis, rheumatoid arthritis, and skeletal metastases, resulting in osteoclast activity in excess of bone deposition by osteoblasts with net bone loss [38].
Osteoclasts likely prime the bone microenvironment for tumor cell growth through bone resorption. Although there is no definitive evidence linking increased bone resorption to increased tumor cell mass, a variety of studies have been carried out with bisphosphonates to investigate this relationship. Bisphosphonates are inorganic pyrophosphates with powerful inhibitory effects on bone resorption. One of their main targets is the osteoclast. Powles et al. showed that the administration of a bisphosphonate, clodronate, was associated with both decreased bone metastasis and death rate in patients with breast cancer [39]. Risedronate was found to reduce tumor burden in addition to osteolytic bone lesions in a nude mouse model [40]. Furthermore, nude mice treated with neutralizing antibodies to PTHrP (key factor involved in the ‘vicious cycle’ of bone metastasis), also experienced a decrease in tumor burden compared to controls [41, 42]. Even though inhibitors to bone resorption seem to reduce tumor burden in bone, the same does not hold true for soft tissues [39]. In the study carried out by Powles et al., the idea that inhibitors of osteolysis slowed tumor growth in soft tissue was refuted [39]. In addition, there are pre-clinical data suggesting that bisphosphonates have no effect on tumor burden in soft tissues if the drugs are administered after the metastases are already formed [3]. Taken together, these studies illustrate the importance of osteoclasts and the uniqueness of the bone microenvironment for tumor cell growth.
In patients with bone metastatic cancer, usually one of two types of lesions predominate: osteolytic or osteoblastic, although mixed lesions may occur especially early on [36]. During osteolytic metastases, typical for breast cancer metastases, osteoclastogenesis and osteoclast activation results from direct and indirect actions by metastatic cancer cells. Increased bone resorption results. Mastro and colleagues additionally found that bone metastatic breast cancer cells suppress osteoblast function, which includes decreased matrix deposition, decreased proliferation, altered adhesion, and loss of osteoblast differentiation [43–45]. These phenomanae together favor a microenvironment of increased bone turnover with decreased bone deposition similar to that seen in osteoporosis.
On the other hand, cancers such as prostate cancer tend to be predominantly osteoblastic in nature. Excess bone deposition occurs but not necessarily in an ordered fashion. While little is known about the exact mechanisms, endothelin-1 has been implicated in osteoblastic breast cancer metastases [46, 47]. Endothelin-1 has been found to stimulate the formation of new bone through osteoblast proliferation [48], and serum endothelin-1 levels were found to be increased in patients with osteoblastic prostate cancer metastases [49]. In addition to endothelin-1, platelet derived growth factor-BB (PDGF-BB) may also be a mediator for osteoblastic metastases. In a study done by Yi et al., increased expression of PDGF-BB by human metastatic breast cancer cells was correlated with increased bone formation [50]. The exact mechanism has yet to be determined. Regardless of the lesion type, these observations underscore the importance of crosstalk between cancer cells and the bone microenvironment which facilitates bone metastases.
Clinically, patients with breast cancer and other osteolytic metastases but also with osteoblastic prostate bone metastases are treated with bisphosphonate drugs that block osteoclast activity. However, therapies utilizing bisphosphonates, such as ibandronate (Boniva™), are not curative [51]. Lesion progression is slowed, but the pre-existing lesions do not heal [52–55]. Severe bone pain, fractures, hypercalcemia, and spinal cord compression may still occur [3, 10, 56, 57]. The inability of bone to regenerate following bisphosphonate treatment supports the in vitro finding that breast cancer cells alter osteoblast function in addition to stimulating osteoclast activity.
Not all bones of the skeleton are equally favored for metastases. The spinal vertebrate, ribs, and the ends of long bones are preferred destinations of metastases. In general, well vascularized areas and areas of the skeleton containing red marrow are the sites of metastatic colonization. In osteoblastic metastases, bone deposition is usually made on trabecular bone surfaces without prior removal of old bone, but it may also be at sites of prior resorption. In myeloma, a malignancy of B cells, the plasma cells accumulate in the bone marrow and lead to osteolysis. Activation of osteoclasts and bone resorption far exceed bone deposition in this disease. Whether the lesions are overall lytic or blastic, the outcome is bone pain, pathological fracture, nerve compression syndromes and hypercalcemia [58].
5 The metaphyseal region of the bone has unique properties that distinguish it from the diaphysis
Scintigrams of humans with advanced disease clearly indicate that highly vascularized metaphyseal bone is a preferred site for secondary metastasis [1]. A trafficking study with a nude mouse model of metastatic breast cancer revealed that within 2 h of an inoculation of a bolus of metastatic breast cancer cells into the left ventricle of the heart, cancer cells were detected throughout the femur. However, by 24 h they had been cleared from the shaft (diaphysis) but remained at the metaphysis where they grew into large tumor masses [59]. The unique properties of the bone metaphysis make it an attractive site for metastatic cancer cells.
6 Metaphyseal bone: structure and vascularity
Metaphyseal bone is a highly vascularized structure found near the ends of long bone, in ribs and in vertebrate. It is composed of a network of thin bone spicules, sometimes referred to as “spongy bone.” In long bones, these spicules appear as mineralized fingers interspersed with red marrow and are in close proximity to the blood supply. The marrow contains hematopoietic, mesenchymal, and stromal cells. The vascular supply is sinusoidal in nature rather than a bed of capillaries. Lining the trabecular bone surfaces are osteoblasts and bone lining cells which share many properties [60]. Bone lining cells are believed to differentiate into osteoblasts when necessary for bone remodeling. The osteoblasts, as well as the marrow cells, provide an environment rich in growth factors, cytokines and chemotactic factors. These factors, and the vascular structure of the trabecular bone, are crucial for metastatic cancer cell colonization and growth.
The interactions of metastatic breast cancer cells with the vasculature has recently been well documented by Glinsky [61] and is only briefly summarized here. Metastatic foci are often seen where the sinusoid microvasculature is abundant [62]. This phenomenon is likely related to the unique anatomic, hemodynamic, and epithelial properties of the metaphyseal vascular bed. For one, the vasculature does not end in capillaries of small diameter as in most tissues. Instead it consists of voluminous sinusoids with lumens many times the diameter of cancer cells [63]. The sinusoids are within a few microns of the trabecular bone [64]. This unique structure leads to a sluggish flow of blood compared to that seen in the capillary networks of most other tissues [63]. For example, Mazo et al. found the blood flow in venous sinusoids of mouse calvaria to be 30 fold lower than the arterial rate. In another animal model, blood flow rates in canine long bone were assessed with microspheres [65]. It was found that metaphyseal and marrow cavity flow rates in sinusoids were 7–14 ml/min/g tissue, much slower than more rapidly metastasizing tissue such as the post-prandial intestine [66]. Thus cells entering the sinusoids are more “in a lake than in a stream.” In addition, sinusoids are specialized to allow easy movement of hematopoietic cells in and out of the marrow. The walls of the sinusoids are trilaminar and their structure helps explain why tumor cells can easily enter and leave [63]. Stromal endothelial cells line the sinusoidal lumen. These cells do not have tight junctions but may overlap or interdigitate. They rest on a basement membrane, the middle layer, which is irregular and discontinuous. The third layer, facing the bone marrow, is composed of adventitial cells, a type of phagocytic cell, which also do not form a tight layer. Thus the nature of the sinusoidal walls allows for easy two-way movement of hematopoietic and lymphoid cells. This structure is used advantageously by cancer cells [67].
Nevertheless, cellular entry into the sinusoids and migration into the marrow are not sufficient to insure colonization by the cancer cells. In a mouse study, it was observed that many more cancer cells entered the marrow cavity of the femur than remained to colonize it [59]. Presumably, many metastatic cancer cells in the blood can circulate through the bone, but few remain. Cancer cells, similar to leukocytes, migrate through the vasculature using a process of attachment-detachment through cell-adhesion molecules. The endothelial cells of the bone sinusoids constitutively and simultaneously express an array of tethering and adhesive proteins including P-selectin, E-selectin, intercellular adhesion molecule (ICAM-1) and VCAM-1. The vasculature of other tissues only express these molecules when stimulated by inflammatory cytokines [63]. Moreover, vasculature in one part of the bone may be different than other parts. Indeed Makuch et al. [68] found expression of P-selectin, E-selectin, ICAM and VCAM by vascular endothelial cells isolated from trabecular bone and from diaphyseal bone. However the endothelial cells from the trabecular bone but not diaphyseal bone showed a significantly increased expression in E-selectin when exposed to conditioned medium from immature osteoblasts. These data can be interpreted to suggest that osteoblasts of immature, metabolically active bone enhance E-selectin expression by nearby endothelial cells. This increase in cell attachment molecules would in turn enhance cancer cell extravasation into the bone marrow. Furthermore as discussed further on, inflammatory cytokines produced by osteoblasts in the presence of breast cancer cells may cause an even greater increase in cancer cell migration. In complementary approaches, others found that prostate cancer cells showed increased adherence to bone marrow microvasculature endothelium than from endothelium of other anatomical sites [69, 70]. Similar findings were reported for breast cancer [71, 72].
7 Adhesion molecules of the vascular endothelium
There has been an ongoing discussion of the roles of adhesion vs. entrapment in the movement of cancer cells into organs. The “leaky” vasculature suggests that entrapment is not the limiting event in bone metastasis. To the contrary, adhesion of metastatic cancer cells to the endothelium appears to play a specific and critical role. Evidence for the role of adhesion molecules has been found, both with prostate and breast cancer cells, which may explain their predilection to the bone [61]. For tumor cells to reach the bone marrow there must be a selective adhesion of the circulatory tumor cells to the endothelium of the bone marrow sinusoids. Therefore the adhesion molecules of the endothelium are of utmost importance.
The movement of cancer cells across the endothelium in the bone marrow has been likened to the movement of leukocytes across the endothelium. While the general patterns are likely the same, the actual molecules involved may differ [73–75]. The reported roles of various adhesion molecules may relate to the particular system, i.e. primary or secondary tumors and the specific organ. For example, selectin-mediated binding of colon cancer cells has been demonstrated to be important for their adhesion to the hepatic microvasculature [76]. This association may not hold for other metastatic tumors [61]. Makuch et al. [68] reported that active osteoblasts influenced E-selectin (but not P or S-selectin) expression on metaphyseal endothelium. The expression of E-selectin depended both on the stage of differentiation of the osteoblasts and the source of the microvasculature endothelium within the bone marrow. Galactin-3 is another molecule that participates in tumor cell, bone microvasculature associations. Galactin-3 and its ligand Thomsen–Friedenreich (TF) antigen are found both on many cancer cells and on microvasculature endothelium. Their interaction appears to be important for the primary arrest of the tumor cells [71, 77]. Another well studied molecule is CD44, the principle cell surface receptor for hyaluronic acid (HA). It is frequently over-expressed on malignant cells. In model systems, its expression correlates with the rate and strength of cancer cell interaction with bone marrow endothelium. CD44 expression on the surface of bone marrow endothelial cells likely acts to bind HA. Cancer cells and bone marrow endothelial cells both appear to express CD44 and HA, and the interaction of the two leads to tethering of the cancer cells to the bone marrow. In addition, there are associated data to suggest that activation of CD44 by HA or by osteopontin is important in downstream signaling through CD44 in bone.
8 Adhesion molecules within the bone marrow cavity
Coordinated bone remodeling involves extensive cell–cell and cell–matrix interactions among osteoblasts, osteoclasts, and bone marrow resident stromal and hematopoietic cells. The sinusoidal endothelium of the bone marrow is a two-way gate, allowing movement in both directions of newly formed and recirculating lymphocytes, hematopoietic stem cells as well as neoplastic cells. The trafficking patterns are organized by adhesion molecules on the circulating cells as well as on the bone marrow reticulocytes. VCAM-1, a member of the immunoglobulin family of cell adhesion molecules, was shown by a radiolabeling technique to be constitutively expressed by bone marrow reticular cells as well as the entire endothelium of the bone marrow sinusoids [78]. Its counter receptor, VLA-4 (α4β1), and ICAM-1, which belongs to a similar family and binds α2β1 integrins, are found on many cancer cells. Thus adhesion molecules which serve normal bone metabolism can be used to the advantage of metastatic tumor cells.
Another integrin member, αvβ3 is associated both with breast cancer [79] and osteoblast function [80, 81]. Interestingly, it is over-expressed in metastatic breast cancer cells once they enter the bone [82]. It is the predominant integrin on osteoclasts and appears to be important for syncytia formation and attaching to the bone matrix [83]. Peptomimetic inhibitors of αvβ3 were found to significantly reduce metastatic cancer formation when injected prior to tumor cells in a mouse model. However, there was less of an effect when administered after tumor inoculation [84]. The expression of adhesion molecules by osteoclasts has been fairly well determined [85]. Three integrins, αvβ3, α2β1, and αvβ1, and CD44 are present on osteoclasts.
The survival of cancer cells in the bone depends on their interactions with other cells. Interactions may be physical, with cell adhesion molecules, or through secreted molecules, such cytokines, chemokines, and other growth factors. Adhesion to various cells in the metastatic site controls anti-apoptotic and proliferative signals (see [84]). Thus the bone marrow displays numerous adhesion molecules that offer opportunities for interactions between cancer cells and normal cells. Some of these interactions do not occur until the cancer cells are in the bone marrow environment after they express new adhesion molecules.
9 Bone remodeling and inflammation
Rodan [86] in an overview of skeletal development and function, points out the similarities between bone remodeling and inflammation. Many of the same cytokines produced by the immune cells as part of an inflammatory response are also produced by osteoblasts. Some of these, IL-1, IL-11, IL-6, PGE and PTHrP, are also osteoclastogenic. Furthermore, both osteoblasts and osteoclasts express toll-like-receptors [87] and respond to trauma, bacterial infection, and metastases with the production of these same molecules [88–90]. In particular, a set of inflammatory stress molecules (IL-6, IL-8, MCP-1, COX-2) appears in normal bone remodeling as well as under these pathogenic conditions [88]. These factors are made by osteoblasts but can also be produced by macrophages. They attract and activate osteoclasts. Osteoclasts degrade bone matrix, leading to the release of many growth factors. This combination of factors creates a very hospitable environment for cancer cells.
10 Cytokines and chemokines
Once established in the bone microenvironment, a ‘vicious cycle’ is created among metastatic tumor cells, osteoblasts, and osteoclasts that facilitates increased bone turnover and metastatic cell survival. Guise et al. developed a model of breast cancer metastasis to the bone, based on breast cancer cell overproduction of PTHrP [3,91] that activates osteoblasts to produce RANK-L. Osteoblast-secreted RANK-L binds the RANK receptor on osteoclasts, inducing osteoclast differentiation and bone matrix degradation. In turn, TGF-β, released from the bone matrix, stimulates the cancer cells to produce more PTHrP [43], thus establishing a positive feedback loop. There is additional evidence that breast cancer-derived IL-8 acts prior to PTHrP to stimulate osteoclastogenesis via both RANK-L dependent and independent mechanisms [92–94]. As a result of constituitive osteoclast activation and an inability of osteoblasts to lay down bone matrix, sustained bone degradation occurs [45, 54]. This feedback establishes a vicious cycle, resulting in continued activation of osteoclasts and breast cancer cells. Ultimately, osteolytic lesions are formed at sites of metastases [10, 57].
It should be noted that the presence of PTHrP is not sufficient for cancer cell metastases to the bone. In a study in which 526 patients with operable breast cancer were examined, it was found that those with PTHrP-positive primary tumors had improved survival and were less likely to develop bone metastases [95]. In those patients with bone metastases, PTHrP presence was found not to be significantly associated with tumor size, vascular invasion, or tumor grade [95]. Thus, it is likely that bone metastases are influenced by other factors in the bone microenvironment besides PTHrP.
In addition to the PTHrP, Bendre et al. found an important role for IL-8. IL-8, the human homolog to murine MIP-2, belongs to the family of CXC chemokines and is naturally constitutively produced by osteoblasts [93, 96, 97]. IL-8 is overexpressed in a bone-homing derivative of MDA-MB-231 human metastatic breast cancer cells suggesting an important role in bone metastasis [94]. IL-8 can stimulate osteoclastogenesis by increasing RANK-L or stimulate the formation of osteoclasts in the absence of RANK-L [92]. It is believed that IL-8 is involved in the early stages of breast cancer metastasis by initiating the bone resorption process [93]. IL-8 also has been shown to increase angiogenesis and suppress osteoblast activity [98, 99]. In addition, IL-8 increases cell motility, invasion, and metastatic potential in breast cancer [93]. If overexpressed in breast cancer cells, IL-8 will lead to increased bone metastasis and osteolytic activity [94]. IL-8 stimulates osteoclast activity independently of RANK-L [92]. Bendre et al. suggested that the vicious cycle with PTHrP is first initiated by breast cancer cells secreting IL-8, thereby stimulating bone resorption by osteoclasts. The release of TGF-β from the bone matrix then stimulates cancer cells to produce more PTHrP, thus continuing the vicious cycle [93].
COX-2 and PGE2 also have been found to contribute to osteoclast activitation and facilitate the creation of a microenvironment favorable for cancer cell metastasis. COX-2 levels and activity correlate with cancer cell metastasis both in vitro and in vivo [100–102]. COX-2 expression also has been implicated in the growth, invasion, apoptosis, and angiogenesis of breast cancer [103–105]. COX-2 expression in patients with cancer has shown to be a negative prognostic factor [106]. Singh et al. recently conducted a study investigating the involvement of COX-2 in breast cancer metastases to the bone [107]. Interestingly, overexpression of COX-2 correlated with increased production of IL-8 [108], which has also been linked to increased metastatic occurrence [94]. Singh et al. found that COX-2 induced both the formation of PGE2 and IL-8 specifically in bone metastatic breast cancer cells. Since PGE2 and IL-8 are mediators of osteoclast activation [109](through direct or indirect mechanisms of stimulation of RANKL [92]), a system in which there is overexpression of COX-2 would favor osteolytic cancer metastases. In addition, Hiraga et al. discovered that bone-derived TGF-β stimulated COX-2 expression, thus enhancing bone metastases in breast cancer [110]. TGF-β, released from the bone during bone resorption, stimulates COX-2 expression and subsequently PGE2 expression in breast cancer cells. PGE2 upregulates RANKL expression on osteoblasts, leading to osteoclast activation and increased bone turnover [110]. Finally, Hall et al. investigated the involvement of Wnts, a family of glycoproteins [111], in the promotion of osteoblastic bone metastases in prostate cancer [112]. They found that promotion of Wnt activity (by blocking the Wnt antagonist DKK-1), led to enhanced osteoblastic bone metastases in typically osteolytic PC-3 prostate cancer cells [112]. These results suggest that the involvement of DKK-1 dictates whether bone metastases are osteoblastic or osteolytic, and once again emphasize the importance of the bone microenvironment.
Cancer cell secreted IL-1, IL-6, and IL-11 have also been found to increase osteoclast activation. IL-6 is a pleiotropic cytokine that is naturally expressed by osteoblasts in low quantities. IL-6 receptors are found on osteoclasts and when stimulated, cause osteoclast differentiation and bone resorption [113]. There is a correlation of poor prognosis with increased IL-6 expression and metastatic breast cancer. IL-6 additionally has been implicated with increased breast cancer cell migration [114–116]. IL-1 is also a potent stimulator of osteoclast activation. To explore the notion that increased bone turnover attracts metastatic cancer cells, Sasaki et al. increased bone resorption by injecting recombinant IL-1β locally over the calvaria of nude mice [117]. Four weeks after cancer cell inoculation, osteolytic metastatic cancer cells were found in the calvariae of IL-1 treated mice. None were seen in the control. IL-11 is an additional key player in osteoclast activation. It has been reported that IL-11 mediates the actions of IL-1 on osteoclast development [118]; however, IL-11 has independent effects on osteoclast activity [119]. IL-11, IL-1, and IL-6 have all been found to be involved in an interacting cascade of cytokines which play a large part in osteoclast development and activity. Increased osteoclast activity subsequently creates a bone microenvironment that favors cancer cell metastasis, growth, and development. IL-1 contributes to the production of IL-11 and IL-6 [118]. IL-6 and IL-11 production are also regulated by IL-1, growth factors such as PDGF, IGF-1, and TGF-β, vitamin D, PTHrP, and PTH [119, 120].
11 Bone matrix is fertile soil for metastatic cancer cells
During bone deposition, osteoblasts secrete a variety of growth factors, such as IGF, TGF-β, FGF, and BMPs, that become incorporated into the bone matrix [121]. As bone resorption occurs, these factors are released into the bone microenvironment, making it an attractive place for cancer cells to metastasize and grow [122]. TGF-β released from the bone, in particular, has been found to stimulate the expression of CTGF, IL-11, and PTHrP by cancer cells [42, 123]. These factors are involved in tumor metastasis to bone and subsequently promote additional bone resorption, leading to the release of more growth factors and further preferential tumor metastasis to bone. Growth factors released during bone remodeling and present in the bone microenvironment may be chemoattractive molecules for the cancer cells. Orr et al. and Mundy et al. using a Boyden Chamber assay both demonstrated that the release of growth factors during bone resorption stimulated the chemotaxis of cancer cells [124, 125]. Additionally, cytokines have been implicated in cancer cell chemotaxis to bone. The SDF-1/CXCR4 axis is a ubiquitous chemotaxis mechanism in normal biology and is used for directed migration of a variety of immune and hematopoetic cells [126–129]. Jung et al. found that SDF-1 is secreted by osteoblasts, and that certain factors, namely IL-1β, PDGF-BB, VEGF, tumor necrosis factor-alpha (TNF-α), and PTH, act on osteoblasts to increase SDF-1 production [130]. Consequently, many of these cytokines play a role in increasing osteoclast activity during bone resorption [119, 120]. Furthermore, the results of this study suggested that osteoblast secretion of SDF-1 may be a chemotactic mechanism for stem cell homing. It goes without saying that the SDF-1/CXCR4 axis may be involved in cancer cell chemoattraction as well. In fact, Muller et al., among others [131, 132], explored the idea that metastatic breast cancer cells were responsive to gradients of chemokines [133]. CXCR4 was found to be highly expressed on metastatic breast cancer cells, and its ligand, SDF-1 was found to be highly expressed in organs to which cancer cells preferentially metastasizes (such as bone) [133]. Treatment with a neutralizing antibody to CXCR4 suppressed bone metastatic breast cancer [133]. Sun et al. conducted a similar study using prostate cancer as a model and found comparable results [134]. Once in bone, cancer cells become tethered by integrins and cell adhesion molecules as previously described [135, 136].
Current models suggest that chemokines and cytokines produced by breast cancer cells are key to breast cancer cell metastasis [92–94, 137]. Elevated levels of IL-8 production by human breast cancer cells have been correlated with increased bone metastasis in vivo and with stimulation of osteoclast differentiation and bone resorption [92, 94]. Tumor-derived IL-1, IL-6, and IL-11, insulin-growth factor-II, TNF-α, and a variety of other factors can also contribute to osteoclast activation and bone destruction [98, 138].
While breast cancer cells undoubtedly play an important role in breast cancer metastasis to the bone, data have shown that osteoblasts can be directed by the breast cancer cells to produce several inflammatory cytokines that have been implicated in osteoclast activation as well as breast cancer cell migration and survival [93, 139–143]. (Table 1 gives a brief summary of some relevant cytokines and their sources in the bone.) Kinder et al. demonstrated that osteoblasts undergo an inflammatory stress response in the presence of human metastatic breast cancer cells and produce elevated levels of IL-6, human IL-8 (murine KC, MIP-2), and MCP-1 [144]. These cytokines are known to attract, differentiate, and activate osteoclasts; thus co-opting osteoblasts into creating a bone microenvironment that exacerbates bone loss [144]. Similar findings were seen with murine osteoblasts and primary calvarial osteoblasts [144]. These results support the idea that cancer metastases create a unique niche in the bone microenvironment by co-opting normal cells of the bone to favor tumor growth and development.
Furthermore, Mastro and colleagues have preliminary in vivo evidence that osteoblasts themselves in the bone naturally produce cytokines that may be chemoattractants for metastatic breast cancer cells. In particular, they showed that metaphyses of bone cleared of bone marrow produced chemokines and cytokines that were different from those in the diaphysis (shaft). Prominent among these were KC (present only in the metaphyses), MIP-2 (murine homolog to human IL-8), and MCP-1 (Fig. 2) (Bussard and Mastro, 2007, unpublished data).

Cytokine expression of murine femur metaphyses ex-vivo following intracardiac inoculation. MDA-MB-231GFP cells (3 × 105 cells) were inoculated into the left cardiac ventricle of 4–6 week old athymic, female mice. Control mice were untreated. Mice were euthanized at 4 weeks and femurs harvested. The bone marrow was removed and the femur metaphyses were fractionated. Isolated metasphyseal bone pieces were crushed and cultured. Media were collected and tested after 24 h. Murine MCP-1 cytokine production was quantified using ELISAs. Murine IL-6, MIP-2, and KC were quantified using a Bio-Rad Bio-Plex™ murine cytokine quantification assay. Shown is a representative experiment. MCP-1 and IL-6 concentrations were multiplied by 10 in order to be shown on the same graph as MIP-2 and KC
These cytokines were strongly observed in cultures of the bone metaphysis alone and not found in cultures of bone marrow from the metaphysis (Bussard and Mastro, 2007, unpublished data). This observation suggests that the cytokines were specifically produced by the cells of the bone (i.e. osteoblasts) and not the stromal cells. Murine IL-6, KC, and MIP-2 located in femur metaphyses were also found to be increased in the presence of human metastatic breast cancer cells compared with femur metaphyses from control mice (Bussard and Mastro, 2007, unpublished data). Finally, a novel experiment was conducted to monitor and quantify the initial stages (arrival, localization, and initial colonization) of breast cancer cell trafficking in the bone [59]. The DNA from femurs of mice inoculated with MDA-MB-435GFP cells via intracardiac injection were isolated at various times, purified, and subjected to quantitative PCR for a human gene, HERV-1, and the number of breast cancer cells calculated. Femurs were separated into metaphyses and diaphyses. Results indicated that breast cancer cells preferentially migrated within days directly to the distal then proximal metaphyses. Few were found in the diaphyses [59]. These results additionally support the idea that metastatic breast cancer cells may follow a gradient of chemoattractant cytokines as well as suggests the importance of the local bone microenvironment.
In addition to IL-6 and IL-8, KC and MCP-1 are osteoblast-derived cytokines that greatly increase in response to metastatic breast cancer cells (Bussard and Mastro, 2007, unpublished data). MCP-1, a member of the CC chemokine family, is naturally produced by osteoblasts [96]. It regulates bone resorption by stimulating the migration of common monocyte-osteoclast progenitor cells from the blood or the bone marrow to the bone. MCP-1 concentrations are increased in metastatic cell lines, and it is associated with angiogenesis and increased cancer cell survival [145–147]. KC is another member of the CXC chemokine family with homology to IL-8 [148]. KC stimulates angiogenesis and is involved in neutrophil chemotaxis and activation [148, 149]. KC is also expressed by osteoblasts [149].
In addition to directing osteoblasts to secrete cytokines which alter the bone microenvironment, cancer cells affect the bone building cells in other ways as well. Mercer et al. demonstrated that culturing mouse osteoblasts with the conditioned medium from a human metastatic breast cancer cell line inhibited expression of osteoblast differentiation and blocked osteoblast ability to mineralize bone matrix [45]. This in vitro observation was confirmed in a mouse study [59]. Since osteoblasts do not differentiate properly in the presence of breast cancer cells, it is possible that the cancer cells may alter the overall protein secretion profile of osteoblasts. This alteration may involve preventing osteoblasts from producing the differentiation proteins necessary for developing into mature, bone-depositing cells, as well as inducing osteoblast production of cytokines that could contribute to progression of bone metastasis, increase activation of osteoclasts, and contribute to the formation of osteolytic lesions.
12 Conclusions
Clearly, the organ microenvironment is extremely influential in cancer cell metastasis to a specific location. Crosstalk between the cancer cell “seed” and the target organ microenvironment “soil” will determine if the cancer cell metastasizes to a specific site and if that microenvironment supports growth and proliferation of the metastatic cancer cell. Only then will the metastatic cancer cell population flourish. Bone provides an especially attractive site for a variety of reasons. Metabolically active areas of bone are well-vascularized with a system that allows various cells to easily enter and exit. The normal remodeling process provides chemotactic and growth factors that attract cancer cells and support them once in place. The bone matrix contains a rich storehouse of growth factors such as TGF-β that are released during bone turnover. Resident cancer cells thrive in the rich cocktail of released cytokines. Finally, both osteoblast and octeoclast activities can be modulated by cancer cells to their advantage. The release of characteristic sets of cytokines by the bone matrix of an osteolytic lesion or osteoblastic lesion (e.g. MCP-1, IL-6, IL-8) will facilitate the chemoattraction and survival of metastatic cancer cells. Understanding the mechanisms behind these events will aid in the development of therapeutics to combat specific metastases and manipulate their target organ microenvironments. While the origin of metastatic variants remains unclear, is it certain that the target organ microenvironment contributes greatly to their metastasis.
13 Unanswered questions
Many unanswered questions remain. One of the most critical is to determine a ‘metastatic signature’ for the primary tumor which would indicate the possibility of metastasis. However, not all tumor cells that arrive in the bone, even from the same primary tumor, will remain or grow there. Dormant metastases in the bone remain a mystery. It is known that individual cells or micrometastases can be found in patients with no evidence of metastasis [150]. These individuals may never exhibit bone metastasis. On the other hand, bone micrometastases can remain dormant for years in spite of the rich microenvironment. What event triggers that cell to begin to grow?
14 Future studies
It is difficult to study bone metastases and the tumor microenvironment for many reasons. (1) The marrow space is relatively inaccessible. (2) It is also a complex space containing not only bone cells, osteoblasts and osteoclasts, but also hematopoietic cells and transient immune cells. Cell lines, particularly osteoblasts, have been developed that recapitulate in culture the stages of osteoblast differentiation. However, these lines have limitations when compared to intact bone.
We have recently developed a specialized bioreactor that allows extended-term culture of osteoblasts. The cells have been grown uninterrupted for up to 10 months. They proliferate and form a multilayer (>6 cell layers) of mature osteoblasts that begin to mineralize and form macroscopic bone chips [151]. By 10 months the morphology of the cells resembles osteocytes. We have inoculated human metastatic breast cancer cells, MDA-MB-231GFP, into these chambers and have seen by microscopy that the cells adhere, grow, and move through the cell layers, mimicking in vivo migration and invasion [152]. We have evidence that the osteoblasts likewise undergo a stress response and produce increased amounts of IL-6, for example. While the bioreactor has been used to study osteoblast-cancer cell interactions, it will allow introduction of other cell types, e.g. macrophages, lymphocytes. Thus, the bioreactor promises to be a useful 3-D culture system to study and to manipulate the bone-tumor microenvironment.
References
Rubens, R. D., & Mundy, G. R. (2000). Cancer and the skeleton. London: Martin Dunitz.
Price, J. E. (2004). The breast comprehensive management of benign and malignant disorders pp. 537–557. St. Louis: Saunders.
Mundy, G. R. (2002). Metastasis to bone: Causes, consequences and therapeutic opportunities. Nature Reviews. Cancer, 2, 584–593.
Chambers, A. F., Groom, A. C., & MacDonald, I. C. (2002). Dissemination and growth of cancer cells in metastatic sites. Nature Reviews. Cancer, 2, 563–572.
Batson, O. V. (1942). Annals of Internal Medicine, 16, 38–45.
Marks, S. C., & Odgren, P. R. (2002). Structure and development of the skeleton. In J. P. Bilezikian, L. G. Raisz, & G. A. Rodan (Eds.) Principles of bone biology (vol. 1 (pp. 3–15). New York: Academic.
Hancox, N. M. (1972). Biology of bone. Cambridge: University Press.
Baron, R. (2003). General principles of bone biology. In M. J. Favus (Ed.) Primer on the metabolic bone diseases and disorders of mineral metabolism (pp. 1–8). Washington, D.C.: American Society for Bone and Mineral Research.
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2002). Molecular biology of the cell p. 1308. New York: Garland Science.
Guise, T. A., & Mundy, G. R. (1998). Cancer and bone. Endocrine Reviews, 19, 18–54.
Price, J. S., Oyajobi, B. O., & Russell, R. G. (1994). The cell biology of bone growth. European Journal of Clinical Nutrition, 48(Suppl 1), S131–S149.
Minguell, J. J., Erices, A., & Conget, P. (2001). Mesenchymal stem cells. Experimental Biology and Medicine, 226, 507–520.
Kanis, J. A., & McCloskey, E. V. (1997). Bone turnover and biochemical markers in malignancy. Cancer, 80, 1538–1545.
Gartner, L. P., & Hiatt, J. L. (1997). Color textbook of histology. Philadelphia: Saunders.
Baron, R. (2003). General principles of bone biology. In J. B. Lian, & S. R. Goldring (Eds.) Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, D.C: American Society for Bone and Mineral Research.
Takahashi, N., Udagawa, N., Takami, M., & Suda, T. (2002). Cells of bone: Osteoclast generation. In J. P. Bilezikian, L. G. Raisz, & G. A. Rodan (Eds.) Principles of bone biology (vol. 1 (pp. 109–126). San Diego: Academic.
Michigami, T., Shimizu, N., Williams, P. J., Miewolna, M., Dallas, S. L., Mundy, G. R., et al. (2000). Cell–cell contact between marrow stromal cells and myeloma cells via VCAM-1 and α4B1 integrin enhances production of osteoclast-stimulating activity. Blood, 96, 1953–1960.
Iyengar, P., Combs, T. P., Shah, S. J., Gouon-Evans, V., Pollard, J. W., Albanese, C., et al. (2003). Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene, 22, 6408–6423.
Maurin, A. C., Chavassieux, P. M., Frappart, L., Delmas, P., Serre, C.-M., & Meunier, P. J. (2000). Influence of mature adipocytes on osteoblast proliferation in human primary cocultures. Bone, 26, 485–489.
Maeda, T., Alexander, C. M., & Friedl, A. (2004). Induction of syndecan-1 expression in stromal fibroblasts promotes proliferation of human breast cancer cells. Cancer Research, 64, 612–621.
Saad, S., Gottlieb, D. J., Bradstock, K. F., Overall, C. M., & Bendall, L. J. (2002). Cancer cell-associated fibronectin induces release of matrix metalloproteinase-2 from normal fibroblasts. Cancer Research, 62, 283–289.
Lau, Y. S., Sabokbar, A., Giele, H., Cerundolo, V., Hofstetter, W., & Athanasou, N. A. (2006). Malignant melanoma and bone resorption. British Journal of Cancer, 94, 1496–1503.
Chavez-Macgregor, M., Aviles-Salas, A., Green, D., Fuentes-Alburo, A., Gomez-Ruiz, C., & Aguayo, A. (2005). Angiogenesis in the bone marrow of patients with breast cancer. Clinical Cancer Research, 11, 5396–5400.
Eberhardt, C., Gray, P. W., & Tjoelker, L. W. (1997). Human lysophosphatidic acid acyltransferase: cDNA cloning, expression, and localization to chromosome 9q34.3. Journal of Biological Chemistry, 272, 20299–20305.
Boucharaba, A., Serre, C. M., Gres, S., Saulnier-Blache, J. S., Bordet, J. C., Guglielmi, J., et al. (2004). Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. Journal of Clinical Investigation, 114, 1714–1725.
Lapumbo, J. S., Talmage, K. E., Massari, J. B., La Jeunesse, C. M., Flick, M. J., Kombrinck, K. W., et al. (2005). Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood, 105, 178–185.
Walsh, M. C., Kim, N., Kadono, Y., Rho, J., Lee, S. Y., Lorenzo, J., et al. (2006). Osteoimmunology: Interplay between the immune system and bone metabolism. In W. E. Paul, C. G. Fathman, & L. H. Glimcher (Eds.) Annual review of immunology (vol. 24 (pp. 33–63). Palo Alto: Annual Reviews.
Sroato, I., Grano, N., Brunetti, G., Colucci, S., Mussa, A., & Bertetto, O. (2005). Mechanisms of spontaneous osteoclastogenesis in cancer with bone involvement. FASEB Journal, 19, 228–230.
Fournier, P. G., Chirgwin, J. M., & Guise, T. A. (2006). New insights into the role of T cells in the vicious cycle of bone metastases. Current Opinion in Rheumatology, 18, 396–404.
Coussens, L. M., & Werb, Z. (2002). Inflammation and cancer. Nature, 420, 860–867.
Brigati, C., Noonan, D. N., Albini, A., & Benelli, R. (2002). Tumors and inflammatory infiltrates: Friends or foes? Clinical & Experimental Metastasis, 19, 247–258.
Schoppmann, S. F., Birner, P., Stockl, J., Kalt, R., Ullrich, R., Caucig, C., et al. (2002). Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. American Journal of Pathology, 161, 947–956.
Torisu, H., Ono, M., Kiryu, H., Furue, M., Ohmoto, Y., Nakayama, J., et al. (2000). Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: Possible involvement of TNF-α and IL-1α. International Journal of Cancer, 85, 182–188.
Jonjic, N., Peri, G., Bernasconi, S., Sciacca, F. L., Colotta, F., Pelicci, P., et al. (1992). Expression of adhesion molecules and chemotactic cytokines in cultured human mesothelial cells. Journal of Experimental Medicine, 176, 1165–1174.
Paget, S. (1889). The distribution of secondary growths in cancer of the breast. Cancer and Metastasis Reviews, 8, 98–101.
Roodman, G. D. (2004). Mechanisms of bone metastasis. New England Journal of Medicine, 350, 1655–1664.
Roodman, G. D. (2001). Biology of osteoclast activation in cancer. Journal of Clinical Oncology, 19, 3562–3571.
Yoneda, T. (1996). Mechanisms of preferential metastasis of breast cancer to bone. Journal of Clinical Oncology, 9, 103–109.
Powles, T., Paterson, S., Kanis, J. A., McCloskey, E. V., Ashley, S., Tidy, A., et al. (2002). Randomized, placebo-controlled trial of clodronate in patients with primary operable breast cancer. Journal of Clinical Oncology, 20, 3219–3224.
Sasaki, A., Boyce, B. F., Story, B., Wright, K. R., Chapman, M., Boyce, R., et al. (1995). Bisphosphonate risedronate reduces metastatic human breast cancer cells and bone metastases development. Journal of Clinical Investigation, 103, 197–206.
Guise, T. A., Yin, J. J., Taylor, S. D., Kumagai, Y., Dallas, M. R., Boyce, B. F., et al. (1996). Evidence for a causal role of parathyroid-hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. Journal of Clinical Investigation, 98, 1544–1549.
Yin, J.-J., Selander, K., Chirgwin, J. M., Dallas, M. R., Grubbs, B. G., Wieser, R., et al. (1999). TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. Journal of Clinical Investigation, 103, 197–206.
Mastro, A. M., Gay, C. V., Welch, D. R., Donahue, H. J., Jewell, J., Mercer, R., et al. (2004). Breast cancer cells induce osteoblast apoptosis: a possible contributor to bone degradation. Journal of Cell Biology, 91, 265–276.
Mastro, A. M., Gay, C. V., & Welch, D. R. (2003). The skeleton as a unique environment for breast cancer cells. Clinical & Experimental Metastasis, 20, 275–284.
Mercer, R., Miyasaka, C., & Mastro, A. M. (2004). Metastatic breast cancer cells suppress osteoblast adhesion and differentiation. Clinical & Experimental Metastasis, 21, 427–435.
Guise, T. A., Yin, J. J., & Mohammad, K. S. (2003). Role of endothelin-1 in osteoblastic bone metastases. Cancer, 97, 779–784.
Yin, J. J., Mohammad, K. S., Käkönen, S. M., Harris, S., Wu-Wong, J. R., Wessale, J. L., et al. (2003). A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proceedings of the National Academy of Sciences, 100, 10954–10959.
Kasperk, C. H., Borcsok, I., Schairer, H. U., Schneider, U., Nawroth, P. P., Niethard, F. U., et al. (1997). Endothelin-1 is a potent regulator of human bone cell metabolism in vitro. Calcified Tissue International, 60, 368–374.
Nelson, J. B., Hedican, S. P., George, D. J., Reddi, A. H., Piantadosi, S., Eisenberger, M. A., et al. (1995). Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nature Medicine, 1, 944–949.
Yi, B., Williams, P. J., Niewolna, M., Wang, Y., & Yoneda, T. (2002). Tumor-derived platelet-derived growth factor-BB plays a critical role in osteosclerotic bone metastasis in an animal model of human breast cancer. Cancer Research, 62, 917–923.
Hiraga, T., Williams, P. J., & Mundy, G. R. (2001). The bisphosphonate ibandronate promotes apoptosis in MDA-MB-231 human breast cancer cells in bone metastases. Cancer Research, 61, 4418–4424.
Delmas, P. D., Demiaux, B., Malaval, L., Chapuy, M. C., Edouard, C., & Meunier, P. J. (1986). Serum bone gamma carboxyglutamic acid-containing protein in primary hyperthyroidism and in malignant hypercalcemia. Journal of Clinical Investigation, 77, 985–991.
Kukreja, S. C., Rosol, T. J., Shevrin, D. H., & York, P. A. (1998). Quantitative bone histomorphometry in nude mice bearing a human squamous cell lung cancer. Journal of Bone and Mineral Research, 3, 341–346.
Stewart, A. F., Vignery, A., Silverglate, A., Ravin, N. D., Livolsi, V., Broadus, A. E., et al. (1982). Quantitative bone histomorphology in humoral hypercalcemia of malignancy: Uncoupling of bone cell activity. Journal of Clinical Endocrinology and Metabolism, 55, 219–227.
Taube, T., Elomaa, I., Blomqvist, C., Benton, N. C., & Kanis, J. A. (1994). Histomorphometric evidence for osteoclast-mediated bone resorption in metastatic breast cancer. Bone, 15, 161–166.
Galasko, C. S. (1982). Mechanisms of lytic and blastic metastatic disease of bone. Clinica Ortopedica, 169, 20–27.
Martin, T. J., & Moseley, J. M. (2000). Mechanisms in the skeletal complications of breast cancer. Endocrine Related Cancer, 7, 271–284.
Mundy, G. R., & Guise, T. A. (2000). Pathophysiology of bone metastasis. In R. D. Rubens, & G. R. Mundy (Eds.) Cancer and the skeleton (pp. 43–64). London: Martin Dunitz Ltd.
Phadke, P. A., Mercer, R. R., Harms, J. F., Jia, Y., Kappes, J. C., Frost, A. R., et al. (2006). Kinetics of metastatic breast cancer cell trafficking in bone. Clinical Cancer Research, 12, 1431–1440.
Everts, V., Delaisse, J. M., Korper, W., Jansen, D. C., Tigchelaar-Gutter, W., Saftig, P., et al. (2002). The bone lining cell: Its role in cleaning Howship’s lacunae and initiating bone formation. Journal of Bone and Mineral Research, 17, 77–90.
Glinsky, V. V. (2006). Intravascular cell-to-cell adhesive interactions and bone metastasis. Cancer and Metastasis Reviews, 25, 531–540.
Sasaki, A., Boyce, B. F., Story, B., Wright, K. R., Chapman, M., Boyce, R., et al. (1995). Bisphosphonate risedronate reduces metastatic human breast cancer burden in bone in nude mice. Cancer Research, 55, 3551–3557.
Mazo, I. B., & von Andrian, U. H. (1999). Adhesion and homing of blood-borne cells in bone marrow microvessels. Journal of Leukocyte Biology, 66, 25–32.
Buckwalter, J. A. (1995). Pharmacological treatment of soft-tissue injuries. Journal of Bone and Joint Surgery. American Volume, 77, 1902–1914.
Schnitzer, J. E., McKinstry, P., Light, T. R., & Ogden, J. A. (1982). Quantitation of regional chondro-osseous circulation in canine tibia and femur. American Journal of Physiology, 242, H365–H375.
Stephenson, R. B. (1989). The splanchnic circulation. In H. D. Patton, A. F. Fuchs, B. Hille, A. M. Scher, & R. Steiner (Eds.) Textbook of physiology (pp. 911–923). Philadelphia: Saunders.
Mastro, A. M., Gay, C. V., & Welch, D. R. (2003). The skeleton as a unique environment for breast cancer cells. Clinical & Experimental Metastasis, 20, 275–284.
Makuch, L. A., Sosnoski, D. M., & Gay, C. V. (2006). Osteoblast-conditioned media influence the expression of E-selectin on bone-derived vascular endothelial cells. Journal of Cellular Biochemistry, 98, 1221–1229.
Lehr, J. E., & Pienta, K. J. (1998). Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. Journal of the National Cancer Institute, 90, 118–123.
Scott, L. J., Clarke, N. W., George, N. J., Shanks, J. H., Testa, N. G., & Lang, S. H. (2001). Interactions of human prostatic epithelial cells with bone marrow endothelium: Binding and invasion. British Journal of Cancer, 84, 1417–1423.
Glinsky, V. V., Glinsky, G. V., Rittenhouse-Olson, K., Huflejt, M. E., Glinskii, O. V., Deutscher, S. L., et al. (2001). The role of Thomsen–Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Research, 61, 4851–4857.
Glinsky, V. V., Huflejt, M. E., Glinsky, G. V., Deutscher, S. L., & Quinn, T. P. (2000). Effects of Thomsen–Friedenreich antigen-specific peptide P-30 on beta-galactoside-mediated homotypic aggregation and adhesion to the endothelium of MDA-MB-435 human breast carcinoma cells. Cancer Research, 60, 2584–2588.
McEver, R. P. (1997). Selectin-carbohydrate interactions during inflammation and metastasis. Glycoconjugate Journal, 14, 585–591.
Cooper, C. R., Bhatia, J. K., Muenchen, H. J., McLean, L., Hayasaka, S., Taylor, J., et al. (2002). The regulation of prostate cancer cell adhesion to human bone marrow endothelial cell monolayers by androgen dihydrotestosterone and cytokines. Clinical & Experimental Metastasis, 19, 25–33.
Glinskii, O. V., Turk, J. R., Pienta, K. J., Huxley, V. H., & Glinsky, V. V. (2004). Evidence of porcine and human endothelium activation by cancer-associated carbohydrates expressed on glycoproteins and tumour cells. Journal of Physiology, 554, 89–99.
Krause, T., & Turner, G. A. (1999). Are selectins involved in metastasis? Clinical & Experimental Metastasis, 17, 183–192.
Khaldoyanidi, S. K., Glinsky, V. V., Sikora, L., Glinskii, A. B., Mossine, V. V., Quinn, T. P., et al. (2003). MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen–Friedenreich antigen-galectin-3 interactions. Journal of Biological Chemistry, 278, 4127–4134.
Jacobsen, K., Kravitz, J., Kincade, P. W., & Osmond, D. G. (1996). Adhesion receptors on bone marrow stromal cells: in vivo expression of vascular cell adhesion molecule-1 by reticular cells and sinusoidal endothelium in normal and gamma-irradiated mice. Blood, 87, 73–82.
Pecheur, I., Peyruchaud, O., Serre, C. M., Guglielmi, J., Voland, C., Bourre, F., et al. (2002). Integrin alpha(v)beta3 expression confers on tumor cells a greater propensity to metastasize to bone. FASEB Journal, 16, 1266–1268.
Faccio, R., Grano, M., Colucci, S., Zallone, A. Z., Quaranta, V., & Pelletier, A. J. (1998). Activation of alphav beta3 integrin on human osteoclast-like cells stimulates adhesion and migration in response to osteopontin. Biochemical and Biophysical Research Communications, 249, 522–525.
Carron, C. P., Meyer, D. M., Engleman, V. W., Rico, J. G., Ruminski, P. G., Ornberg, R. L., et al. (2000). Peptidomimetic antagonists of alphavbeta3 inhibit bone resorption by inhibiting osteoclast bone resorptive activity, not osteoclast adhesion to bone. Journal of Endocrinology, 165, 587–598.
Liapis, H., Flath, A., & Kitazawa, S. (1996). Integrin alpha V beta 3 expression by bone-residing breast cancer metastases. Diagnostic Molecular Pathology, 5, 127–135.
Chellaiah, M., Kizer, N., Silva, M., Alvarez, U., Kwiatkowski, D., & Hruska, K. A. (2000). Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. Journal of Cell Biology, 148, 665–678.
Harms, J. F., Welch, D. R., Samant, R. S., Shevde, L. A., Miele, M. E., Babu, G. R., et al. (2003). A small molecule antagonist of the alpha v beta 3 integrin suppresses MDA-MB-435 skeletal metastasis. Clinical & Experimental Metastasis, 21, 119–128.
Horton, M. A., Nesbitt, S. A., Bennett, J. H., & Stenbeck, G. (2002). Integrins and other cell surface attachment molecules of bone cells. In J. P. Bilezikian, L. G. Raisz, & G. A. Rodan (Eds.) Principles of bone biology (vol. 1 (pp. 265–286). San Diego: Academic.
Rodan, G. A. (2003). The development and function of the skeleton and bone metastases. Cancer, 97, 726–732.
Kikuchi, T., Matsuguchi, T., Tsuboi, N., Mitani, A., Tanaka, S., Matsuoka, M., et al. (2001). Gene expression of osteoclast differentiation factor is induced by lipopolysaccharide in mouse osteoblasts via Toll-like receptors. Journal of Immunology, 166, 3574–3579.
Fritz, E., Jacobs, J., Glant, T., & Roebuck, K. (2005). Chemokine IL-8 induction by particulate wear debris in osteoblasts is mediated by NF-kappaB. Journal of Orthopaedic Research, 23, 1249–1257.
Lisignoli, G., Toneguzzi, S., Grassi, F., Piacentini, A., Tschon, M., Cristino, S., et al. (2002). Different chemokines are expressed in human arthritic bone biopsies: IFN-gamma and IL-6 differently modulate IL-8, MCP-1 and rantes production by arthritic osteoblasts. Cytokine, 20, 231–238.
Fritz, E., Glant, T., Vermes, C., Jacobs, J., & Roebuck, K. (2005). Chemokine gene activation in human bone marrow-derived osteoblasts following exposure to particulate wear debris. Journal of Biomedical Materials Research A, 77, 192–201.
Guise, T. A., Yin, J. J., Taylor, S. D., Kumagai, Y., Dallas, M., Boyce, B. F., et al. (1996). Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast-cancer-mediated osteolysis. Journal of Clinical Investigation, 98, 1544–1549.
Bendre, M., Montague, D. C., Peery, T., Akel, N. S., Gaddy, D., & Suva, L. J. (2003). Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone, 33, 28–37.
Bendre, M., Gaddy, D., Nicholas, R. W., & Suva, L. J. (2003). Breast cancer metastasis to bone. Clinical Orthopaedics and Related Research, 415S, S39–S45.
Bendre, M., Gaddy-Kurten, D., Foote-Mon, T., Akel, N. S., Skinner, R. A., Nicholas, R. W., et al. (2002). Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Research, 62, 5571–5579.
Henderson, M. A., Danks, J. A., Slavin, J. L., Bryrnes, G. B., Choong, P. F. M., Spillane, J. B., et al. (2006). Parathyroid hormone-related protein localization in breast cancers predict improved prognosis. Cancer Research, 66, 2250–2256.
Horowitz, M. C., & Lorenzo, J. A. (2002). Principles of bone biology. San Diego: Academic.
Graves, D. T., Jiang, Y., & Valente, A. J. (1999). The expression of monocyte chemoattractant protein-1 and other chemokines by osteoblasts. Frontiers in Bioscience, 4, 571–580.
Guise, T. A., & Chirgwin, J. M. (2003). Transforming growth factor-beta in osteolytic breast cancer bone metastases. Clinical Orthopaedics and Related Research, 4155, 532–538.
Dovio, A., Sartori, M. L., Masera, R. G., Peretti, L., Perotti, L., & Angeli, A. (2004). Effects of physiological concentrations of steriod hormones and interleukin-11 on basal and stimulated production of interleukin-8 by human osteoblast-like cells with different functional profiles. Clinical and Experimental Rheumatology, 22, 79–84.
Kundu, N., Yang, Q., Dorsey, R., & Fulton, A. M. (2001). Increased cyclooxygenase-2 (COX-2) expression and activity in a murine model of metastatic breast cancer. International Journal of Cancer, 94, 681–686.
Liu, C. H., Chang, S.-H., Narko, K., Trifan, O. C., Wu, M.-T., Smith, E., et al. (2001). Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. Journal of Biological Chemistry, 276, 18563–18569.
Ristimaki, A., Sivula, A., Lundin, J., Lundin, M., Salminen, T., Haglund, C., et al. (2002). Prognostic significance of elevated cycloozygenase-2 expression in breast cancer. Cancer Research, 62, 632–635.
Rozic, J. G., Chakraborty, C., & Lala, P. K. (2001). Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. International Journal of Cancer, 93, 497–506.
Witters, L. M., Crispino, J., Fraterrigo, T., Green, J., Lipton, A. (2003). Effects of the combination of docetaxel, zoledronic acid, and a COX-2 inhibitor on the growth of human breast cancer cell line. American Journal of Clinical Oncology, 26.
Davies, G., Salter, J., Hills, M., Martin, L. A., Sacks, N., & Dowsett, M. (2003). Correlation between cyclooxygenase-2 expression and angiogenesis in human breast cancer. Clinical Cancer Research, 9, 2651–2656.
Denkert, C., Winzer, K. J., Muller, B. M., Weichert, W., Pest, S., Kobel, M., et al. (2003). Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer, 97, 2978–2987.
Singh, B., Berry, J. A., Vincent, L. E., & Lucci, A.(2006). Involvement of IL-8 in COX-2-mediated bone metastases from breast cancer. Journal of Surgical Research.
Benoy, I. H., Salgado, R., Van Dam, P., Geboers, K., Van Marck, E., Scharpe, S., et al. (2004). Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clinical Cancer Research, 10, 7157–7162.
Li, X., Pilbeam, C. C., Pan, L., Breyer, R. M., & Raisz, L. G. (2002). Effects of prostaglandin E2 on gene expression in primary osteoblastic cells from prostaglandin receptor knockout mice. Bone, 30, 567–573.
Hiraga, T., Myoui, A., Choi, M. E., Yoshikawa, H., & Yoneda, T. (2006). Stimulation of cyclooxygenase-2 expression by bone-derived transforming growth factor-β enhances bone metastases in breast cancer. Cancer Research, 66, 2067–2073.
Westendorf, J. J., Kahler, R. A., & Schroeder, T. M. (2004). Wnt signaling in osteoblasts and bone diseases. Gene, 341, 19–39.
Hall, C. L., Bafico, A., Dai, J., Aaronson, S. A., & Keller, E. T. (2005). Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Research, 65, 7554–7560.
Manolagas, S. C. (1995). Role of cytokines in bone resorption. Bone, 17, 63S–67S.
Morinaga, Y., Fujita, N., Ohishi, K., & Tsurot, T. (1997). Stimulation of interleukin-11 production from osteoclast-like cells by transforming growth factor-beta and tumor cell factors. International Journal of Cancer, 71, 422–428.
Zhang, G. J., & Adachi, I. (1999). Serum interleukin-6 levels correlate to tumor progression and prognosis in metastatic breast carcinoma. Anticancer Research, 19, 1427–1432.
Yoneda, T., Sasaki, A., & Mundy, G. R. (1994). Osteolytic bone metastasis in breast cancer. Breast Cancer Research and Treatment, 32, 273–284.
Sasaki, A., Williams, P., Mundy, G. R., & Yoneda, T. (1994). Osteolysis and tumor growth are enhanced in sites of increased bone turnover in vivo. Journal of Bone and Mineral Research, 9, S294.
Manolagas, S. C. (1995). Role of cytokines in bone resorption. Bone, 17, 63S–67S.
Girasole, G., Passeri, G., Jilka, R. L., & Monolagas, S. C. (1994). Interleukin-11: A new cytokine critical for osteoclast development. Journal of Clinical Investigation, 93, 1516–1524.
Manolagas, S. C., Jilka, R. L., Girasole, G., Passeri, G., & Bellido, T. (1994). Estrogens, cytokines, and the pathophysiology of osteoporosis. In P. O. Kohler (Ed.) Current opinion in endocrinology and diabetes (pp. 275–281). Philadelphia: Current Science.
Hauschka, P. V., Mavrakos, A. E., Iafrati, M. D., Doleman, S. E., & Klagsburn, M. (1986). Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-Sepharose. Journal of Biological Chemistry, 261, 12665–12674.
Pfeilschifter, J., & Mundy, G. R. (1987). Modulation of transforming growth factor beta activity in bone cultures by osteotropic hormones. Proceedings of the National Academy of Sciences, 84, 2024–2028.
Kang, Y., Siegel, P. M., Shu, W., Drobnjak, M., Kakonen, S. M., Cordon-Cardo, C., et al. (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Research, 3, 537–549.
Mundy, G. R., DeMartino, S., & Rowe, D. W. (1981). Collagen and collagen fragments are chemotactic for tumor cells. Journal of Clinical Investigation, 68, 1102–1105.
Orr, F. W., Varani, J., Gondek, M. D., Ward, P. A., & Mundy, G. R. (1979). Chemotactic response of tumor cells to productions of resorbing bone. Science, 203, 176–179.
Zou, Y.-R., Kottmann, A. H., Kuroda, M., Tainwchi, I., & Littman, D. R. (1998). Function of the chemokine receptor CXCR4 in hematopoiesis and in cerebellar development. Nature, 393, 595–599.
Nagasawa, T., Hirota, S., Tachibana, K., Takakura, N., Nishikawa, S., Kitamura, Y., et al. (1996). Defects of B-cell lymphopoiesis and bone marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature, 382, 635–638.
Bluel, C. C., Fuhlbrigge, R. C., Casanovas, J. M., Aiuiti, A., & Springer, T. A. (1996). Highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). Journal of Experimental Medicine, 184, 1101–1109.
D’Apuzzo, M., Rolink, A., Loetscher, M., Hoxie, J. A., Clark-Lewis, I., Melchers, F., et al. (1997). The chemokine SDF-1, stromal cell-derived factor-1, attracts early stage B cell precursors via the chemokine receptor CXCR4. European Journal of Immunology, 27, 1788–1793.
Jung, Y., Wang, J., Schneider, A., Sun, Y.-X., Koh-Paige, A. J., Osman, N. I., et al. (2006). Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone, 38, 497–508.
Wang, J., Wang, J., Sun, Y.-X., Song, W., Nor, J. E., Wang, C. Y., et al. (2005). Diverse signaling pathways through the SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to altered patterns of cytokine secretion and angiogenesis. Cellular Signalling, 17, 1578–1592.
Luker, K. E., & Luker, G. D. (2005). Functions of CXCL12 and CXCR4 in breast cancer. Cancer Letters.
Muller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M. E., et al. (2001). Involvement of chemokine receptors in breast cancer metastasis. Nature, 410, 50–56.
Sun, Y.-X., Schneider, A., Jung, Y., Wang, J., Dai, J., Wang, J., et al. (2005). Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. Journal of Bone and Mineral Research, 20, 318–329.
Siclari, V. A., Guise, T. A., & Chirgwin, J. M. (2006). Molecular interactions between breast cancer cells and the bone microenvironment drive skeletal metastases. Cancer and Metastasis Reviews, 25, 621–633.
Yoneda, T. (2000). Cellular and molecular basis of preferential metastasis of breast cancer to bone. Journal of Orthopaedic Science, 5, 75–81.
Guise, T. A. (2000). Molecular mechanisms of osteolytic bone metastases. Cancer, 88, 2892–2898.
Pederson, L., Winding, B., Foged, N. T., Spelsberg, T. C., & Oursler, M. J. (1999). Identification of breast cancer cell line-derived paracrine factors that stimulate osteoclast activity. Cancer Research, 59, 5849–5855.
Badache, A., & Hynes, N. E. (2001). Interleukin 6 inhibits proliferation and, in cooperation with an epidermal growth factor receptor autocrine loop, increases migration of T47D breast cancer cells. Cancer Research, 61, 383–391.
Kotake, S., Sato, K., Kim, K. J., Takahashi, N., Udagawa, N., Nakamura, I., et al. (1996). Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. Journal of Bone and Mineral Research, 11, 88–95.
Scapini, P., Morini, M., Tecchio, C., Minghelli, S., Di Carlo, E., Tanghetti, E., et al. (2004). CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. Journal of Immunology, 172, 5034–5040.
Goede, V., Brogelli, L., Ziche, M., & Augustin, H. G. (1999). Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. International Journal of Cancer, 82, 765–770.
Fitzgerald, K. A., O’Neill, L. A. J., Gearing, A. J. H., & Callard, R. E. (2001). The cytokine facts book. New York: Academic.
Kinder, M. , Chislock, E. M., Bussard, K. M., Shuman, L. A., & Mastro, A. M. (2007). Metastatic breast cancer induces an osteoblast inflammatory response. Experimental Cell Research, Oct. 4 (in press).
Neumark, E., Cohn, M. A., Lukanidin, E., Witz, I. P., & Ben-Baruch, A. (2002). Possible co-regulation of genes associated with enhanced progression of mammary adenocarcinomas. Immunology Letters, 82, 111–121.
Neumark, E., Sagi-Assif, O., Shalmon, B., Ben-Baruch, A., & Witz, I. P. (2003). Progression of mouse mammary tumors: MCP-1-TNF-alpha cross regulatory pathway and clonal expression of promalignancy and antimalignancy factors. International Journal of Cancer, 106, 879–886.
Ueno, T., Toi, M., Saji, J., Muta, M., Bando, H., Kuroi, K., et al. (2000). Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clinical Cancer Research, 6, 3282–3289.
Mukaida, N., Ketlunsky, S. A., & Matsushima, K. (2003). The cytokine handbook. Amsterdam: Academic.
Bischoff, D. S., Zhu, J. H., Makhijani, N. S., & Yamaguchi, D. T. (2005). KC chemokine expression by TGF-B in C3H10T1/2 cells induced towards osteoblasts. Biochemical and Biophysical Research Communications, 326, 364–370.
Wolfle, U., Muller, V., & Pantel, K. (2006). Disseminated tumor cells in breast cancer: detection, characterization and clinical relevance. Future Oncology, 2, 553–561.
Dhurjati, R., Liu, X., Gay, C. V., Mastro, A. M., & Vogler, E. A. (2006). Extended-term culture of bone cells in a compartmentalized bioreactor. Tissue Engineering, 12, 3045–3054.
Dhurjati, R., Shuman, L. A., Krishnan, V., Mastro, A. M., Gay, C. V., & Vogler, E. A. (2006). Compartmentalized bioreactor: In vitro model for osteobiology and osteopathology. 28th Annual Meeting of the American Society for Bone and Mineral Research, 21, 349.
Deyama, Y., Takeyama, S., Suzuki, K., Yoshimura, Y., Nishikata, M., & Matsumoto, A. (2001). Inactivation of NF-KB involved in osteoblast development through interleukin-6. Biochemical and Biophysical Research Communications, 282, 1080–1084.
Ishimi, Y., Miyaura, C., Jin, C. H., Akatsu, T., Abe, E., Nakamura, Y., et al. (1990). IL-6 is produced by osteoblasts and induces bone resorption. Journal of Immunology, 145, 3297–3303.
Bendre, M., Gaddy, D., Nicholas, R. W., & Suva, L. J. (2003). Breast cancer metastasis to bone: It is not all about PTHrP. Clinical Orthopaedics and Related Research, 415S, S39–S45.
Girasole, G., Jilka, R. L., Passeri, G., Boswell, S., Boder, G., Williams, D. C., et al. (1992). 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: A potential mechanism for the antiosteoporotic effect of estrogens. Journal of Clinical Investigation, 89, 883–891.
Linkhart, T. A., Linkhart, S. G., MacCharles, D. C., Long, D. L., & Strong, D. D. (1991). Interleukin-6 messenger RNA expression and interleukin-6 protein secretion in cells isolated from normal human bone. Journal of Bone and Mineral Research, 6, 1285–1294.
O’Keefe, R. J., Teot, L. A., Singh, D., Puzas, J. E., Rosier, R. N., & Hicks, D. G. (1997). Osteoclasts constitutively express regulators of bone resorption: An immunohistochemical and in situ hybridization study. Laboratory Investigation, 76, 457–465.
Chaudhary, L. R., & Avioli, L. V. (1994). Dexamethasone regulates IL-1 beta and TNF-alpha-induced interleukin-8 production in human bone marrow stromal and osteoblast-like cells. Calcified Tissue International, 55, 16–20.
Bendre, M. S., Margulies, A. G., Walser, B., Akel, N. S., Bhattacharrya, S., Skinner, R. A., et al. (2005). Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Research, 65, 11001–11009.
Rothe, L., Collin-Osdoby, P., Chen, Y., Sunyer, T., Chaudhary, L., Tsay, A., et al. (1998). Human osteoclasts and osteoclast-like cells synthesize and release high basal and inflammatory stimulated levels of the potent chemokine interleukin-8. Endocrinology, 139, 4353–4363.
Sozzani, S., Locati, M., Allavena, P., Van Damme, J., & Mantovani, A. (1996). Chemokines: A superfamily of chemotactic cytokines. International Journal of Clinical & Laboratory Research, 26, 69–82.
Lipton, A. (2000). Bisphosphonates and breast carcinoma: Present and future. Cancer, 88, 3033–3037.
Oslen, N. J., Gu, X., & Kovacs, W. J. (2001). Bone marrow stromal cells mediate androgenic suppression of B lymphocyte development. Journal of Clinical Investigation, 108, 1697–1704.
Schultz-Cherry, S., Ribeiro, S., Gentry, L., & Murphy-Ullrich, J. E. (1994). Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. Journal of Biological Chemistry, 269, 26775–26782.
Acknowledgments
KMB was supported by a predoctoral traineeship from the U.S. Army Medical and Materiel Research Command Breast Cancer Program (W81XWH-06-1-0363). The authors’ work was supported by was supported by the U.S. Army Medical and Material Research Command Breast Cancer Program; (DAMD 17-02-1-0358 and W81XWH-06-1-0432 to AMM,); National Foundation for Cancer Research, Center for Metastasis Research; The Susan G. Komen Breast Cancer Foundation, BCTR104406; The American Institute of Cancer Research, #06A027-REV2; The authors thank Richard Ball of the Immunomodulation Core, Penn State GCRC (work supported by NIHM01RR10732) for technical advice regarding ELISAs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bussard, K.M., Gay, C.V. & Mastro, A.M. The bone microenvironment in metastasis; what is special about bone?. Cancer Metastasis Rev 27, 41–55 (2008). https://doi.org/10.1007/s10555-007-9109-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-007-9109-4