
Abstract
S100A6 protein belongs to the A group of the S100 protein family of Ca2+-binding proteins. It is expressed in a limited number of cell types in adult normal tissues and in several tumor cell types. As an intracellular protein, S100A6 has been implicated in the regulation of several cellular functions, such as proliferation, apoptosis, the cytoskeleton dynamics, and the cellular response to different stress factors. S100A6 can be secreted/released by certain cell types which points to extracellular effects of the protein. RAGE (receptor for advanced glycation endproducts) and integrin β1 transduce some extracellular S100A6’s effects. Dosage of serum S100A6 might aid in diagnosis in oncology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
S100A6 belongs to the S100 family of Ca2+-binding proteins of the EF-hand type [1]. It is also known as 2A9, 5B10, CABP, Cacy, Calcyclin, growth factor-inducible protein 2A9, PRA—prolactin receptor associated protein, and S100 calcium binding protein A6, S100a6. As a member of the A group of S100 proteins, human S100A6 gene maps to chromosome 1q21 [2], a locus where frequent chromosomal rearrangements occur in neoplasia. S100A6 is expressed as an 89-amino-acid protein in mouse and rat and a 90-amino-acid protein in human and rabbit. Two chicken isoforms, A (92 amino acids) and B (91 amino acids), probably result from alternative mRNA splicing [3]. S100A6 from all these sources differs in only a few amino acids and in the length of the carboxy terminus. The S100A6 gene was initially identified in growth-arrested rodent fibroblasts stimulated with serum [4] and suggested to have a role in cell-cycle progression as inferred by its upregulation in several tumors [5–7]. S100A6 was found to interact with calcyclin-binding protein/Siah-1-interacting protein (CacyBP/SIP) [8]. Because CacyBP/SIP is a component of the ubiquitin ligase complexes, S100A6 was suggested to be involved in the ubiquitination of β-catenin [9], thus supporting the possibility that S100A6 might play a role in the control of cell-cycle progression. S100A6’s ability to inhibit the interaction between the heat shock proteins (Hsp70 and Hsp90) and Sgt1 [10] and Hop [11], suggested a potential role for S100A6 in the cellular response to different stress factors. In this respect, S100A6 was found to favor apoptosis in some cell types [12, 13], but to limit it in others [14]. The in vitro interaction of S100A6 with caldesmon [15], calponin [16], tropomyosin [17], and kinesin light chain [11] suggested that S100A6 might be involved in the regulation of cytoskeleton dynamics, particularly microfilament dynamics [18], and in vesicular transport. As an extracellular factor, S100A6 was shown to be involved in the release of lactogen II [19], insulin [20], and histamine [21]. By binding to the transmembrane receptor for advanced glycation endproducts (RAGE), S100A6 induced neuronal apoptosis by causing reactive oxygen species (ROS)-dependent activation of JNK and of caspases 3 and 7 [22]. RAGE transduces extracellular effects of several S100 proteins (23). Integrin β1 is another potential membrane protein transducing extracellular effects of S100A6 [24]. The present review seeks to critically summarize information about functional roles of S100A6 also in light of results of recent studies of S100A6 in cancer (Table 1).
Regulation of expression
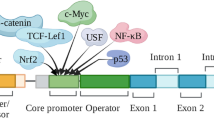
Several factors have been shown to increase S100A6 mRNA and protein levels such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and serum [25], retinoic acid [26], estrogen [27], palmitate [28], vasopressin [29], and gastrin [30] (Fig. 1). S100A6 levels are also upregulated upon stress conditions such as ischemia [31], mechanical force [32], irradiation [33], and oxidative stress [34].

Regulation of S100A6 expression. Several extracellular factors can upregulate S100A6 expression via the indicated transcription factors. The tumor suppressor, p53, decreases SP1 and NF-κB binding to the S100A6 promoter
In vivo, S100A6 protein levels are elevated in myocardial disease [35] and in many types of tumor cells (see below). However, the primary cause(s) of this increase remain(s) to be fully elucidated. A decrease in S100A6 levels was observed during the course of TRAIL (tumor necrosis factor-related apoptosis-inducing ligand)- and etoposid-induced apoptosis in human breast cancer cells [36]. At the transcriptional level, USF (upstream transcription factor), NF-κB (nuclear transcription factor κB), Sp1 (specificity protein 1), and Nrf2 (Nf-E2-related factor 2) have been shown to activate the S100A6 gene promoter [13, 34, 37–39], whereas the tumor suppressor, p53, acts indirectly to suppress transcription via interference with Sp1 and NF-κB function on the S100A6 promoter [40]. Insufficient suppression of S100A6 gene by p53 mutants might thus be responsible for S100A6 overexpression and cell-cycle deregulation in cancer.
Regulation of activity
Like other members of the S100 protein family (excepting S100G) [1], S100A6 exists within cells in the form of a homodimer in which the two subunits are arranged in an antiparallel fashion [41]. Each subunit consists of two EF-hand (helix–loop–helix) motifs tethered by a flexible hinge domain and a C-terminal tail [1, 42, 43]. Each S100A6 subunit binds two calcium ions. Ca2+-binding induces conformational changes in the C-terminal half of each subunit of the S100A6 homodimer [41] as observed for other S100 proteins [1]. The Ca2+ binding causes hydrophobic residues of the hinge region, helices III and IV of each subunit to become exposed which enables S100A6 to interact with many target proteins such as glyceraldehyde-3-phosphate dehydrogenase, annexin II, annexin XI, annexin VI, and tropomyosin [44]. Additional intracellular interacting proteins are caldesmon, calponin and lysozyme, CacyBP/SIP, Sgt1, and melusin [44], p53 [13, 45], and the Hsp90/Hsp70-organizing protein (Hop) and kinesin light chain [10, 11, 43]. A Ca2+- or Zn2+-dependent S100A6/S100B heterodimer was identified in a yeast two-hybrid assay and confirmed in vivo [46, 47]. However, no functional correlates have been reported for these interactions with the exception of CacyBP/SIP (see subheading S100A6 and cell proliferation and cancer).
S100A6 also binds Zn2+ [48]. The binding of Zn2+ induces conformational changes in the S100A6 molecule that are different from those observed following Ca2+ binding. At present, there are no data showing a potential zinc-dependent activity of S100A6.
S100A6 and cell proliferation and cancer
S100A6 affects cell proliferation and cancer development by acting from within and from outside cells. S100A6 is overexpressed in breast, stomach, pancreas and colon cancer and in melanoma, whereas it is underexpressed in prostate and oral cancer [49]. It is considered a diagnostic marker or prognostic factor in pancreatic, gastric and prostate cancer, melanoma, non-small cell lung carcinoma, and hepatocellular carcinoma. S100A6 is also overexpressed in thyroid carcinoma [50, 51] and is suggested to play a key role in the progression and development of papillary thyroid carcinoma [52]. S100A6 affects murine models of cancer; however, its contribution to promoting a cancerous phenotype has only been examined in a limited number of model systems and the mechanistic basis for the observed effects on tumor progression has not been fully delineated [43]. In gastric cancer cells, S100A6 negatively regulates its partner-CacyBP/SIP-mediated inhibition of cell proliferation and tumorigenesis by affecting β-catenin degradation (Fig. 2) [53]. In addition, S100A6 enhanced migration and invasion of pancreatic ductal adenocarcinoma cells and promoted epithelial-mesenchymal transition via activation of β-catenin [54] (Fig. 2). The tumorigenic activity of overexpressed S100A6 was reported for clear cell renal cell carcinoma in which the protein inhibited the expression of the anti-tumor chemokine, CXCL14, and CXCL14-induced apoptosis [55]. It has been hypothesized that S100A6 may regulate CXCL14 through estrogen receptor 1 [55], but no experimental evidence for this has been presented. Additional evidence for S100A6-CacyBP/SIP interactions comes from the observation that S100A6 inhibits CacyBP/SIP phosphorylation by casein kinase II similar to the CacyBP/SIP phosphorylation inhibitor, DRB, which results in reduced phosphatase CacyBP/SIP activity towards the mitogen-activated protein kinases (MAPKs), ERK1/2 [56], which in turn might sustain cell proliferation and/or tumorigenesis.

Implications of S100A6 interaction with CacyBP/SIP. S100A6 inhibits CacyBP/SIP thereby stimulating cell proliferation and migration, tumorigenesis, and epithelial-mesenchymal transition via inhibition of β-catenin degradation, inhibiting cell differentiation, and participating in aging and neurodegeneration
S100A6 is reported to regulate endothelial cell cycle and senescence. In primary human endothelial cells, depletion of S100A6 caused increased cell-cycle arrest in G2/M phase. S100A6 depletion caused a decrease in both cyclin-dependent kinase 1 (CDK1), phosphorylated CDK1 levels, and cyclin A1 and cyclin B genes with effects on cell-cycle progression [57] likely via inhibition of antiproliferative signal transducers and activators of transcription (STAT) 1 signaling [58] (Fig. 3a). A role for S100A6 as an intracellular regulator of cell proliferation and differentiation is suggested by the finding that S100A6 becomes downregulated at the beginning of keratinocyte differentiation and that S100A6 overexpression in these cells causes accelerated proliferation, enhanced adhesive properties, and reduced differentiation [59] (Fig. 3b). Downregulation of S100A8 and S100A9 proteins upon differentiation of monocytes into mature macrophages has been reported [60, 61]. Transient downregulation has also been reported for S100B protein at the beginning of myoblast, neuronal, chondrocyte, and astrocyte differentiation and this event has been shown to be permissive for the differentiation of these cell types [62–67]. Whether transient downregulation at the beginning of cell differentiation is a general property of S100 proteins remains to be established.

Roles of S100A6 in cell proliferation and differentiation. a S100A6 is suggested to stimulate cell-cycle progression and to inhibit differentiation and senescence in endothelial cells. b S100A6 becomes downregulated at the beginning of keratinocyte and astrocyte differentiation, and S100A6 overexpression in these cells causes accelerated proliferation, enhanced adhesive properties, and reduced differentiation. c S100A6 is upregulated in and secreted by colorectal carcinoma cells. Secreted S100A6 stimulates cell proliferation and migration in a receptor-mediated manner
Transfection with recombinant S100A6 of or administration of recombinant S100A6 to HCT116, a colorectal carcinoma cell line with relative low S100A6 expression, resulted in enhanced cell proliferation and migration, MAPK activation in vitro, and tumor growth in vivo. Conversely, RNAi-mediated knockdown of S100A6 in LoVo, a colorectal carcinoma cell line with relatively high S100A6 expression, resulted in reduced cell proliferation, migration, and MAPK activity. S100A6-induced proliferation was partially attenuated by an ERK1/2 inhibitor, while migration was suppressed by a p38 MAPK inhibitor [68] (Fig. 3c). These results suggest that S100A6 might act as an extracellular signaling molecule affecting cancer cells in a receptor-mediated manner. RAGE is a candidate receptor as it transduces certain effects of S100A6 on responsive cells [22, 69]. However, whether RAGE transduces S100A6 effects on colorectal carcinoma cells remains to be determined.
Regarding S100A6-RAGE interaction, one study reported that S100A6 binds the C1 and C2 domains of RAGE [22], whereas another study showed that S100A6 binds RAGE V domain similar to other S100 proteins [1, 70]. Recent work [71] suggests that the primary S100A6 binding site is formed by the RAGE C1 domain and that S100A6 adopts a dimeric conformation different from all known S100 dimers; the N-terminus of helix H1 from one S100A6 subunit inserts into the hydrophobic cleft formed between helices H3 and H4 from the opposite subunit in the presence of Ca2+. Incidentally, this cleft binds RAGE [71] and CacyBP/SIP [72].
Contrasting effects have been reported regarding effects of S100A6 on cell proliferation. S100A6 increases adhesion and inhibits proliferation of mesenchymal stem cells isolated from Wharton’s jelly of the umbilical cord [24]. Integrin β1 appears to be the membrane protein (receptor) transducing these S100A6 effects because neutralization of integrin β1, but not RAGE blunted them. On the other hand, exogenous expression of S100A6 in mesenchymal stem cells increased proliferation and inhibited osteogenic differentiation, and stimulated osteosarcoma growth in vivo [73]. Whether these latter S100A6’s effects result from intracellular regulatory activities, receptor-mediated mechanism(s), or both remains to be determined.
Interaction of certain S100 proteins with RAGE and with receptors other than RAGE is not unusual [1]. For example, S100A4, S100A8/S100A9, and S100B engage RAGE in several cell types but can act independently of RAGE on other cell types [1, 23]. Thus, S100A4 stimulates neurite outgrowth by binding to heparan sulphate proteoglycans and a putative Gαq-coupled receptor [74] and stimulates tumor progression by interacting with EGFR ligands [75]. S100A8 and S100A9 can activate toll-like receptor 4 in phagocytes [76], S100A12 activates a G-protein coupled receptor in mast cells and monocytes [77], and S100B binds bFGF and enhances bFGF/FGFR1 signaling and simultaneously blocks RAGE in high-density myoblast cultures thereby promoting cell proliferation [78].
S100A6 modulates RAGE-dependent survival of neuroblastoma cells by triggering apoptosis and generation of ROS through c-Jun NH2 terminal protein kinase activation [22]. S100A6 may regulate secretory processes in some cells. It stimulates secretion of lactogen II by trophoblasts [19] and insulin release from pancreatic islet cells [20], and may modulate allergic responses by inhibiting histamine release by mast cells [21, 79]. The receptor(s) transducing these effects remain(s) to be determined.
The role of S100A6 as an intracellular regulator of cell proliferation/apoptosis is further complicated by its effects on the anti-tumor, p53 [80], and by the finding that p53 acts indirectly to suppress S100A6 transcription [40] (Fig. 1). S100A6 competes with MDM2, an ubiquitin E3 ligase that degrades p53, for binding to p53, and with p300 acetyltransferase. Once acetylated, p53 loses the ability to bind S100A6, suggesting that high S100A6 concentrations might interfere with p53 acetylation, and thus, that S100A6 might protect p53 against untimely degradation and/or acetylation thus resulting in the promotion of p53 nuclear translocation and, likely, p53 transcriptional activity. In this perspective, S100A6 might aid in cell proliferation arrest and/or apoptosis. However, the opposite has also been observed in mixed-lineage leukemia/AF4-positive acute lymphoblastic leukemia, where IL-24-induced inhibition of S100A6 expression was shown to exert pro-apoptotic effects, which points to an anti-apoptotic, tumorigenic role of S100A6 in these cells [81, 82] (Fig. 4). For S100A6’s anti-apoptotic effects, also see subheading S100A6 and stem cells in the following.

S100A6 exerts anti-apoptotic effects in mixed-lineage leukemia (MLL)/AF4-positive leukemia cells. In MLL/AF4-positive acute lymphoblastic leukemia cells, IL-24-induced inhibition of S100A6 expression exerts pro-apoptotic effects, which points to an anti-apoptotic function of S100A6 in these cells
Other S100 proteins have been implicated in tumorigenesis and metastasis [1, 42, 43]. Intracellular S100A4 has pro-metastatic activity [83]. Once released by stromal/epithelial cancer cells, S100A4 stimulates cancer cell invasiveness [84], cooperates with the chemochine, RANTES (CCL5), in promoting tumor progression [85], and interacts with EGF receptor ligands, thereby enhancing EGFR/ErbB2 receptor signaling and cell proliferation [74]. S100A7 overexpression is seen in invasive breast cancer [86] and enhances mammary tumorigenesis and breast cancer metastasis RAGE dependently [87, 88]. The S100A8/S100A9 heterodimer (calprotectin) facilitates tumor cell invasion [89, 90]. Overexpression of S100B is associated with and exerts a pathogenic role in malignant melanoma [91–93] and glioma [62, 94–96]. Thus, several S100 proteins have a role in tumor development and progression with a certain specificity in terms of mechanism of action and cell type.
S100A6 and cytoskeleton
Intracellular S100A6 has been functionally linked to changes in cellular motility and cytoskeletal reorganization [42, 79], but a clear mechanistic picture is still lacking. Knockdown of S100A6 in NIH-3T3 fibroblastic cells causes a reorganization of the actin cytoskeleton with an extensive cortical network of actin filaments and tropomyosin structures and increase in the number of focal adhesions at the cell periphery [18, 97]. Thus, S100A6 effects on actin filaments and tropomyosin structures might be responsible at least in part for the large increase in lamellipodia and possibly for the enhancement of cellular motility seen when S100A6 levels are reduced by siRNA techniques [98]. The involvement of S100A6 in the motility of cancer cells has also been reported, albeit with contradictory outcomes. Down- or upregulating of S100A6 expression in osteosarcoma cells led to increased or decreased migration, respectively, suggesting a role for S100A6 as an inhibitor of cell motility in cultured cells [98, 99]. However, S100A6 has also been shown to promote cellular motility in pancreatic cancer cells [100, 101] by a mechanism that is dependent on the presence of annexin 2. Elevated levels of intracellular S100A6 have been shown to be associated with tumorigenesis (reviewed in [22]) and the ability of colorectal adenocarcinoma cells [102] and Ras-transformed NIH 3T3 cells [103] to metastasize to form secondary lesions. Yet, the molecular mechanisms underpinning S100A6’s ability to regulate cell motility have remained elusive. Direct interaction between S100A6 and the tropomyosin–actin complex has been shown in vitro [17], but remains to be confirmed in vivo; the only current evidence suggests that S100A6 acts as a downregulator of tropomyosin expression [18]. S100A6 interacts in vitro with other components of the actin cytoskeletal architecture, such as the myosin ATPase inhibitors, caldesmon [104, 105], and calponin [16], but no mechanistic link to cell motility has been demonstrated. Other S100 proteins interact with the cytoskeleton. S100B binds to tubulin and inhibits its polymerization into microtubules [106, 107], associates with microtubules and intermediate filaments in cultured cells [108, 109], increases the Ca2+-sensitivity of microtubules [110], and promotes stress fibers formation in and migration of proliferating astrocytes via a Src/PI3K/RhoA/ROCK pathway [65]. S100A1 interferes with the assembly of desmin intermediate filaments [111] and interacts with the giant sarcomeric protein, titin [112] thereby reducing sarcomeric passive tension before contraction [113]. Indeed, S100A1 gene delivery rescues failing myocardium [114]. Interaction with non-muscular myosin heavy chain IIA, tropomyosin, and actin is causally related to the pro-metastatic activity of intracellular S100A4 [42, 83, 115]. S100A8, S100A9, and the heterocomplex S100A8/S100A9 (also known as calprotectin) associate with vimentin intermediate filaments during Ca2+ transients in monocytes [116] and with keratin intermediate filaments in keratinocytes [117], and stimulate microtubule assembly during transendothelial migration of phagocytes [118]. Thus, regulation of the cytoskeleton and cytoskeleton-associated activities seems to be one intracellular function shared by several S100 proteins with each of them showing a preferential molecular target among cytoskeletal elements likely due to the unique length and primary sequence of the hinge region and C-terminal tail of individual S100 members and the cell type(s), where they are expressed.
S100A6 and stem cells
S100A6 was shown to be expressed in neural stem cells in the subgranular zone of the dentate gyrus in adult hippocampus [119]—a major neurogenic niche. These S100A6-expressing cells were recognized as astrocyte precursors. The finding that S100A6 was not detected in mature astrocytes suggested that S100A6 might play an important, yet unknown role during astrocytic differentiation of neural stem cells. Possibly, S100A6 has to be downregulated for astrocyte precursors to undergo differentiation, as observed for keratinocyte differentiation [59] (Fig. 3b) and with other S100 proteins [60–67]. S100A6 also marks glial precursor cells in neuroblastoma [120].
S100A6 expression is increased in the peri-infarct zone of rat heart postinfarction and functions as a global negative regulator of the induction of cardiac genes by trophic stimuli [35]. S100A6 is induced in cardiomyocytes by TNF-α via NF-κB activation and protects cardiomyocytes from TNF-α-induced apoptosis by associating with p53 and interfering with p53 phosphorylation [14] (Fig. 5). Similar to S100A6, S100B is induced in cardiomyocytes surviving an infarct, and can limit the hypertrophic response by inhibiting expression of α-actin and β-myosin [121]. However, extracellular S100B causes cardiomyocyte apoptosis in a RAGE-mediated manner [122] and induces myofibroblast proliferation in a RAGE-VEGF-mediated manner potentially contributing to the scar formation observed in infarcted myocardium [123]. At present, there is no information about potential extracellular effects of S100A6 in the context of cardiac infarction.

Induction of S100A6 expression in peri-infarct cardiomyocytes. The TNF-α/NF-κB axis induces S100A6 in peri-infarct cardiomyocytes. Induced S100A6 reduces the cardiomyocyte hypertrophic response and inhibits the pro-apoptotic effect of p53
S100A6 and neurodegenerative diseases
In Alzheimer’s disease mouse models, astrocytic S100A6 protein was shown to be homogeneously upregulated within the white matter, whereas within the grey matter, almost all S100A6 immunoreactivity was found to be concentrated in astrocytes surrounding the Aβ amyloid deposits of senile plaques [124]. S100A6 is also overexpressed in astrocytes located near impaired axons of motoneurons in amyotrophic lateral sclerosis (ALS) [125]. These findings suggest that S100A6 might participate in the pathophysiology of Alzheimer’s disease and ALS, respectively. Mechanistically, S100A6 was shown to form oligomers and amyloid-like fibrils, an event negatively regulated by Ca2+, and to potentiate in vitro the aggregation of superoxide dismutase-1 (SOD1) [126], that forms cytoplasmic aggregates in ALS-affected neurons. Although S100A6 oligomers but not fibrils proved toxic to neuronal cell in culture [126], there are no data to demonstrate that S100A6 plays a role in the promotion of SOD1 aggregates in ALS neurons. Other S100 proteins (e.g., S100B, S100A8, S100A9, and S100A12) are known to take part in the pathophysiology of neurodegenerative disorders by affecting neurons, astrocytes, and/or microglia mostly as extracellular signals [1, 127–140]. Likely, accumulation of S100 proteins, including S100A6, in the brain extracellular space might be a consequence rather than a cause of the underlying neurodegenerative disorder, and represents one molecular means by which astrocytes and microglia respond to noxious stimuli (such as disturbances of the local circulation, changes in the redox status, metabolic disorders, etc) to bring about a complex inflammatory response that cannot, however, progress through the canonical phases, i.e., an early proinflammatory, defense phase, and a late reparative phase, due to the persistence of the underlying noxious stimulus and the intervention of several membrane receptors. Overall, the available information about the involvement of several S100 proteins in the pathophysiology of neurodegenerative disorders [124, 127–140] suggests that targeting one single S100 protein might not be sufficient to reverse the pathology.
S100A6 as a serum marker of disease
Serum levels of S100A6 are significantly elevated in early stage non-small cell lung cancer [141], gastric cancer [142], urinary bladder urothelial carcinoma [143] and ovarian cancer [144] as well as in acute coronary syndrome and myocardial infarction [145]. Elevation of serum levels of other S100 proteins (e.g., S100B, S100A4, S100A7, S100A8, and S100A9) has been reported in several cancers [146–150]. However, pending additional work on larger cohorts of patients, usage of serum levels of S100A6 in diagnosis of the above tumors [141–144] is promising.
Conclusions
Studies of S100A6’s interaction with and inhibition of its partner, CacyBP/SIP—an inhibitor of cell proliferation and tumorigenesis by virtue of its ability to promote degradation of β-catenin—support a role for S100A6 as a positive regulator of cell proliferation in the epidermis, endothelial cells, and tumor cells and as an anti-apoptotic factor in certain leukemias. In addition, upregulation of S100A6 in peri-infarct cardiomyocytes results in reduction of p53-induced apoptosis via interference with p53 phosphorylation and inhibition of induction of fetal genes responsible for cardiomyocyte hypertrophy. On the other hand, interaction with the tumor suppressor, p53, implicates S100A6 in apoptosis, with high concentrations of S100A6, as is typical of certain tumor cells, protecting p53 from inactivation by p300 acetyltransferase and degradation by MDM2. S100A6 can be secreted/released by certain cell types which points to extracellular effects of the protein. RAGE and integrin β1 might transduce extracellular S100A6’s effects, but further analyses in physiological and pathological contexts are required. Finally, dosage of serum S100A6 might aid in diagnosis in oncology and acute coronary syndrome. The growing interest on S100A6 in cancer makes this protein a potential therapeutic target.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- CacyBP/SIP:
-
Calcyclin-binding protein/Siah-1-interacting protein
- CDK:
-
Cyclin-dependent kinase
- EGF:
-
Epidermal growth factor
- Hsp:
-
Heat shock protein
- IL:
-
Interleukin
- MDM:
-
Transformed mouse 3T3 cell double minute
- NF-κB:
-
Nuclear transcription factor κB
- Nrf2:
-
Nf-E2 related factor 2
- PDGF:
-
Platelet-derived growth factor
- RAGE:
-
Receptor for advanced glycation end product
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- Sp1:
-
Specificity protein 1
- TNF:
-
Tumor necrosis factor
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- USF:
-
Upstream transcription factor
- VEFG:
-
Vascular endothelial growth factor
References
Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL (2013) Functions of S100 proteins. Curr Mol Med 13:24–57
Marenholz I, Heizmann CW, Fritz G (2004) S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 322:1111–1122
Allen BG, Andrea JE, Sutherland C, Schönekess BO, Walsh MP (1997) Molecular cloning of chicken calcyclin (S100A6) and identification of putative isoforms. Biochem Cell Biol 75:733–738
Hirschhorn RR, Aller P, Yuan Z-A, Gibson CW, Baserga R (1984) Cell-cycle specific cDNAs from mammalian cells temperature sensitive for growth. Proc Natl Acad Sci USA 87:6004–6008
Calabretta B, Kaczmarek L, Mars W, Ochoa D, Gibson CW, Hirschhorn RR, Baserga R (1985) Cell-cycle-specific genes differentially expressed in human leukemias. Proc Natl Acad Sci USA 82:4463–4467
Calabretta B, Battini R, Kaczmarek L, de Riel JK, Baserga R (1986) Molecular cloning of the cDNA for a growth factor-inducible gene with strong homology to S-100, a calcium-binding protein. J Biol Chem 261:12628–12632
Weterman MA, Stoopen GM, van Muijen GN, Kuznicki J, Ruiter DJ, Bloemers HP (1992) Expression of calcyclin in human melanoma cell lines correlates with metastatic behavior in nude mice. Cancer Res 52:1291–1296
Filipek A, Kuźnicki J (1998) Molecular cloning and expression of a mouse brain cDNA encoding a novel protein target of calcyclin. J Neurochem 70:1793–1798
Matsuzawa SI, Reed JC (2001) Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell 7:915–926
Spiechowicz M, Zylicz A, Bieganowski P, Kuznicki J, Filipek A (2007) Hsp70 is a new target of Sgt1—an interaction modulated by S100A6. Biochem Biophys Res Commun 357:1148–1153
Shimamoto S, Takata M, Tokuda M, Oohira F, Tokumitsu H, Kobayashi R (2008) Interactions of S100A2 and S100A6 with the tetratricopeptide repeat proteins, Hsp90/Hsp70-organizing protein and kinesin light chain. J Biol Chem 283:28246–28258
Joo JH, Yoon SY, Kim JH, Paik SG, Min SR, Lim JS, Choe IS, Choi I, Kim JW (2008) S100A6 (calcyclin) enhances the sensitivity to apoptosis via the upregulation of caspase-3 activity in Hep3B cells. J Cell Biochem 103:1183–1197
Słomnicki ŁP, Nawrot B, Leśniak W (2009) S100A6 binds p53 and affects its activity. Int J Biochem Cell Biol 41:784–790
Tsoporis JN, Izhar S, Parker TG (2008) Expression of S100A6 in cardiac myocytes limits apoptosis induced by tumor necrosis factor-alpha. J Biol Chem 283:30174–33083
Mani RS, Kay CM (1992) Purification and characterization of a novel 12,000-Da calcium binding protein from smooth muscle. Arch Biochem Biophys 296:442–449
Wills FL, McCubbin WD, Kay CM (1994) Smooth muscle calponin–caltropin interaction: effect on biological activity and stability of calponin. Biochemistry 33:5562–5569
Golitsina NL, Kordowska J, Wang CL, Lehrer SS (1996) Ca2+-dependent binding of calcyclin to muscle tropomyosin. Biochem Biophys Res Commun 220:360–365
Breen EC, Tang K (2003) Calcyclin (S100A6) regulates pulmonary fibroblast proliferation, morphology, and cytoskeletal organization in vitro. J Cell Biochem 88:848–854
Thordarson G, Southard JN, Talamantes F (1991) Purification and characterization of mouse decidual calcyclin: a novel stimulator of mouse placental lactogen-II secretion. Endocrinology 129:1257–1265
Okazaki K, Niki I, Iino S, Kobayashi S, Hidaka H (1994) A role of calcyclin, a Ca2+-binding protein, on the Ca2+-dependent insulin release from the pancreatic beta cell. J Biol Chem 269:6149–6152
Fujii T, Kuzumaki N, Ogoma Y, Kondo Y (1994) Effects of calcium-binding proteins on histamine release from permeabilized rat peritoneal mast cells. Biol Pharm Bull 17:581–585
Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A (2007) S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem 282:31317–31331
Donato R (2007) RAGE: a single receptor for several ligands and different cellular responses. The case of certain S100 proteins. Curr Mol Med 7:711–724
Jurewicz E, Góral A, Filipek A (2014) S100A6 is secreted from Wharton’s jelly mesenchymal stem cells and interacts with integrin β1. Int J Biochem Cell Biol 55:298–303
Ghezzo F, Lauret E, Ferrari S, Baserga R (1988) Growth factor regulation of the promoter for calcyclin, a growth-regulated gene. J Biol Chem 263:4758–4763
Tonini GP, Casalaro A, Cara A, Di Martino D (1991) Inducible expression of calcyclin, a gene with strong homology to S-100 protein, during neuroblastoma cell differentiation and its prevalent expression in Schwann-like cell lines. Cancer Res 51:1733–1737
Hong EJ, Park SH, Choi KC, Leung PC, Jeung EB (2006) Identification of estrogen-regulated genes by microarray analysis of the uterus of immature rats exposed to endocrine disrupting chemicals. Reprod Biol Endocrinol 29(4):49
Busch AK, Cordery D, Denyer GS, Biden TJ (2002) Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic beta-cell function. Diabetes 51:977–987
Courtois-Coutry N, Le Moellic C, Boulkroun S, Fay M, Cluzeaud F, Escoubet B, Farman N, Blot-Chabaud M (2002) Calcyclin is an early vasopressin-induced gene in the renal collecting duct. Role in the long term regulation of ion transport. J Biol Chem 277:25728–25734
Kucharczak J, Pannequin J, Camby I, Decaestecker C, Kiss R, Martinez J (2001) Gastrin induces over-expression of genes involved in human U373 glioblastoma cell migration. Oncogene 20:7021–7028
Lewington AJ, Padanilam BJ, Hammerman MR (1997) Induction of calcyclin after ischemic injury to rat kidney. Am J Physiol 273:F380–F385
Breen EC, Fu Z, Normand H (1999) Calcyclin gene expression is increased by mechanical strain in fibroblasts and lung. Am J Respir Cell Mol Biol 21:746–752
Orre LM, Pernemalm M, Lengqvist J, Lewensohn R, Lehtiö J (2007) Up-regulation, modification, and translocation of S100A6 induced by exposure to ionizing radiation revealed by proteomics profiling. Mol Cell Proteomics 6:2122–2131
Leśniak W, Szczepańska A, Kuźnicki J (2005) Calcyclin (S100A6) expression is stimulated by agents evoking oxidative stress via the antioxidant response element. Biochim Biophys Acta 1744:29–37
Tsoporis JN, Marks A, Haddad A, O’Hanlon D, Jolly S, Parker TG (2005) S100A6 is a negative regulator of the induction of cardiac genes by trophic stimuli in cultured rat myocytes. Exp Cell Res 303:471–481
Leong S, Christopherson RI, Baxter RC (2007) Profiling of apoptotic changes in human breast cancer cells using SELDI-TOF mass spectrometry. Cell Physiol Biochem 20:579–590
Leśniak W, Jezierska A, Kuźnicki J (2000) Upstream stimulatory factor is involved in the regulation of the human calcyclin (S100A6) gene. Biochim Biophys Acta 1517:73–81
Joo JH, Kim JW, Lee Y, Yoon SY, Kim JH, Paik SG, Choe IS (2003) Involvement of NF-kappaB in the regulation of S100A6 gene expression in human hepatoblastoma cell line HepG2. Biochem Biophys Res Commun 307:274–280
Thaisuchat H, Baumann M, Pontiller J, Hesse F, Ernst W (2011) Identification of a novel temperature sensitive promoter in CHO cells. BMC Biotechnol 11:51
Króliczak W, Pietrzak M, Puzianowska-Kuznicka M (2008) P53-dependent suppression of the human calcyclin gene (S100A6): the role of Sp1 and of NFkappaB. Acta Biochim Pol 55:559–570
Otterbein LR, Kordowska J, Witte-Hoffmann C, Wang CL, Dominguez R (2002) Crystal structures of S100A6 in the Ca2+-free and Ca2+-bound states: the calcium sensor mechanism of S100 proteins revealed at atomic resolution. Structure 10:557–567
Gross SR, Sin CG, Barraclough R, Rudland PS (2014) Joining S100 proteins and migration: for better or for worse, in sickness and in health. Cell Mol Life Sci 71:1551–1579
Bresnick AR, Weber DJ, Zimmer DB (2015) S100 proteins in cancer. Nat Rev Cancer 15:96–109
Filipek A, Michowski W, Kuznicki J (2008) Involvement of S100A6 (calcyclin) and its binding partners in intracellular signaling pathways. Adv Enzyme Regul 48:225–239
Fernandez-Fernandez MR, Rutherford TJ, Fersht AR (2008) Members of the S100 family bind p53 in two distinct ways. Protein Sci 17:1663–1670
Yang Q, O’Hanlon D, Heizmann CW, Marks A (1999) Demonstration of heterodimer formation between S100B and S100A6 in the yeast two-hybrid system and human melanoma. Exp Cell Res 246:501–509
Deloulme JC, Assard N, Mbele GO, Mangin C, Kuwano R, Baudier J (2000) S100A6 and S100A11 are specific targets of the calcium- and zinc-binding S100B protein in vivo. J Biol Chem 275:35302–35310
Filipek A, Heizmann CW, Kuźnicki J (1990) Calcyclin is a calcium and zinc binding protein. FEBS Lett 264:263–266
Chen H, Xu C, Jin Q, Liu Z (2014) S100 protein family in human cancer. Am J Cancer Res 4:89–115
Cross SS, Hamdy FC, Deloulme JC, Rehman I (2005) Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology 46:256–269
Ito Y, Yoshida H, Tomoda C, Uruno T, Miya A, Kobayashi K, Matsuzuka F, Kakudo K, Kuma K, Miyauchi A (2005) Expression of S100A2 and S100A6 in thyroid carcinomas. Histopathology 46:569–575
Zhao M, Wang KJ, Tan Z, Zheng CM, Liang Z, Zhao JQ (2016) Identification of potential therapeutic targets for papillary thyroid carcinoma by bioinformatics analysis. Oncol Lett 11:51–58
Ning X, Sun S, Zhang K, Liang J, Chuai Y, Li Y, Wang X (2012) S100A6 protein negatively regulates CacyBP/SIP-mediated inhibition of gastric cancer cell proliferation and tumorigenesis. PLoS One 7:e30185
Chen X, Liu X, Lang H, Zhang S, Luo Y, Zhang J (2015) S100 calcium-binding protein A6 promotes epithelial-mesenchymal transition through β-catenin in pancreatic cancer cell line. PLoS One 10:e0121319
Lyu XJ, Li HZ, Ma X, Li XT, Gao Y, Ni D, Shen DL, Gu LY, Wang BJ, Zhang Y, Zhang X (2015) Elevated S100A6 (Calcyclin) enhances tumorigenesis and suppresses CXCL14-induced apoptosis in clear cell renal cell carcinoma. Oncotarget 6:6656–6669
Wasik U, Kadziolka B, Kilanczyk E, Filipek A (2016) Influence of S100A6 on CacyBP/SIP Phosphorylation and Elk-1 Transcriptional Activity in Neuroblastoma NB2a Cells. Cell Biochem 117:126–131
Bao L, Odell AF, Stephen SL, Wheatcroft SB, Walker JH, Ponnambalam S (2012) The S100A6 calcium-binding protein regulates endothelial cell-cycle progression and senescence. FEBS J 279:4576–4588
Lerchenmüller C, Heißenberg J, Damilano F, Bezzeridis VJ, Krämer I, Bochaton-Piallat ML, Hirschberg K, Busch M, Katus HA, Peppel K, Rosenzweig A, Busch H, Boerries M, Most P (2016) S100A6 regulates endothelial cell cycle progression by attenuating antiproliferative signal transducers and activators of transcription 1 signaling. Arterioscler Thromb Vasc Biol 36:1854–1867
Graczyk A, Leśniak W (2014) S100A6 expression in keratinocytes and its impact on epidermal differentiation. Int J Biochem Cell Biol 57:135–141
Zwadlo G, Briiggen J, Gerhards G, Schlegel R, Sorg C (1988) Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin Exp Immunol 72:510–515
Lagasse E, Weissman IL (1992) Mouse MRP8 and MRP14, two intracellular calcium-binding proteins associated with the development of the myeloid lineage. Blood 79:1907–1915
Arcuri C, Bianchi R, Brozzi F, Donato R (2005) S100B increases proliferation in PC12 neuronal cells and reduces their responsiveness to NGF via Akt activation. J Biol Chem 280:4402–4414
Saito T, Ikeda T, Nakamura K et al (2007) S100A1 and S100B, transcriptional targets of SOX trio, inhibit terminal differentiation of chondrocytes. EMBO Rep 8:504–509
Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C, Deloulme JC (2007) S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia 55:165–677
Brozzi F, Arcuri C, Giambanco I, Donato R (2009) S100B protein regulates astrocyte shape and migration via interaction with Src kinase: implications for astrocyte development, activation and tumor growth. J Biol Chem 284:8797–8811
Tubaro C, Arcuri C, Giambanco I, Donato R (2011) S100B in myoblasts regulates the transition from activation to quiescence and from quiescence to activation, and reduces apoptosis. Biochim Biophys Acta Mol Cell Res 1813:1092–1104
Beccafico S, Riuzzi F, Puglielli C, Mancinelli R, Fulle S, Sorci G, Donato R (2011) Human muscle satellite cells show age-related differential expression of S100B protein and RAGE. Age 33:523–541
Duan L, Wu R, Zou Z, Wang H, Ye L, Li H, Yuan S, Li X, Zha H, Sun H, Zhang Y, Chen X, Zhou L (2014) S100A6 stimulates proliferation and migration of colorectal carcinoma cells through activation of the MAPK pathways. Int J Oncol 44:781–790
Meghnani V, Wagh A, Indurthi VS, Koladia M, Vetter SW, Law B, Leclerc E (2014) The receptor for advanced glycation end products influences the expression of its S100 protein ligands in melanoma tumors. Int J Biochem Cell Biol 57:54–62
Mohan SK, Gupta AA, Yu C (2013) Interaction of the S100A6 mutant (C3S) with the V domain of the receptor for advanced glycation end products (RAGE). Biochem Biophys Res Commun 434:328–333
Yatime L, Betzer C, Jensen RK, Mortensen S, Jensen PH, Andersen GR (2016) The structure of the RAGE: S100A6 complex reveals a unique mode of homodimerization for S100 proteins. Structure 24:2043–2052
Lee YT, Dimitrova YN, Schneider G, Ridenour WB, Bhattacharya S, Soss SE, Caprioli RM, Filipek A, Chazin WJ (2008) Structure of the S100A6 complex with a fragment from the C-terminal domain of Siah-1 interacting protein: a novel mode for S100 protein target recognition. Biochemistry 47:10921–10932
Li Y, Wagner ER, Yan Z, Wang Z, Luther G, Jiang W, Ye J, Wei Q, Wang J, Zhao L, Lu S, Wang X, Mohammed MK, Tang S, Liu H, Fan J, Zhang F, Zou Y, Song D, Liao J, Haydon RC, Luu HH, He TC (2015) The calcium-binding protein S100A6 accelerates human osteosarcoma growth by promoting cell proliferation and inhibiting osteogenic differentiation. Cell Physiol Biochem 37:2375–2392
Kiryushko D, Novitskaya V, Soroka V et al (2006) Molecular mechanisms of Ca2+ signaling in neurons induced by the S100A4 protein. Mol Cell Biol 26:3625–3638
Klingelhöfer J, Møller HD, Sumer EU, Berg CH, Poulsen M, Kiryushko D, Soroka V, Ambartsumian N, Grigorian M, Lukanidin EM (2009) Epidermal growth factor receptor ligands as new extracellular targets for the metastasis-promoting S100A4 protein. FEBS J 276:5936–5948
Vogl T, Tenbrock K, Ludwig S et al (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13:1042–1049
Yan WX, Armishaw C, Goyette J et al (2008) Mast cell and monocyte recruitment by S100A12 and its hinge domain. J Biol Chem 283:13035–13043
Riuzzi F, Sorci G, Donato R (2011) S100B protein regulates myoblast proliferation and differentiation by activating FGFR1 in a bFGFdependent manner. J Cell Sci 124:2389–2400
Leśniak W, Słomnicki ŁP, Filipek A (2009) S100A6—new facts and features. Biochem Biophys Res Commun 390:1087–1092
Graczyk A, Słomnicki LP, Leśniak W (2013) S100A6 competes with the TAZ2 domain of p300 for binding to p53 and attenuates p53 acetylation. J Mol Biol 425:3488–3494
Tamai H, Miyake K, Yamaguchi H, Takatori M, Dan K, Inokuchi K, Shimada T (2012) AAV8 vector expressing IL24 efficiently suppresses tumor growth mediated by specific mechanisms in MLL/AF4-positive ALL model mice. Blood 119:64–71
Tamai H, Miyake K, Yamaguchi H, Shimada T, Dan K, Inokuchi K (2014) Inhibition of S100A6 induces GVL effects in MLL/AF4-positive ALL in human PBMC-SCID mice. Bone Marrow Transplant 49:699–703
Ismail TM, Fernig DG, Rudland PS, Terry CJ, Wang G, Barraclough R (2008) The basic C-terminal amino acids of calcium binding protein S100A4 promote metastasis. Carcinogenesis 29:2259–2266
Schmidt-Hansen B, Ornås D, Grigorian M et al (2004) Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene 23:5487–5495
Forst B, Hansen MT, Klingelhöfer J et al (2010) Metastasis-inducing S100A4 and RANTES cooperate in promoting tumor progression in mice. PLoS One 5:e10374
Al-Haddad S, Zhang Z, Leygue E, Snell L, Huang A, Niu Y et al (1999) Psoriasin (S100A7) expression and invasive breast cancer. Am J Pathol 155:2057–2066
Nasser MW, Qamri Z, Deol YS, Ravi J, Powell CA, Trikha P et al (2012) S100A7 enhances mammary tumorigenesis through upregulation of inflammatory pathways. Cancer Res 72:604–615
Nasser MW, Wani NA, Ahirwar DK, Powell CA, Ravi J, Elbaz M et al (2015) RAGE mediates S100A7-induced breast cancer growth and metastasis by modulating the tumor microenvironment. Cancer Res 75:974–985
Hiratsuka S, Watanabe A, Aburatani H, Maru Y (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermine lung metastasis. Nat Cell Biol 8:1369–1375
Saha A, Lee YC, Zhang Z, Chandra G, Su SB, Mukherjee AB (2010) Lack of an endogenous anti-inflammatory protein in mice enhances colonization of B16F10 melanoma cells in the lungs. J Biol Chem 285:10822–10831
Gaynor R, Irie R, Morton D, Herschman HR (1980) S100 protein is present in cultured human malignant melanomas. Nature 286:400–401
Hauschild A, Engel G, Brenner W, Gläser R, Mönig H, Henze E, Christophers E (1999) S100B protein detection in serum is a significant prognostic factor in metastatic melanoma. Oncology 56:338–344
Lin J, Yang Q, Yan Z, Markowitz J, Wilder PT, Carrier F, Weber DJ (2004) Inhibiting S100B restores p53 levels in primary malignant melanoma cancer cells. J Biol Chem 279:34071–34077
Zhang L, Liu W, Alizadeh D, Zhao D, Farrukh O, Lin J, Badie SA, Badie B (2011) S100B attenuates microglia activation in gliomas: possible role of STAT3 pathway. Glia 59:486–498
Wang H, Zhang L, Zhang IY, Chen X, Da Fonseca A, Wu S, Ren H, Badie S, Sadeghi S, Ouyang M, Warden CD, Badie B (2013) S100B promotes glioma growth through chemoattraction of myeloid-derived macrophages. Clin Cancer Res 19:3764–3775
Holla FK, Postma TJ, Blankenstein MA, van Mierlo TJ, Vos MJ, Sizoo EM, de Groot M, Uitdehaag BM, Buter J, Klein M, Reijneveld JC, Heimans JJ (2016) Prognostic value of the S100B protein in newly diagnosed and recurrent glioma patients: a serial analysis. J Neurooncol 129:525–532
Slomnicki LP, Lesniak W (2010) S100A6 (calcyclin) deficiency induces senescence-like changes in cell cycle, morphology and functional characteristics of mouse NIH 3T3 fibroblasts. J Cell Biochem 109:576–584
Luo X, Sharff KA, Chen J, He TC, Luu HH (2008) S100A6 expression and function in human osteosarcoma. Clin Orthop Relat Res 466:2060–2070
Luu HH, Zhou L, Haydon RC, Deyrup AT, Montag AG, Huo D, Heck R, Heizmann CW, Peabody TD, Simon MA, He TC (2005) Increased expression of S100A6 is associated with decreased metastasis and inhibition of cell migration and anchorage independent growth in human osteosarcoma. Cancer Lett 229:135–148
Nedjadi T, Kitteringham N, Campbell F, Jenkins RE, Park BK, Navarro P, Ashcroft F, Tepikin A, Neoptolemos JP, Costello E (2009) S100A6 binds to annexin 2 in pancreatic cancer cells and promotes pancreatic cancer cell motility. Br J Cancer 101:1145–1154
Ohuchida K, Mizumoto K, Ishikawa N, Fujii K, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M, Tanaka M (2005) The role of S100A6 in pancreatic cancer development and its clinical implication as a diagnostic marker and therapeutic target. Clin Cancer Res 11:7785–7793
Komatsu K, Kobune-Fujiwara Y, Andoh A, Ishiguro S, Hunai H, Suzuki N, Kameyama M, Murata K, Miyoshi J, Akedo H, Tatsuta M, Nakamura H (2000) Increased expression of S100A6 at the invading fronts of the primary lesion and liver metastasis in patients with colorectal adenocarcinoma. Br J Cancer 83:769–774
Guo XJ, Chambers AF, Parfett CL, Waterhouse P, Murphy LC, Reid RE, Craig AM, Edwards DR, Denhardt DT (1990) Identification of a serum-inducible messenger RNA (5B10) as the mouse homologue of calcyclin: tissue distribution and expression in metastatic, ras-transformed NIH 3T3 cells. Cell Growth Differ 1:333–338
Mani RS, Kay CM (1990) Isolation and characterization of a novel molecular weight 11,000 Ca2+-binding protein from smooth muscle. Biochemistry 29:1398–1404
Filipek A, Zasada A, Wojda U, Makuch R, Dabrowska R (1996) Characterization of chicken gizzard calcyclin and examination of its interaction with caldesmon. Comp Biochem Physiol B: Biochem Mol Biol 113:745–752
Donato R (1983) Effect of S-100 protein on assembly of brain microtubule proteins in vitro. FEBS Lett 162:310–313
Donato R (1987) Quantitative analysis of the interaction between S-100 proteins and brain tubulin. Cell Calcium 8:283–297
Bianchi R, Giambanco I, Donato R (1993) S-100 protein, but not calmodulin, binds to and inhibits the polymerization of the glial fibrillary acidic protein in a Ca2+-dependent manner. J Biol Chem 268:12669–12674
Sorci G, Agneletti AL, Bianchi R, Donato R (1998) Association of S100B with intermediate filaments and microtubules in glial cells. Biochim Biophys Acta 1448:277–289
Sorci G, Agneletti AL, Donato R (2000) Effects of S100A1 and S100B on microtubule stability. An in vitro study using triton-cytoskeletons from astrocyte and myoblast cell lines. Neuroscience 99:773–783
Garbuglia M, Verzini M, Giambanco I, Spreca A, Donato R (1996) Effects of calcium-binding proteins (S100a0, S100a, S100b) on desmin assembly in vitro. FASEB J 10:317–324
Heierhorst J, Kobe B, Feil SC, Parker MW, Benian GM, Weiss KR, Kemp BE (1996) Ca2+/S100 regulation of giant protein kinases. Nature 380:636–639
Yamasaki R, Berri M, Wu Y, Trombitás K, McNabb M, Kellermayer MS, Witt C, Labeit D, Labeit S, Greaser M, Granzier H (2001) Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J 81:2297–2313
Most P, Pleger ST, Völkers M et al (2004) Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Investig 114:1550–1563
Malashkevich VN, Dulyaninova NG, Ramagopal UA et al (2010) Phenothiazines inhibit S100A4 function by inducing protein oligomerization. Proc Natl Acad Sci USA 107:8605–8610
Roth J, Burwinkel F, van den Bos C, Goebeler M, Vollmer E, Sorg C (1993) MRP8 and MRP14, S-100-like proteins associated with myeloid differentiation, are translocated to plasma membrane and intermediate filaments in a calcium-dependent manner. Blood 82:1875–1883
Goebeler M, Roth J, van den Bos C, Ader G, Sorg C (1995) Increase of calcium levels in epithelial cells induces translocation of calcium-binding proteins migration inhibitory factor-related protein 8 (MRP8) and MRP14 to keratin intermediate filaments. Biochem J 309:419–424
Vogl T, Ludwig S, Goebeler M, Strey A, Thorey IS, Reichelt R, Foell D, Gerke V, Manitz MP, Nacken W, Werner S, Sorg C, Roth J (2004) MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 104:4260–4268
Yamada J, Jinno S (2014) S100A6 (calcyclin) is a novel marker of neural stem cells and astrocyte precursors in the subgranular zone of the adult mouse hippocampus. Hippocampus 24:89–101
Acosta S, Mayol G, Rodríguez E, Lavarino C, de Preter K, Kumps C, Garcia I, de Torres C, Mora J (2011) Identification of tumoral glial precursor cells in neuroblastoma. Cancer Lett 312:73–81
Tsoporis JN, Marks A, Haddad A, Dawood F, Liu PP, Parker TG (2005) S100B expression modulates left ventricular remodeling after myocardial infarction in mice. Circulation 111:598–606
Tsoporis JN, Izhar S, Leong-Poi H, Desjardins JF, Huttunen HJ, Parker TG (2010) S100B interaction with the receptor for advanced glycation end products (RAGE): a novel receptor mediated mechanism for myocyte apoptosis postinfarction. Circ Res 106:93–101
Tsoporis JN, Izhar S, Proteau G, Slaughter G, Parker TG (2011) S100B-RAGE dependent VEGF secretion by cardiac myocytes induces myofibroblast proliferation. J Mol Cell Cardiol 52:464–473
Boom A, Pochet R, Authelet M, Pradier L, Borghgraef P, Van Leuven F, Heizmann CW, Brion JP (2004) Astrocytic calcium/zinc binding protein S100A6 overexpression in Alzheimer’s disease and in PS1/APP transgenic mice models. Biochim Biophys Acta 1742:161–168
Hoyaux D, Boom A, Van den Bosch L, Belot N, Martin JJ, Heizmann CW, Kiss R, Pochet R (2002) S100A6 overexpression within astrocytes associated with impaired axons from both ALS mouse model and human patients. J Neuropathol Exp Neurol 61:736–744
Botelho HM, Leal SS, Cardoso I, Yanamandra K, Morozova-Roche LA, Fritz G, Gomes CM (2012) S100A6 amyloid fibril formation is calcium-modulated and enhances superoxide dismutase-1 (SOD1) aggregation. J Biol Chem 287:42233–42242
Akiyama H, Ikeda K, Katoh M, McGeer EG, McGeer PL (1994) Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol 50:195–201
Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, Tubaro C, Giambanco I (2009) S100B’s double life: intracellular regulator and extracellular signal. Biochim Biophys Acta 1793:1008–1022
de Souza DF, Leite MC, Quincozes-Santos A, Nardin P, Tortorelli LS, Rigo MM, Gottfried C, Leal RB, Gonçalves CA (2009) S100B secretion is stimulated by IL-1beta in glial cultures and hippocampal slices of rats: likely involvement of MAPK pathway. J Neuroimmunol 206:52–57
Roltsch E, Holcomb L, Young KA, Marks A, Zimmer DB (2010) PSAPP mice exhibit regionally selective reductions in gliosis and plaque deposition in response to S100B ablation. J Neuroinflammation 7:78
Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Yu AC (2011) Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 8:67–68
Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T (2010) Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 58:300–314
Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, Halliday GM (2006) Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol Aging 27:1554–1563
Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE (2006) Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol 79:596–610
Chang KA, Kim HJ, Suh YH (2012) The role of S100a9 in the pathogenesis of Alzheimer’s disease: the therapeutic effects of S100a9 knockdown or knockout. Neurodegener Dis 10:27–29
Zhang C, Liu Y, Gilthorpe J, van der Maarel JR (2012) MRP14 (S100A9) protein interacts with Alzheimer beta-amyloid peptide and induces its fibrillization. PLoS One 7:e32953
Kummer MP, Vogl T, Axt D, Griep A, Vieira-Saecker A, Jessen F, Gelpi E, Roth J, Heneka MT (2012) Mrp14 deficiency ameliorates amyloid β burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J Neurosci 32:17824–17829
Wilcock DM, Griffin WS (2013) Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation 10:84
Kim HJ, Chang KA, Ha TY, Kim J, Ha S, Shin KY, Moon C, Nacken W, Kim HS, Suh YH (2014) S100A9 knockout decreases the memory impairment and neuropathology in crossbreed mice of Tg2576 and S100A9 knockout mice model. PLoS One 9:e88924
Gruden MA, Davydova TV, Wang C, Narkevich VB, Fomina VG, Kudrin VS, Morozova-Roche LA, Sewell RD (2016) The misfolded pro-inflammatory protein S100A9 disrupts memory via neurochemical remodelling instigating an Alzheimer’s disease-like cognitive deficit. Behav Brain Res 306:106–116
Wang T, Liang Y, Thakur A, Zhang S, Yang T, Chen T, Gao L, Chen M, Ren H (2016) Diagnostic significance of S100A2 and S100A6 levels in sera of patients with non-small cell lung cancer. Tumor Biol 37:2299–2304
Zhang J, Zhang K, Jiang X, Zhang J (2014) S100A6 as a potential serum prognostic biomarker and therapeutic target in gastric cancer. Dig Dis Sci 59:2136–2144
Nishi M, Matsumoto K, Kobayashi M, Yanagita K, Matsumoto T, Nagashio R, Ishii D, Fujita T, Sato Y, Iwamura M (2014) Serum expression of S100A6 is a potential detection marker in patients with urothelial carcinoma in the urinary bladder. Biomed Res 35:351–356
Wei BR, Hoover SB, Ross MM, Zhou W, Meani F, Edwards JB, Spehalski EI, Risinger JI, Alvord WG, Quiñones OA, Belluco C, Martella L, Campagnutta E, Ravaggi A, Dai RM, Goldsmith PK, Woolard KD, Pecorelli S, Liotta LA, Petricoin EF, Simpson RM (2009) Serum S100A6 concentration predicts peritoneal tumor burden in mice with epithelial ovarian cancer and is associated with advanced stage in patients. PLoS One 4:e7670
Cai XY, Lu L, Wang YN, Jin C, Zhang RY, Zhang Q, Chen QJ, Shen WF (2011) Association of increased S100B, S100A6 and S100P in serum levels with acute coronary syndrome and also with the severity of myocardial infarction in cardiac tissue of rat models with ischemia-reperfusion injury. Atherosclerosis 217:536–5342
Mocellin S, Zavagno G, Nitti D (2008) The prognostic value of serum S100B in patients with cutaneous melanoma: a meta-analysis. Int J Cancer 123:2370–2376
McIlroy M, McCartan D, Early S et al (2010) Interaction of developmental transcription factor HOXC11 with steroid receptor coactivator SRC-1 mediates resistance to endocrine therapy in breast cancer [corrected]. Cancer Res 70:1585–1594
Gautam P, Nair SC, Gupta MK, Sharma R, Polisetty RV, Uppin MS, Sundaram C, Puligopu AK, Ankathi P, Purohit AK, Chandak GR, Harsha HC, Sirdeshmukh R (2012) Proteins with altered levels in plasma from glioblastoma patients as revealed by iTRAQ-based quantitative proteomic analysis. PLoS One 7:e46153
Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL (2013) Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol 190:794–804
Sun W, Xing B, Guo L, Liu Z, Mu J, Sun L, Wei H, Zhao X, Qian X, Jiang Y, He F (2016) Quantitative proteomics analysis of tissue interstitial fluid for identification of novel serum candidate diagnostic marker for hepatocellular carcinoma. Sci Rep 6:26499
Acknowledgements
This work was supported by Associazione Italiana Ricerca sul Cancro (Project No. Project 17581). The authors wish to thanks the reviewers for criticism and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Donato, R., Sorci, G. & Giambanco, I. S100A6 protein: functional roles. Cell. Mol. Life Sci. 74, 2749–2760 (2017). https://doi.org/10.1007/s00018-017-2526-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2526-9