
Abstract
The insulin-secreting beta cells in the endocrine pancreas regulate blood glucose levels, and loss of functional beta cells leads to insulin deficiency, hyperglycemia (high blood glucose) and diabetes mellitus. Current treatment strategies for type-1 (autoimmune) diabetes are islet transplantation, which has significant risks and limitations, or normalization of blood glucose with insulin injections, which is clearly not ideal. The type-1 patients can lack insulin counter-regulatory mechanism; therefore, hypoglycemia is a potential risk. Hence, a cell-based therapy offers a better alternative for the treatment of diabetes. Past research was focused on attempting to generate replacement beta cells from stem cells; however, recently there has been an increasing interest in identifying mechanisms that will lead to the conversion of pre-existing differentiated endocrine cells into beta cells. The goal of this review is to provide an overview of several of the key factors that regulate new beta cell formation (neogenesis) and beta cell proliferation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is associated with a progressive loss of beta cell mass and function, including the inability to generate new functional pancreatic beta cells through neogenesis or proliferation of existing beta cells in response to glycemic challenges. Type 2 diabetes is the predominant form found in the adult population worldwide, arising due to insulin resistance, while type 1 diabetes is an autoimmune disease entailing beta cell destruction. A 2014 report by World Health Organization showed that diabetes has reached an epidemic proportion, approximately 9 % of the world’s adult population over 18 are obese and diabetic, and this number is expected to increase exponentially soon, making it the seventh leading cause of death by 2030. This massive increase in the number of diabetic patients has been an immense strain on other effective therapeutic strategies, such as pancreas transplant, due to lack of sufficient available donor islet tissue.
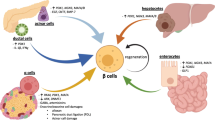
However, the past two decades have seen significant advances in pancreas research, particularly identifying several key factors that regulate pancreas development, opening the possibility of generating functionally suitable engineered beta cells. These studies have produced vital knowledge regarding several key factors that regulate endocrine development in the embryonic pancreas (through endocrine cell neogenesis from progenitor cells) and subsequently regulate proliferation to expand the beta cell mass. There has been an attempt to apply this embryonic developmental program to the adult pancreas as a way to generate new functional beta cells for the treatment of diabetes, either through neogenesis from stem cells or transdifferentiation of non-beta cells within the pancreas. Many studies have been encouraging, and have identified the significant plasticity of differentiated cells within the pancreas (Fig. 1). Recent studies [1–3] found that following an extreme loss of beta cells, the alpha cells could reprogram to become functional insulin-positive cells capable of maintaining blood glucose (Fig. 1). Other studies have shown that a single molecule (Pax-4) can convert pancreatic progenitor cells into alpha cells, and then subsequently into beta cells [1]. Alternatively, a combination of the transcription factors Maf-A, neurogenin-3 (Ngn-3) and Pdx-1, ectopically expressed, can induce reprogramming of differentiated pancreatic acinar cells into beta cells (Fig. 1) [4], demonstrating that cell phenotypes in the pancreas can be changed by ectopic expression of transcription factors. This review provides an overview of several of the factors that have been shown to have an essential role in beta cell neogenesis, both during development and in the adult islet.

A schematic representation of potential sources for beta cell proliferation and neogenesis. Accumulating data now suggest that in the early embryonic pancreas glucagon or GLP-1 directly influence progenitor cells to become endocrine cells. Alpha cells have now been shown to be a source for beta cells regeneration following an extreme loss of beta cells, where alpha cells transdifferentiate to become functional beta cells, and maintain blood glucose in mice. In most other scenarios, where the loss of beta cells is only partial, the recovery seems to be through the proliferation of existing beta cells. However, some studies have shown that ductal cells are the source of regenerating beta cells following partial loss of beta cells. New findings show that growth factors, insulinotrophic factors, or a combination of beta cell specific transcription factors can convert progenitor cells or differentiated cells within the pancreas into insulin-producing cells
Transcription factors regulating pancreas development
Pancreatic development and subsequent pancreatic function are highly regulated by a series of transcription factors, incretins and growth factors. The pancreatic duodenal homeobox-1 (Pdx1), a transcription factor that has a crucial role in the adult islet for beta cell function, was identified as one of the master regulators of pancreas development [5, 6]. Pdx-1 regulates the early development and commitment of certain progenitor cells of the foregut to form a pancreatic bud. Pdx-1-expressing progenitor cells give rise to all pancreatic cell types in the pancreas [4] (Fig. 2). The absence of pdx-1 expression during development results in pancreas agenesis and the pups die soon after they are born [7, 8]. Similarly, pancreas-specific transcription factor-1a (Ptf1A) is expressed in early progenitor cells within the pancreas, and in the mice loss of ptf1A expression during development produced pancreas agenesis, although endocrine islets were found in the spleen [9, 10]. Downstream of pdx-1 expression, paired box 6 (pax6) has been identified as an early marker of endocrine-committed cells in the embryonic mouse pancreas, and lineage-tagging studies have shown that early pax6 expression is a marker of all islet endocrine cell progenitors. Loss of pax6 expression led to the development of a pancreas without alpha cells, and subsequently formation of a highly disorganized islet, and an overall reduction in the number of other islet cells [11, 12]. Beyond pax6, endocrine pancreatic commitment and β-cell commitment entail expression of a relatively well-defined hierarchy of transcription factors, including the basic helix loop helix transcription factor neurogenin-3 (ngn3), and neuroD, pax4, nkx2.2, mafB, mafA, nkx6.1, nkx6.2, etc., each affecting downstream developmental and functional aspects of the endocrine pancreas (Fig. 2). Lack of ngn3 during development led to the formation of a pancreas without an endocrine component, and the embryos die soon after birth [13]. Targeted disruption of neuroD in mice caused perinatal lethality, and further analysis revealed an impaired islet formation and maturation with a significant reduction in the number of beta cells [14]. Loss of Pax4 expression results in a lack of beta and delta (somatostatin-producing) cell development in the pancreas, and pups die after birth due to diabetes [15]. Many other transcription factors regulate the expansion and maturation of beta cells specifically. Nkx2.2 mutants failed to develop beta cells and die soon after birth due to severe hyperglycemia [16], whereas nkx6.1 led to reduced mature beta cell neogenesis from progenitor cells in the embryo, as well as poor expansion of beta cells later postnatally [17]. The Maf family proteins, MafA, and MafB also have a central role in later development and maturation of endocrine cells [18–20]. In the embryonic pancreas, a significant portion of insulin-positive cells express Maf-B, and as part of the beta cell maturation process, these cells transitioned from MafB+/MafA−/Ins+ intermediate cells to MafB−/MafA+/Ins+ mature beta cells, and this transition also coincided with a high level of pdx1 expression in these now mature cells [21]. Mature beta cells express Pdx-1, Pax4, Nkx 2.2, Nkx 6.1 and MafA (Fig. 2) [19]. Although the MafB(−/−) null mutant embryonic pancreas had reduced numbers of insulin and glucagon-positive cells, the total number of endocrine cells appeared to remain the same [19, 20]. MafA−/− mutants, on the other hand, had apparently normal endocrine development, and some studies have suggested that MafA functions and works in conjunction with MafB, and the primary role for MafA is in the maturation of beta cells [19, 20, 22].

The pancreas develops from the foregut endoderm as a dorsal and ventral buddings between gestational days 8.5 and 9.5. There is a downregulation of sonic hedgehog and an upregulation of pdx-1 in the pancreatic anlage within the foregut endoderm during this budding process. From gestational day 9.5 onwards, the dorsal bud contains multipotent progenitors that are ptf1A and pdx1 positive, and can give rise to all cell types in the mature pancreas. Around gestational days 11 and 12, the dorsal bud begins to branch out and segregate the progenitors into tip and trunk progenitors. The tip progenitors, under the control of acinar-specific transcription factors, can give rise to acinar cells. Some trunk progenitors transiently express neurogenin-3 and form the endocrine compartments of the mature pancreas, consisting of alpha, beta, delta, PP and ghrelin-expressing cells. The neurogenin-3 negative trunk progenitors, on the other hand, form the ductal network in the pancreas
TGF-β pathways regulating pancreas development and beta cell mass
TGF-β signaling in the pancreas
Transforming growth factor-β (TGF-β) superfamily proteins regulate cell function over a broad spectrum of cell and organ development. TGF-β signaling can either induce or inhibit cell proliferation [23–25]. The canonical signaling is mediated through activation of the cytoplasmic Smad proteins by phosphorylation and translocation of the resulting phopho-smads into the nucleus, where they regulate transcription of downstream targets (Fig. 3) [26, 27]. Expansion and renewal of pancreatic beta cells are crucial for both normal development of the pancreas and maintenance of function in the adult islet. Increased workload on the beta cell has been shown to increase beta cell DNA synthesis in vitro in cultured islets, as well in in vivo models [28] (Fig. 1). Also, there is a large volume of evidence that indicate that paracrine regulation by trophic factors can stimulate growth and expansion of beta cells. Recently, several studies have identified some of the key roles for TGF-β signaling in the developing pancreas. Specifically, TGF-β signaling promotes the endocrine commitment of progenitor cells and their subsequent maturation [29, 30].

TGFβ receptor signaling is initiated by the binding of TGFβ ligands to the TGFβ type II receptor (TGFbRII) to recruit and catalyze phosphorylation of the TGFβ type I receptor (TGFbRI), which subsequently phosphorylates the transcription factors smad2 and/or smad3. Phosphorylated smad2 and/or smad3 generate a complex with the common (CO-) smad, smad4, and then the smad-CO-smad4 complex translocates to the nucleus and modulates gene expression to regulate cellular functions, e.g., cell cycle control. Smad7 is a potent inhibitor of TGFβ receptor signaling. Smad7 can block R-smad phosphorylation, degrade type I receptors, and even exert an inhibitory effect on the nucleus. Smad2/3 nuclear translocation can similarly be affected by other non-superfamily pathways, e.g., activation of epidermal growth factor receptor (EGFR) signaling pathway by M2 macrophages in inflammation following an injury. Macrophages can directly boost smad7 expression which, in turn, can inhibit smad2/3 translocation and induce beta cell proliferation
TGF-β superfamily ligands like TGF-β1, activins, and bone morphogenetic proteins (BMPs) bind to Type II surface receptors, which then recruit Type I receptors, which then phosphorylate the intracellular mediators, the smads (Fig. 3). Phosphorylation leads to activation of receptor-associated-smads (R-smads), which then form a complex with the common smad (Co-smad, smad4) which then translocates to the nucleus to alter gene transcription, often in association with other transcriptional modifiers (Fig. 3). R-smads 2 and 3 have been shown to directly control endocrine differentiation in the pancreas [31, 32]. Inhibitory smads 6 and 7 suppress phosphorylation of the smads. Smad6 targets the BMP-associated R-smads 1,5, and 8, whereas smad7 targets all receptor-activated smads, including smads 2 and 3, implicated in endocrine differentiation (Fig. 3).
TGF-β signaling during pancreas development
In the developing pancreas, the presence of activated components of TGF-β signaling has been reported throughout the embryonic period. During early stages of development several TGF-β isoform receptors (RI or Alk5, RII, and AlK1) were localized to both the pancreatic epithelium and mesenchyme, but then with increasing age localized to the pancreatic islets and ducts [33]. The mesenchymal compartment of the embryonic pancreas expressed TGF-β receptor antagonists such as follistatin, while the epithelium of the pancreas predominantly expressed receptor ligands [34–36]. In vitro culture of the epithelium without mesenchyme produced only endocrine cells. Culturing epithelium and mesenchyme together, but asymmetrically induced acinar cells in the part of the epithelium that was in direct contact with the mesenchyme, while endocrine cells formed in the epithelium away from the mesenchyme [37, 38]. Similarly, adding exogenous activin, or the BMP inhibitor noggin in vitro into the culture medium induced endocrine differentiation in the embryonic epithelium [39], while follistatin in a similar in vitro culture system inhibited endocrine differentiation [35].
TGF-β2 and TGF-β3 homozygous mutants develop severe embryonic developmental defects and 100 % embryonic lethality, making it difficult to identify the roles of these molecules in vivo [40, 41]. Furthermore, disrupting the TGF-β1 gene leads to severe multifocal inflammatory diseases, thus confounding analyzes of different tissues [42]. The use of a dominant negative form of TGF-beta receptor 2, the DNTβRII transgene, allowed for a less complete inactivation of the receptor and helped identify a possible role for TGF-β signaling in regulating pancreatic endocrine expansion and maturation during development [43–45]. Using mice overexpressing the dominant negative form of TGF-β type II receptor Tulachan et al. [45] inhibited TGF-β signaling at the receptor level and found an increase in the number of endocrine precursors, as well as a proliferation of endocrine cells. At E11.5 and E12.5, there was no gross morphological or a histological difference between the DNTβRII transgenic embryos and wild-type littermates. However, by mid-gestation between E14.5 and E16.5, when there is normally a robust expansion and differentiation of both exocrine and endocrine cells in the normal embryonic pancreas, the transgenic pancreata showed an exaggerated expansion of the endocrine cells. At E16.5, the phenotype of the DNTβRII embryonic pancreas diverged markedly from wild-type littermates, with an increase in the insulin- and glucagon-positive cells. BrdU incorporation assay at E16.5 revealed an enhanced proliferation of endocrine cells in the transgenic pancreas compared to littermate wild-type controls. The study showed an important role for TGF-β signaling in maintaining the differentiated state of endocrine cells in the pancreas.
Also with TGF-β type 2 receptor inhibition, there was an increase in the accumulation of endocrine cells around the developing ducts during mid-late stage gestation. BrdU proliferation assay again showed that the periductal cells were proliferating at a significantly higher rate in the transgenic pancreas at E16.5 compared with controls [45]. The majority of the periductal cells were endocrine, mostly expressing insulin or glucagon, and many of these periductal cells were positive for the pro-endocrine markers neurogenin-3 and neuroD1. In vitro inhibition of TGF-β signaling in an explant culture using a pan-neutralizing antibody against the type II receptor produced similar results, with an increase in mature endocrine cells, including beta cells as evidenced by co-localization with pdx1 [45].
In vitro, recombining wild-type epithelium with wild-type mesenchyme led to normal differentiation, and immunostaining showed the expected amount of insulin-positive cells in a wild-type pancreas with a large amount of amylase staining. However, when the transgenic (TGF-βRII blocked) epithelium was recombined with either wild-type or transgenic mesenchyme, there was an augmentation in the number of insulin-positive cells. A further enhancement in the augmentation of the insulin-positive area was seen when transgenic (TGF-βRII blocked) mesenchyme was recombined with wild-type epithelium. These mix-and-match studies using wild-type and transgenic epithelium and mesenchyme suggest that TGF-β receptor II signaling is important in both the epithelium and the mesenchyme (and perhaps more so in the mesenchyme) for restraining the growth and differentiation of pancreatic endocrine cells.
Expression of smads and their role in pancreas development
Downstream transcription factor mediators of TGF-β signaling include smads 2 and 3, which are expressed in the developing pancreas [46]. The activated phosphorylated forms (p-smad2/3) showed diffuse expression in the gestational day 11.5 embryonic mouse pancreas, both in the mesenchyme and epithelium. Interestingly, a day later at E12.5 p-smad2/3 was found to localize specifically in the nucleus of newly formed glucagon and insulin-positive cells, that pattern became persistent, and more prominent by E18.5, and persisted in the adult 10-week-old mouse pancreas. The nuclear localization of the p-smads in these endocrine-committed, hormone-positive, non-proliferating cells suggests a role for activated p-smads in endocrine maturation in the early embryo [47]. In vitro, blocking either smad2 or smad3 with antisense treatment significantly enhanced the insulin- and glucagon-positive cell numbers, to 1.7-fold with smad2 inhibition, and 2-fold with smad3 inhibition [47]. BrdU incorporation assay showed that the percentage of endocrine cells positive for BrdU was doubled after treatment with smad2 or smad3 antisense. A similar expansion of endocrine cells containing a large proportion of BrdU and Nkx2.2-positive cells was found with short term inhibition of smads2/3 in culture, suggesting that smad2 and smad3 may have a significant role in both suppressing recruitment of new progenitors, and in promoting endocrine cell maturation. In vivo, after deleting smad2 expression in pdx-1 positive cells using a tamoxifen-inducible pdx1-cre-ERT;R26R-tomato;smad2 fx/fx mouse [4], at E13.5, greater than 95 % of pancreatic epithelial and endocrine cells were lineage tagged after a single E10.5 tamoxifen injection, and therefore likely smad2−/−. By gestational day 17 and 18, there was a 20 % increase in the number of insulin-positive cells, and a 40 % increase in the number of glucagon-positive cells. In global smad3 null mutant mice there was an even greater (50 %) increase in the number of endocrine cells, with a relatively equal percentage increase in insulin and glucagon-positive cells. Similarly, Nodal, a TGF-β superfamily member, is expressed and shown to lead to phosphorylation of smad2 in the embryonic pancreas and adult islets. Blocking nodal in the Ins-1 cell line inhibited cell proliferation and induced apoptosis, suggesting that TGF-β signaling in endocrine progenitor cells is antiproliferative [48]. The expansion and proliferation of endocrine cells after inhibition of smad2/3 suggests a potential mechanism for expansion of endocrine hormone-positive cells during development or when needed.
A role for Smad7 during development
Inhibitory smad7 negatively regulates TGF-β signaling, but recent evidence also suggests that Smad7 can interact with TGF-β-independent signaling pathways, potentially expanding its role. In the pancreas, Smad7 was present throughout the early (E13.5) pancreatic epithelium, mostly co-localizing with E-cadherin-positive epithelial cells. Only a few hormone-positive endocrine cells co-localized with smad-7, those possibly representing immature newly formed endocrine cells. The smad7 co-localization became weaker within hormone-positive endocrine cells by mid-gestation (E15.5), but persisted in the E-cadherin+/hormone− cells. This transition of smad7 expression from being positive in a few early hormone-positive, immature endocrine cells at E13.5, to being reduced in late gestational, more mature hormone-positive cells, is consistent with a role for smad7 in suppressing smad2/3 in immature endocrine cells. In the embryonic pancreas, Smad7 positive cells are BrdU+ and often express ngn3, suggesting a role for smad7 in maintaining or expanding the endocrine progenitor pool. Then, in the late gestational pancreas at E16.5, smad7 expression is predominantly localized to the acinar tissue, and by postnatal day 7, smad7 is almost exclusively localized to the acinar tissue. The delicate role smad7 plays in regulating smad2/samd3 signaling during pancreas development is evidenced from both smad7 overexpression and smad7 inhibitory studies. Smad7 overexpression in pdx-1-positive cells led to a greater than 85 % reduction in beta cells, and an associated increase in the number of alpha cells at birth P1 [32]. On the other hand, smad7 inhibition also resulted in the suppression of endocrine development [49]. In vitro, inhibition of smad7 in the early E11.5 embryonic pancreas grown in an explant culture significantly reduced the endocrine cell number. BrdU incorporation assay revealed that there was a significant drop in the percentage of proliferating endocrine cells after smad7 inhibition in vitro. Similarly, deleting smad7 expression in the pdx1 lineage by crossing smad7fx/fx mice with pdx1-cre-ERT mice and giving tamoxifen at E10.5 resulted in a marked reduction in hormone-positive cells at late gestation (E17.5). Thus, smad7-mediated inhibition of smad2/3 may have a role in preventing premature endocrine maturation, which is evident from the fact that blocking smad7 expression in pdx1-positive cells resulted in a significant decrease in the mafB/insulin double-positive immature β-cell population. Normally as beta cells mature, there is a shift in the expression of MafB+/insulin+ cells to a MafB−/mafA+/insulin+ cells [18].
TGF-β signaling in the adult islet
In the adult islet, all three TGF-β isoforms are expressed in the endocrine cells in a diffuse pattern. However, the intensity was higher for TGF-β2 and TGF-β3 in insulin-positive cells. In the exocrine pancreas, most of the acinar cells were positive for TGF-β1, while all three ligands appeared to be equally expressed in the ductal cells [50]. Adult beta cells have very low turnover and a low proliferation rate. However, replacement beta cells form in response to a loss of beta cell mass, either through self-replication of beta cells or through neogenesis. In contrast to its known role during pancreatic development and endocrine cell lineage selection, normal beta cell turnover in the adult islet may be independent of TGF-β superfamily signaling. However, TGF-β has a major role in beta cell maturation during postnatal growth. Disruption of TGF-β signaling in mice lacking the TGF-β ligand Gdf11 prevented terminal differentiation of beta cells [32]. The islets in these mice had an increase in Nkx6.1+/insulin− cells and a greater than 50 % reduction in pancreatic insulin+ cell mass. There was also a 50 % reduction in MafA+ cells in Gdf11 null mice, suggesting defective maturation of differentiating beta cells.
Fifty to sixty percent partial pancreatectomy is a known method to induce a normoglycemic workload increase for existing beta cells. Normally, islet nuclei are strongly positive for phospho-smad2 and 3 and negative for BrdU. However, following partial pancreatectomy, there is a loss of phospho-smad2/3 in many islet cells, and many of these cells are BrdU-positive [51]. Partial pancreatectomy in wild-type mice is known to induce beta cell proliferation compared to sham controls. Partial pancreatectomy in a TGF-β receptor signaling defective mutant mouse with loss of both TGF-β-R1 and TGF-β-R2 (ptf1a-cre;TBR1fx/fx;TBR2fx/fx) in pancreatic cells led to increased proliferation of beta cells compared to wild-type mice that have undergone the same procedure. Thus, this workload-induced beta cell proliferation is clearly not dependent on the TGF-β signaling pathway. In another model, increased workload in existing beta cells by feeding 10 % sucrose to normal mice induced beta cell proliferation in both wild-type and ptf1a-cre;TBR1fx/fx;TBR2fx/fx mice, again suggesting that workload-induced beta cell proliferation is TGFβ signaling independent [28]. Following partial pancreatectomy, there was significant downregulation of phosphorylated TGF-β receptor II in insulin-positive cells, and many insulin-positive cells became positive for smad-7 expression [51]. These cells were also typically BrdU-positive, suggesting that smad7 may be downregulating the smad2/3 expression in beta cells in response to an increased workload induced by partial pancreatectomy. To further demonstrate the role of TGF-β signaling in maintaining a mature endocrine cell status, and its effect on beta cell proliferation in the adult islet, El-Gohary et al. [51] used two separate animal models in which smad2 and/or smad3 signaling are inhibited. First, they performed partial pancreatectomy in pdx1-cre-ERT;smad2fx/fx mice, and found that there was a 50 % increase in BrdU-positive cells within 1 week of partial pancreatectomy, compared to wild-type mice that underwent partial pancreatectomy. The same partial pancreatectomy performed in global smad3−/− mutant mice tripled the number of BrdU+/insulin+ cells. Interestingly, this study found that double smad2/3 mutant mice did not show an additive increase in islet cell proliferation, but was similar to the smad3 mutant mice, suggesting that smad2 may in some way work partially via smad3, explaining the greater effect of the smad3 mutation on islet cell replication.
In contrast to partial pancreatectomy, pancreatic duct ligation (PDL) causes local inflammation in the pancreas and BrdU analysis during the recovery period showed that beta cell proliferation increased significantly after PDL compared to controls [28]. This beta cell proliferation following PDL is TGF-beta signaling dependent. When PDL was performed on ptf1a-cre;TBR1fx/fx;TBR2fx/fx mice and analyzed for beta cell proliferation, there was a nearly complete loss of beta cell proliferation, further confirming that inflammation-induced beta cell proliferation is TGF-beta signaling dependent and opposite to workload increase models [28]. Macrophages have been reported to release cell type-specific signals required for pancreatic regeneration in mice [52, 53]. Following PDL, invading macrophages induce smad7-mediated beta cell proliferation [53]. Xiao et al. [53] showed that the macrophages, specifically M2 macrophages release TGFβ1 to stimulate smad7-mediated beta cell proliferation. Further, they also release EGF to activate EGF receptor signaling, which inhibits TGFβ1-activated smad2 nuclear translocation, resulting in the specific inhibition of cytosolic TGFβ signaling and promoting smad-7 mediated beta cell proliferation [53].
A role for Smad7 in beta cell mass restoration in the adult islet
The normal adult islet continues to express TGFβ receptors and Smad2 and 3, while Smad7 expression is not detectable in glucagon, insulin, PDX-1, somatostatin, nor Glut2-positive cells. Smad7 expression re-emerged in cells in the regenerating islets, and often co-localized with BrdU-positive cells. Interestingly, several pancreatic polypeptide-positive cells, apparently dedifferentiated beta cells, were smad7-positive [51]. One week after pancreatectomy, permanently lineage tagged beta cells (RIP-Cre;R26Rtomato reporter mice) had turned off insulin expression, but then became smad-7-positive, supporting the possibility that pre-existing beta cells de-differentiate (lose insulin) before undergoing proliferation. In a parallel experiment, where alpha cells are permanently labeled using glucagon-cre;R26Rtomato reporter mice, alpha cells did not dedifferentiate or express smad7 following pancreatectomy [51]. Forced, constitutive smad7 expression in the developing pancreas or in otherwise normal islets appears to have a negative effect on growth, differentiation and function of the pancreas [32]. In the embryo, overexpression of smad7 led to pancreatic hypoplasia, while conditional transgenic overexpression of smad7 in pdx-1 positive adult islet cells led to downregulated phospho-smad expression and resulted in disrupted TGFβ signal transduction [32]. Within a few weeks after smad7 expression in the adult islet, the mice developed hyperglycemia and showed impaired glucose tolerance. Interestingly, this beta cell dysfunction completely reversed after withdrawing the smad7. However, the smad7 upregulation seen following partial pancreatectomy was found to be necessary for beta cell proliferation. Inhibiting smad7 expression in adult pdx-1-positive cells led to a complete absence of proliferation of islet cells following pancreatectomy [51]. A similar lack of proliferation of islet cells after pancreatectomy was demonstrated after smad7 inhibition in both ptf1-a lineage and ngn-3 lineage cells [51].
Growth factors and extracellular signaling in islet growth and function
The role of alpha cells in beta cell mass and function
Pancreatic alpha cells have long been thought to be simply modulators of glucose homeostasis. However, several recent studies have shown a significant role for glucagon signaling [54] in regulating pancreatic endocrine cell neogenesis, both in the embryo and from ductal progenitor cells [55] in the adult pancreas. Inhibition of glucagon signaling in the embryonic pancreas, either in vitro or in vivo, will inhibit insulin-positive differentiation [54, 56]. A likely scenario is that glucagon in the embryonic pancreas is an important regulator of the differentiation of other endocrine lineages, particularly insulin cells. Several studies now show the influence of glucagon signaling in the development of other islet cell types, and on pancreatic progenitor cells during normal pancreas development, and during pancreatic regeneration.
Regulation of beta cell development and mass by the glucagon family of peptide signaling molecules
Glucagon and its related peptides are essential regulators of early insulin-positive differentiation, and recent studies have identified glucagon as a key regulator of a pancreatic endocrine progenitor cell population from the pax6-positive lineage. Constitutive transgenic expression of ngn-3 in pdx-1-positive progenitor cells resulted in the development of an endocrine pancreas consisting only of a few alpha cells and no other endocrine cells [57], suggesting that alpha cells are the default endocrine pathway during development. Glucagon-expression in the early embryo is several hundredfold greater than that of insulin, suggesting that the early progenitor cell recruitment, differentiation, and growth of other endocrine cells in the embryonic pancreas may be regulated by this early glucagon-expression [58]. It has been proposed that these early glucagon cells in the embryonic pancreas are immature alpha cells, and they express prohormone convertase 1/3 (PC1/3) [59, 60], which converts proglucagon to glucagon-like peptide (GLP)-1 and GLP-2. PC1/3 is usually expressed in the beta cells, and its expression in early embryonic alpha cells suggests that GLP1 may have a role in recruiting progenitor cells into the endocrine developmental program. Blocking preproglucagon in the embryonic pancreas with antisense in vitro led to inhibition of endocrine differentiation in cultured explants [54]. However, adding a GLP-1 agonist, exendin-4, to these cultures rescued the preproglucagon antisense-mediated inhibition of endocrine differentiation. These results suggest that the glucagon family of peptides has an important role in the recruitment of progenitor cells during early development in the pancreas.
The glucagon family of peptide hormones, which includes glucagon and glucagon-like peptide-1 (GLP-1), is highly conserved in its amino acid sequences throughout evolution. This high degree of conservation of glucagon and GLP-1 amino acid sequences indicates the importance of physiological processes regulated by these peptide hormones. The major known function of glucagon is to maintain blood glucose levels during fasting, whereas GLP-1 functions primarily during feeding as an incretin to stimulate insulin release and to lower blood glucose levels. Cell specific processing of preproglucagon in pancreatic alpha cells primarily leads to the production of glucagon, but immunoreactive GLP-1 is also detectable in rat pancreatic alpha cells [61]. However, in the pancreas, immature or progenitor cells committed to an endocrine lineage primarily expressing proglucagon are also known to express GLP-1 [62]. The role of GLP-1 as an insulinotropic hormone has been discussed in detail [63–66]. Insulin-producing cells generated from embryonic stem cells, when treated with exendin-4, augmented their insulin expression more than fivefold [67], suggesting the possibility that embryonic stem cells may express GLP-1 receptor and that GLP1 may have a role in recruitment and differentiation of progenitor cells during pancreatic development. AR42J cells, which are derived from a chemically induced pancreatic tumor, retain both exocrine and neuroendocrine properties, but normally lack endocrine hormone expression. GLP-1 can induce AR42J cells to differentiate into insulin-, glucagon-, and pancreatic polypeptide-producing cells in vitro [68, 69]. A detailed analysis of this GLP-1 mediated conversion of AR42J cells into insulin-positive cells showed that it is dependent on endogenous TGFβ isoform release by the AR42J cells as a mechanism to promote beta cell formation [68].
Adult glucagon receptor null mutant (GCGR−/−)mice have a decreased fasting blood glucose level and an improved glucose tolerance. These mice have alpha cell hyperplasia [56, 70], and it is believed that the alpha cells are immature, still expressing GLP-1 receptor and GLP-1 [71–73]. Analysis of these mice showed a markedly elevated level of GLP-1 peptide (1–37 form) in adult pancreas (25-fold) and blood (10-fold) [70], suggesting activation of potential compensatory pathways. In addition, GCGR−/− mice are known to have a lower baseline plasma glucose [56, 74] and are resistant to the development of high fat diet-induced hyperinsulinemia, hyperleptinemia, and hepatic steatosis [75]. When the beta cells were destroyed with streptozotocin in GCGR−/− mice, the mice did not develop hyperglycemia [75]. Similar findings were reported in other models, including treatment with high-affinity glucagon-neutralizing antibodies [76, 77], or glucagon receptor antisense oligonucleotides. In all cases, there was improved blood glucose levels for different diabetic models [78, 79]. However, a recent study reported that near total elimination of alpha cells in mice using diphtheria toxin (DT) did not prevent streptozotocin-induced diabetes [80]. Unlike the GCGR null mutant mice, the DT-treated mice with no alpha cells did not have high circulating GLP-1 or glucagon. It is not clear whether the insulin-enhancing effect of GLP-1 is, in fact, responsible for the improved glucose tolerance in GCGR−/− mice. The GLP-1 receptor null mutants have elevated plasma insulin levels and impaired glucose tolerance, and Ali et al. [77] reported in double knockout mice for GCGR and GLP-1 receptor that they have improved or normalized glucose tolerance and plasma insulin levels. Hence, Safina Ali et al. [77] suggest the possibility that the elevated levels of GLP-1 leading to increased GLP-1R signaling are primarily responsible for enhanced beta cell function and improved glucose clearance after i.p. glucose challenge in GCGR−/− mice.
In addition, the GLP-1 receptor null mutant mice showed an increased number of smaller islets, with a centralization of the glucagon cells. This latter finding suggests that GLP-1 may play a role in the proliferation of beta cells after the formation of an early islet [81], but perhaps a lesser role in the generation of new beta cells from ducts or other progenitors. However, transgenic mice overexpressing GLP1R in pancreatic exocrine acinar cells, when treated with gastrin and exendin-4, synergistically promoted beta cell neogenesis, and those new beta cells expressed mature beta cell specific transcription factors, PDX1, NKX6.1 and MafA [82]. Although the level of insulin generated in those new beta cells was comparatively low [83], the GLP1 overexpression in the exocrine pancreas did not cause any negative consequences such as pancreatitis, as has been reported in other models of transdifferentiation [84, 85].
GIP expression in the pancreas
Glucose-dependent insulinotropic polypeptide (GIP) is another incretin, and is a member of a structurally related group of hormones that includes glucagon, glucagon-like peptides, and secretin. GIP and GIP receptor mRNA were both detected in the embryonic pancreas by embryonic day 9.5, and then persisted throughout gestation. GIP was co-expressed with glucagon by immunostaining. The GIP receptor was typically co-expressed with insulin. The prohormone Pro-GIP is expressed in mouse alpha cells, and PC2 activity yields the bioactive form that has a possible role in islet development and survival [86]. Blocking either GIP receptor or GIP ligand in the embryonic pancreas in vitro using antisense led to inhibition of insulin-positive differentiation, as well as a significant decrease in pdx-1 and sox9-positive cells [87]. Transgenic mice expressing a dominant negative form of GIPr (GIPRdn) in beta cells developed severe diabetes with significantly higher blood glucose and insulin, and accompanied by a reduction in beta cell mass [88]. In a separate finding, similar transgenic expression of GIPRdn in beta cells also led to a severe reduction in the postnatal expansion of total islet and beta cell mass due to a decrease in islet neogenesis [89]. Transgenic expression of a similar dominant negative form of GIP receptor in Pigs also led to reduced oral glucose tolerance and delayed insulin secretion [90]. In the transgenic pigs, the beta cell proliferation was reduced significantly, and the beta cell mass was reduced by 35 % at 5 months of age and by 58 % at 16 months [90]. These studies provide direct evidence for a role for GIP-mediated signaling in postnatal islet and beta cell development and neogenesis. Interestingly, Safina Ali et al. [77] reported that the insulinotropic response to GIP was significantly increased in GCGR and GLP-1R double knockout mice.
Beta cell restoration from transdifferentiation and neogenesis
Unlike 60 % partial pancreatectomy, or oral glucose stimulation of beta cell proliferation, where the enhanced workload is not extreme, several studies [1, 3] now show that near complete loss of beta cells is a strong enough trigger to initiate transdifferentiation of existing endocrine cells within the islet to become insulin-positive. Thorel et al. found that within a few weeks after nearly all beta cells were destroyed, new beta cells appeared in the islet and blood glucose normalized in mice [3]. Lineage-tagging of the alpha cells confirmed that the newly formed beta cells were derived from alpha cells through a direct conversion (transdifferentiation) (Fig. 1). Unlike adult beta cells, however, DT-mediated ablation and complete loss of alpha cells did not induce neogenesis of an alpha cell or transdifferentiation of existing endocrine cells into alpha cells in the adult islet [91]. It appears that the number of alpha cells required to maintain glucose homeostasis is very low, and in mice less than 5 % of alpha cells is sufficient [92], perhaps explaining why alpha cell loss is not a trigger for neogenesis of alpha cells. Unlike the other models, where complete or near total alpha cell ablation in adult islets did not induce new alpha cell formation [80, 91, 92] alpha cell loss due to permanent lineage alterations beginning in the embryonic pancreas appears to initiate an ongoing neogenesis program that continues into adult life. Interestingly, unlike the direct conversion of alpha cells into beta cells due to extreme loss of beta cells, the alpha cell neogenesis induced by pax-4 ectopic expression appears to derive from ductal cells [93]. Pancreatic ductal epithelium has long been considered to be a source of progenitor cells for islet cell neogenesis and increased numbers of pdx-1-positive cells within the pancreatic ducts have been described in the regenerating pancreas [55, 94]. Collombat et al. [93] found that following ectopic Pax-4 expression in alpha cells, most of the newly forming endocrine cells that were “over-recruited” due to low glucagon levels were specifically localized to the duct end of the islet, and often these cells co-expressed either glucagon and PP, or glucagon and somatostatin, suggesting an immature endocrine progenitor phenotype. Again, this accumulation of periductal cells was reversed by exogenous glucagon injection, further supporting a role for glucagon in regulating recruitment of ductal progenitor cells to become endocrine cells.
Endothelial factors in regulating pancreas development and islet mass
It is widely accepted that the process of organogenesis and vascularization are intimately related. The endothelium and vascular endothelial growth factor A (VEGF-A) have an important regulatory role in the early developing pancreas. It has been shown that aortic endothelial cells in the embryonic pancreas foster expression of pdx1 and ptf1 in the endothelium [95, 96]. These two molecules are markers of early endoderm commitment to become pancreas. Lammert et al. [96] showed that direct contact with endothelial cells is required for early pancreatic endocrine differentiation. It is not clear whether the direct contact with endothelial cells is the only required stimulus or whether other factors may be necessary for this early differentiation. The early mammalian embryo is growing in a very hypoxic environment, but is still able to undergo rapid growth and organogenesis. The endothelium-induced pancreatic differentiation in vitro in embryonic recombination experiments of aortic endothelium and foregut suggest endothelial cells alone may be sufficient to induce organ development. However, other studies point to the fact that the inflow of blood, with the ensuing increase in oxygen tension, may be a control point for differentiation [97–99]. Like the pancreas, many other developing tissues have been shown to use oxygen as a control point for differentiation [97, 100].
In the adult islet, VEGF is essential for islet vascularization and insulin secretion [101, 102]. In the adult pancreas, VEGF is thought to be predominantly secreted by beta cells, affecting islet function and physiology [101, 102]. In vivo, induction of a persistent hypoglycemia in mice led to the loss of VEGF-A expression and was followed by increased apoptosis of endothelial cells and beta cells, resulting in a significant reduction in beta cell mass [28]. Continuous treatment with exogenous VEGF-A prevented the endothelial and beta cell apoptosis, suggesting a role for VEGF-A in maintaining islet vasculature and beta cell mass [28]. In the human pancreas, duct cells have been shown to secrete VEGF at angiogenic levels in culture [103], and Xiao et al. [104] found that, in contrast to beta cells, the duct cells had a much higher ratio of secreted to intracellular VEGF. Purified duct cells co-transplanted with islets showed a significantly improved vascularization and function compared with islets transplanted alone or islets transplanted with purified duct cells pretreated with VEGF-A shRNA, suggesting that the presence of duct cells in the islet preparation may improve islet transplantation [104]. However, several studies have reported that either inhibiting or overexpressing VEGF signaling in mouse beta cells using the insulin promoter (rat insulin promoter) both led to a loss of blood vessels and hypoxia/ischemia in the islet, but did not affect β-cell proliferation or mass [105, 106]. This paradox may well be due to the expression of non-physiological levels of VEGF using a strong beta cell specific promoter.
Discussion and summary
Pancreas development follows a strict regimen of events regulated by a series of transcription factors and growth factors. Research over the past decade has identified many key factors that regulate pancreas development, and those findings have facilitated several attempts to recapitulate the embryonic program of pancreatic endocrine differentiation in stem cells, non-endocrine cells, as well as in adult islet cells, with some success. For example, a combination of transcription factors, pdx1, ngn3, and mafA, when ectopically expressed in acinar cells, can convert those cells into differentiated beta cells. Other molecules such as GLP1 or its agonist exendin-4 have also been shown to induce conversion of a variety of cells into beta cells. Similar plasticity is found within islet endocrine cells, and recent findings show a recruitment and conversion of alpha cells into functional beta cells after extreme loss of insulin-producing cells.
In the adult islet, beta cell mass is maintained primarily by the proliferation of existing beta cells [107–111]. In all cases reported, when beta cells are challenged by glucose-induced workload, or by the partial loss of beta cells, the primary means of maintaining the beta cell mass is proliferation of existing beta cells [28] (Fig. 1). Interestingly, though the end goal and the means are the same, the factors that regulate this process appear to be different. While inflammation-induced beta cell proliferation is TGF-β signaling dependent, the proliferation of beta cells after a partial pancreatectomy is not. Similarly, any increase in demand for beta cell function due to hyperglycemia also appears to be independent of TGF-β signaling. The studies described in this review showed that the TGF-β signaling pathway plays a major role in beta cell pancreatic development, proliferation, and function [51] (Fig. 3). In the adult islet, beta cell recovery following an inflammation-induced injury is TGF-β receptor signaling dependent [28] (Fig. 2). A complex interaction among intracellular TGF-β regulators, consisting of several smads (smads 7, 2, and 3), appears to control beta cell proliferation after both beta cell loss and in the recovery from an inflammation-induced injury. Specifically and uniquely, smad7 is necessary for beta cell proliferation in both cases [28].
The role of other non-transcription factor intracellular signals, as well as other extracellular signals in control of the differentiation of embryonic pancreatic endocrine cells from progenitor endodermal cell lineages is gaining attention. In general, major developmental signaling pathways such as a notch, hedgehog, FGF’s, TGF-β’s, etc. have all been found to play contributory roles. However, the role of these signaling pathways in embryonic development does not appear to be recapitulated in the mature pancreas during the generation of new pancreatic endocrine cells.
Glucagon hormone expressed in the early embryonic pancreas [112] appears to be necessary for the early differentiation of other pancreatic endocrine cells, especially beta cells [54, 56], whereas loss of glucagon signaling led to the recruitment and differentiation of endocrine progenitor cells, both in the embryonic pancreas and in the developing and regenerating postnatal pancreas [93, 113–115]. Here, the recruited progenitor population of cells was derived from the pax6 lineage (i.e., pancreatic endocrine-specific progenitor cells) [93]. A key finding was that the administration of exogenous glucagon hormone to these mice abrogated the recruitment, confirming that diminished or absent glucagon was the key to inducing endocrine progenitor recruitment [93]. Thus, glucagon appears to be a critical factor whose levels, both in the embryo and in the adult mouse, likely regulate the recruitment of all pancreatic endocrine cells [28]. In vitro inhibition of proglucagon signaling in the embryonic pancreas (E11–13) [54], loss of alpha cells in a Pax6 null mutant mice [12]), or the absence of glucagon function due to absence of prohormone convertase-2 (PC2/pcsk2) [116] or absence of functional glucagon receptor all led to a lack of formation of early insulin-positive cells [56, 70]. Other related members of the glucagon family of proteins such as GIP [87, 88, 90] and GLP-1 [77] have also been shown to have a similar role in endocrine differentiation.
VEGF-A expressed in the adult islet is another factor now shown to play a significant role in maintaining beta cell mass and function. Loss of VEGF-A expression in the islet led to the loss of islet vasculature and a subsequent reduction in beta cell mass and development of hyperglycemia in mice. Here, exogenous VEGF-A restored the beta cell mass and function.
In summary, significant plasticity exists within the islet, and there are several key pathways that may be successfully targeted to maintain beta cell mass as a therapeutic strategy.
Abbreviations
- BMP:
-
Bone morphogenetic proteins
- DT:
-
Diphtheria toxin
- EGFR:
-
Epidermal growth factor receptor
- FGF:
-
Fibroblast growth factor
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- PDL:
-
Pancreatic duct ligation
- VEGF:
-
Vascular endothelial growth factor
- BrdU:
-
Bromodeoxyuridine (5-bromo-2′-deoxyuridine)
- EGF:
-
Epidermal growth factor
- GCGR:
-
Glucagon receptor
- MafA:
-
V-maf musculoaponeurotic fibrosarcoma oncogene homolog A
- MafB:
-
V-maf musculoaponeurotic fibrosarcoma oncogene homolog B
- Ngn3:
-
Neurogenin-3
- Nkx2.2:
-
NK2 homeobox 2
- Nkx6.1:
-
NK6 transcription factor related, locus 1
- Nkx6.2:
-
NK6 transcription factor related, locus 2
- Pax4:
-
Paired box gene 4
- Pax6:
-
Paired box gene 6
- Pdx1:
-
Pancreatic duodenal homeobox-1
- PP:
-
Pancreatic polypeptide
- Ptf1A:
-
Pancreas-specific transcription factor-1a
- TGF:
-
Transforming growth factor
- TGFbRI:
-
Transforming growth factor-β type I receptor
- TGFbRII:
-
Transforming growth factor-β type II receptor
References
Chung CH, Hao E, Piran R, Keinan E, Levine F (2010) Pancreatic beta-cell neogenesis by direct conversion from mature alpha-cells. Stem Cells 28(9):1630–1638
Chung CH, Levine F (2010) Adult pancreatic alpha-cells: a new source of cells for beta-cell regeneration. Rev Diabet Stud 7(2):124–131
Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL (2010) Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464(7292):1149–1154
Gu G, Dubauskaite J, Melton DA (2002) Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129(10):2447–2457
Gradwohl G, Dierich A, LeMeur M, Guillemot F (2000) Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 97(4):1607–1611
Larsson LI, Madsen OD, Serup P, Jonsson J, Edlund H (1996) Pancreatic-duodenal homeobox 1—role in gastric endocrine patterning. Mech Dev 60(2):175–184
Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF (1997) Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 15(1):106–110
Li H, Arber S, Jessell TM, Edlund H (1999) Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet 23(1):67–70
Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK, Goodwin G, Houlston RS (2004) Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 36(12):1301–1305
Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, Kawaguchi M, Terao M, Doi R, Wright CV, Hoshino M, Chiba T, Uemoto S (2008) Reduction of Ptf1a gene dosage causes pancreatic hypoplasia and diabetes in mice. Diabetes 57(9):2421–2431
Dohrmann C, Gruss P, Lemaire L (2000) Pax genes and the differentiation of hormone-producing endocrine cells in the pancreas. Mech Dev 92(1):47–54
St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P (1997) Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 387(6631):406–409
Jenny M, Uhl C, Roche C, Duluc I, Guillermin V, Guillemot F, Jensen J, Kedinger M, Gradwohl G (2002) Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J 21(23):6338–6347
Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ (1997) Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev 11(18):2323–2334
Brink C, Chowdhury K, Gruss P (2001) Pax4 regulatory elements mediate beta cell specific expression in the pancreas. Mech Dev 100(1):37–43
Sussel L, Kalamaras J, Hartigan-O’Connor DJ, Meneses JJ, Pedersen RA, Rubenstein JL, German MS (1998) Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 125(12):2213–2221
Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M (2000) Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 127(24):5533–5540
Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R (2010) MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 59(10):2530–2539
Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, Sharma A (2006) A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev Biol 293(2):526–539
Abdellatif AM, Ogata K, Kudo T, Xiafukaiti G, Chang YH, Katoh MC, El-Morsy SE, Oishi H, Takahashi S (2015) Role of large MAF transcription factors in the mouse endocrine pancreas. Exp Anim 64(3):305–312
Nishimura W, Bonner-Weir S, Sharma A (2009) Expression of MafA in pancreatic progenitors is detrimental for pancreatic development. Dev Biol 333(1):108–120
Nishimura W, Rowan S, Salameh T, Maas RL, Bonner-Weir S, Sell SM, Sharma A (2008) Preferential reduction of beta cells derived from Pax6-MafB pathway in MafB deficient mice. Dev Biol 314(2):443–456
Moses HL, Arteaga CL, Alexandrow MG, Dagnino L, Kawabata M, Pierce DF Jr, Serra R (1994) TGF beta regulation of cell proliferation. Princess Takamatsu Symp 24:250–263
Moses HL, Coffey RJ Jr, Leof EB, Lyons RM, Keski-Oja J (1987) Transforming growth factor beta regulation of cell proliferation. J Cell Physiol Suppl Suppl 5:1–7
Moses HL, Yang EY, Pietenpol JA (1990) TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell 63(2):245–247
Wrighton KH, Lin X, Feng XH (2009) Phospho-control of TGF-beta superfamily signaling. Cell Res 19(1):8–20
Matsuzaki K (2013) Smad phospho-isoforms direct context-dependent TGF-beta signaling. Cytokine Growth Factor Rev 24(4):385–399
Xiao X, Wiersch J, El-Gohary Y, Guo P, Prasadan K, Paredes J, Welsh C, Shiota C, Gittes GK (2013) TGFbeta receptor signaling is essential for inflammation-induced but not beta-cell workload-induced beta-cell proliferation. Diabetes 62(4):1217–1226
Ungefroren H, Groth S, Sebens S, Lehnert H, Gieseler F, Fandrich F (2011) Differential roles of Smad2 and Smad3 in the regulation of TGF-beta1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: control by Rac1. Mol Cancer 10:67
Ungefroren H, Sebens S, Groth S, Gieseler F, Fandrich F (2011) The Src family kinase inhibitors PP2 and PP1 block TGF-beta1-mediated cellular responses by direct and differential inhibition of type I and type II TGF-beta receptors. Curr Cancer Drug Targets 11(4):524–535
Goulley J, Dahl U, Baeza N, Mishina Y, Edlund H (2007) BMP4-BMPR1A signaling in beta cells is required for and augments glucose-stimulated insulin secretion. Cell Metab 5(3):207–219
Smart NG, Apelqvist AA, Gu X, Harmon EB, Topper JN, MacDonald RJ, Kim SK (2006) Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol 4(2):e39
Gittes GK (2009) Developmental biology of the pancreas: a comprehensive review. Dev Biol 326(1):4–35
Maldonado TS, Kadison AS, Crisera CA, Grau JB, Alkasab SL, Longaker MT, Gittes GK (2000) Ontogeny of activin B and follistatin in developing embryonic mouse pancreas: implications for lineage selection. J Gastrointest Surg 4(3):269–275
Miralles F, Czernichow P, Scharfmann R (1998) Follistatin regulates the relative proportions of endocrine versus exocrine tissue during pancreatic development. Development 125(6):1017–1024
Szabat M, Johnson JD, Piret JM (2010) Reciprocal modulation of adult beta cell maturity by activin A and follistatin. Diabetologia 53(8):1680–1689
Gittes GK, Galante PE, Hanahan D, Rutter WJ, Debase HT (1996) Lineage-specific morphogenesis in the developing pancreas: role of mesenchymal factors. Development 122(2):439–447
Rose MI, Crisera CA, Colen KL, Connelly PR, Longaker MT, Gittes GK (1999) Epithelio-mesenchymal interactions in the developing mouse pancreas: morphogenesis of the adult architecture. J Pediatr Surg 34(5):774–779 (discussion 780)
Zhang YQ, Cleary MM, Si Y, Liu G, Eto Y, Kritzik M, Dabernat S, Kayali AG, Sarvetnick N (2004) Inhibition of activin signaling induces pancreatic epithelial cell expansion and diminishes terminal differentiation of pancreatic beta-cells. Diabetes 53(8):2024–2033
Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J (1995) Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial–mesenchymal interaction. Nat Genet 11(4):415–421
Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T (1997) TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124(13):2659–2670
Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D et al (1992) Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359(6397):693–699
Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM (1997) Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor beta receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res 57(24):5564–5570
Bottinger EP, Jakubczak JL, Roberts IS, Mumy M, Hemmati P, Bagnall K, Merlino G, Wakefield LM (1997) Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J 16(10):2621–2633
Tulachan SS, Tei E, Hembree M, Crisera C, Prasadan K, Koizumi M, Shah S, Guo P, Bottinger E, Gittes GK (2007) TGF-beta isoform signaling regulates secondary transition and mesenchymal-induced endocrine development in the embryonic mouse pancreas. Dev Biol 305(2):508–521
Brorson M, Hougaard DM, Nielsen JH, Tornehave D, Larsson LI (2001) Expression of SMAD signal transduction molecules in the pancreas. Histochem Cell Biol 116(3):263–267
Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD (2000) Control of endodermal endocrine development by Hes-1. Nat Genet 24(1):36–44
Boerner BP, George NM, Targy NM, Sarvetnick NE (2013) TGF-beta superfamily member Nodal stimulates human beta-cell proliferation while maintaining cellular viability. Endocrinology 154(11):4099–4112
El-Gohary Y, Tulachan S, Guo P, Welsh C, Wiersch J, Prasadan K, Paredes J, Shiota C, Xiao X, Wada Y, Diaz M, Gittes G (2013) Smad signaling pathways regulate pancreatic endocrine development. Dev Biol 378(2):83–93
Yamanaka Y, Friess H, Buchler M, Beger HG, Gold LI, Korc M (1993) Synthesis and expression of transforming growth factor beta-1, beta-2, and beta-3 in the endocrine and exocrine pancreas. Diabetes 42(5):746–756
El-Gohary Y, Tulachan S, Wiersch J, Guo P, Welsh C, Prasadan K, Paredes J, Shiota C, Xiao X, Wada Y, Diaz M, Gittes G (2014) A smad signaling network regulates islet cell proliferation. Diabetes 63(1):224–236
Criscimanna A, Coudriet GM, Gittes GK, Piganelli JD, Esni F (2014) Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and beta-cell regeneration in mice. Gastroenterology 147(5):1106–18 e11
Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, Song Z, El-Gohary Y, Prasadan K, Shiota C, Gittes GK (2014) M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci USA 111(13):E1211–E1220
Prasadan K, Daume E, Preuett B, Spilde T, Bhatia A, Kobayashi H, Hembree M, Manna P, Gittes GK (2002) Glucagon is required for early insulin-positive differentiation in the developing mouse pancreas. Diabetes 51(11):3229–3236
El-Gohary Y, Wiersch J, Tulachan S, Xiao X, Guo P, Rymer C, Fischbach S, Prasadan K, Shiota C, Gaffar I, Song Z, Galambos C, Esni F, Gittes GK (2016) Intraislet pancreatic ducts can give rise to insulin-positive cells. Endocrinology 157(1):166–175
Vuguin PM, Kedees MH, Cui L, Guz Y, Gelling RW, Nejathaim M, Charron MJ, Teitelman G (2006) Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology 147(9):3995–4006
Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS (2000) Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127(16):3533–3542
Rall LB, Pictet RL, Williams RH, Rutter WJ (1973) Early differentiation of glucagon-producing cells in embryonic pancreas: a possible developmental role for glucagon. Proc Natl Acad Sci USA 70(12):3478–3482
Lee YC, Damholt AB, Billestrup N, Kisbye T, Galante P, Michelsen B, Kofod H, Nielsen JH (1999) Developmental expression of proprotein convertase 1/3 in the rat. Mol Cell Endocrinol 155(1–2):27–35
Wilson ME, Kalamaras JA, German MS (2002) Expression pattern of IAPP and prohormone convertase 1/3 reveals a distinctive set of endocrine cells in the embryonic pancreas. Mech Dev 115(1–2):171–176
Whalley NM, Pritchard LE, Smith DM, White A (2011) Processing of proglucagon to GLP-1 in pancreatic alpha-cells: is this a paracrine mechanism enabling GLP-1 to act on beta-cells? J Endocrinol 211(1):99–106
Masur K, Tibaduiza EC, Chen C, Ligon B, Beinborn M (2005) Basal receptor activation by locally produced glucagon-like peptide-1 contributes to maintaining beta-cell function. Mol Endocrinol 19(5):1373–1382
Ryan AS, Egan JM, Habener JF, Elahi D (1998) Insulinotropic hormone glucagon-like peptide-1-(7–37) appears not to augment insulin-mediated glucose uptake in young men during euglycemia. J Clin Endocrinol Metab 83(7):2399–2404
Parkes DG, Pittner R, Jodka C, Smith P, Young A (2001) Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 50(5):583–589
Kemp DM, Habener JF (2001) Insulinotropic hormone glucagon-like peptide 1 (GLP-1) activation of insulin gene promoter inhibited by p38 mitogen-activated protein kinase. Endocrinology 142(3):1179–1187
Abraham EJ, Leech CA, Lin JC, Zulewski H, Habener JF (2002) Insulinotropic hormone glucagon-like peptide-1 differentiation of human pancreatic islet-derived progenitor cells into insulin-producing cells. Endocrinology 143(8):3152–3161
Bai L, Meredith G, Tuch BE (2005) Glucagon-like peptide-1 enhances production of insulin in insulin-producing cells derived from mouse embryonic stem cells. J Endocrinol 186(2):343–352
Yew KH, Prasadan KL, Preuett BL, Hembree MJ, McFall CR, Benjes CL, Crowley AR, Sharp SL, Li Z, Tulachan SS, Mehta SS, Gittes GK (2004) Interplay of glucagon-like peptide-1 and transforming growth factor-beta signaling in insulin-positive differentiation of AR42J cells. Diabetes 53(11):2824–2835
Zhou J, Wang X, Pineyro MA, Egan JM (1999) Glucagon-like peptide 1 and exendin-4 convert pancreatic AR42J cells into glucagon- and insulin-producing cells. Diabetes 48(12):2358–2366
Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L, Obici S, Tang B, Holst JJ, Fledelius C, Johansen PB, Rossetti L, Jelicks LA, Serup P, Nishimura E, Charron MJ (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA 100(3):1438–1443
Stanojevic V, Habener JF (2015) Evolving function and potential of pancreatic alpha cells. Best Pract Res Clin Endocrinol Metab 29(6):859–871
Habener JF, Stanojevic V (2013) Alpha cells come of age. Trends Endocrinol Metab 24(3):153–163
Kedees MH, Grigoryan M, Guz Y, Teitelman G (2009) Differential expression of glucagon and glucagon-like peptide 1 receptors in mouse pancreatic alpha and beta cells in two models of alpha cell hyperplasia. Mol Cell Endocrinol 311(1–2):69–76
Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I, de Chavez V, Vuguin PM, Charron MJ, Powers AC, Drucker DJ (2013) Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes 62(4):1196–1205
Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, Ronan J, Liu F, Roy RS, Zhu L, Charron MJ, Zhang BB (2007) Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50(1):142–150
Sloop KW, Cao JX, Siesky AM, Zhang HY, Bodenmiller DM, Cox AL, Jacobs SJ, Moyers JS, Owens RA, Showalter AD, Brenner MB, Raap A, Gromada J, Berridge BR, Monteith DK, Porksen N, McKay RA, Monia BP, Bhanot S, Watts LM, Michael MD (2004) Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest 113(11):1571–1581
Ali S, Lamont BJ, Charron MJ, Drucker DJ (2011) Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest 121(5):1917–1929
Postic C, Magnuson MA (2000) DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26(2):149–150
Chen M, Mema E, Kelleher J, Nemechek N, Berger A, Wang J, Xie T, Gavrilova O, Drucker DJ, Weinstein LS (2011) Absence of the glucagon-like peptide-1 receptor does not affect the metabolic phenotype of mice with liver-specific G(s)alpha deficiency. Endocrinology 152(9):3343–3350
Steenberg VR, Jensen SM, Pedersen J, Madsen AN, Windelov JA, Holst B, Quistorff B, Poulsen SS, Holst JJ (2016) Acute disruption of glucagon secretion or action does not improve glucose tolerance in an insulin-deficient mouse model of diabetes. Diabetologia 59:363–370
Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, Lacas-Gervais S, Ambrosetti D, Benizri E, Hecksher-Sorensen J, Gounon P, Ferrer J, Gradwohl G, Heimberg H, Mansouri A, Collombat P (2013) Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell 26(1):86–100
Sasaki S, Miyatsuka T, Matsuoka TA, Takahara M, Yamamoto Y, Yasuda T, Kaneto H, Fujitani Y, German MS, Akiyama H, Watada H, Shimomura I (2015) Activation of GLP-1 and gastrin signalling induces in vivo reprogramming of pancreatic exocrine cells into beta cells in mice. Diabetologia 58(11):2582–2591
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA (2008) In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455(7213):627–632
Li L, Shen J, Bala MM, Busse JW, Ebrahim S, Vandvik PO, Rios LP, Malaga G, Wong E, Sohani Z, Guyatt GH, Sun X (2014) Incretin treatment and risk of pancreatitis in patients with type 2 diabetes mellitus: systematic review and meta-analysis of randomised and non-randomised studies. BMJ 348:g2366
Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC (2012) Chronic GLP-1 receptor activation by exendin-4 induces expansion of pancreatic duct glands in rats and accelerates formation of dysplastic lesions and chronic pancreatitis in the Kras(G12D) mouse model. Diabetes 61(5):1250–1262
Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T, Zhang T, Wang R, Ao Z, Warnock GL, Kwok YN, Kieffer TJ (2010) Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology 138(5):1966–1975
Prasadan K, Koizumi M, Tulachan S, Shiota C, Lath N, Paredes J, Guo P, El-Gohary Y, Malek M, Shah S, Gittes GK (2011) The expression and function of glucose-dependent insulinotropic polypeptide in the embryonic mouse pancreas. Diabetes 60(2):548–554
Herbach N, Goeke B, Schneider M, Hermanns W, Wolf E, Wanke R (2005) Overexpression of a dominant negative GIP receptor in transgenic mice results in disturbed postnatal pancreatic islet and beta-cell development. Regul Pept 125(1–3):103–117
Herbach N, Bergmayr M, Goke B, Wolf E, Wanke R (2011) Postnatal development of numbers and mean sizes of pancreatic islets and beta-cells in healthy mice and GIPR(dn) transgenic diabetic mice. PLoS One 6(7):e22814
Renner S, Fehlings C, Herbach N, Hofmann A, von Waldthausen DC, Kessler B, Ulrichs K, Chodnevskaja I, Moskalenko V, Amselgruber W, Goke B, Pfeifer A, Wanke R, Wolf E (2010) Glucose intolerance and reduced proliferation of pancreatic beta-cells in transgenic pigs with impaired glucose-dependent insulinotropic polypeptide function. Diabetes 59(5):1228–1238
Shiota C, Prasadan K, Guo P, El-Gohary Y, Wiersch J, Xiao X, Esni F, Gittes GK (2013) Alpha-cells are dispensable in postnatal morphogenesis and maturation of mouse pancreatic islets. Am J Physiol Endocrinol Metab 305(8):E1030–E1040
Thorel F, Damond N, Chera S, Wiederkehr A, Thorens B, Meda P, Wollheim CB, Herrera PL (2011) Normal glucagon signaling and beta-cell function after near-total alpha-cell ablation in adult mice. Diabetes 60(11):2872–2882
Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A (2009) The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 138(3):449–462
Criscimanna A, Speicher JA, Houshmand G, Shiota C, Prasadan K, Ji B, Logsdon CD, Gittes GK, Esni F (2011) Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 141(4):1451–1462, 1462 e1–6
Yoshitomi H, Zaret KS (2004) Endothelial cell interactions initiate dorsal pancreas development by selectively inducing the transcription factor Ptf1a. Development 131(4):807–817
Lammert E, Cleaver O, Melton D (2001) Induction of pancreatic differentiation by signals from blood vessels. Science 294(5542):564–567
Heinis M, Simon MT, Ilc K, Mazure NM, Pouyssegur J, Scharfmann R, Duvillie B (2010) Oxygen tension regulates pancreatic beta-cell differentiation through hypoxia-inducible factor 1alpha. Diabetes 59(3):662–669
Heinis M, Simon MT, Duvillie B (2010) New insights into endocrine pancreatic development: the role of environmental factors. Horm Res Paediatr 74(2):77–82
Shah SR, Esni F, Jakub A, Paredes J, Lath N, Malek M, Potoka DA, Prasadan K, Mastroberardino PG, Shiota C, Guo P, Miller KA, Hackam DJ, Burns RC, Tulachan SS, Gittes GK (2011) Embryonic mouse blood flow and oxygen correlate with early pancreatic differentiation. Dev Biol 349(2):342–349
Fraker CA, Alvarez S, Papadopoulos P, Giraldo J, Gu W, Ricordi C, Inverardi L, Dominguez-Bendala J (2007) Enhanced oxygenation promotes beta-cell differentiation in vitro. Stem Cells 25(12):3155–3164
Brissova M, Shostak A, Shiota M, Wiebe PO, Poffenberger G, Kantz J, Chen Z, Carr C, Jerome WG, Chen J, Baldwin HS, Nicholson W, Bader DM, Jetton T, Gannon M, Powers AC (2006) Pancreatic islet production of vascular endothelial growth factor-a is essential for islet vascularization, revascularization, and function. Diabetes 55(11):2974–2985
Dai C, Brissova M, Reinert RB, Nyman L, Liu EH, Thompson C, Shostak A, Shiota M, Takahashi T, Powers AC (2013) Pancreatic islet vasculature adapts to insulin resistance through dilation and not angiogenesis. Diabetes 62(12):4144–4153
Movahedi B, Gysemans C, Jacobs-Tulleneers-Thevissen D, Mathieu C, Pipeleers D (2008) Pancreatic duct cells in human islet cell preparations are a source of angiogenic cytokines interleukin-8 and vascular endothelial growth factor. Diabetes 57(8):2128–2136
Xiao X, Prasadan K, Guo P, El-Gohary Y, Fischbach S, Wiersch J, Gaffar I, Shiota C, Gittes GK (2014) Pancreatic duct cells as a source of VEGF in mice. Diabetologia 57(5):991–1000
Cai Q, Brissova M, Reinert RB, Pan FC, Brahmachary P, Jeansson M, Shostak A, Radhika A, Poffenberger G, Quaggin SE, Jerome WG, Dumont DJ, Powers AC (2012) Enhanced expression of VEGF-A in beta cells increases endothelial cell number but impairs islet morphogenesis and beta cell proliferation. Dev Biol 367(1):40–54
D’Hoker J, De Leu N, Heremans Y, Baeyens L, Minami K, Ying C, Lavens A, Chintinne M, Stange G, Magenheim J, Swisa A, Martens G, Pipeleers D, van de Casteele M, Seino S, Keshet E, Dor Y, Heimberg H (2013) Conditional hypovascularization and hypoxia in islets do not overtly influence adult beta-cell mass or function. Diabetes 62(12):4165–4173
Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK (2013) No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest 123(5):2207–2217
Georgia S, Bhushan A (2004) Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 114(7):963–968
Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC (2008) Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57(6):1584–1594
Teta M, Rankin MM, Long SY, Stein GM, Kushner JA (2007) Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 12(5):817–826
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429(6987):41–46
Gittes GK, Rutter WJ (1992) Onset of cell-specific gene expression in the developing mouse pancreas. Proc Natl Acad Sci USA 89(3):1128–1132
Brennand K, Huangfu D, Melton D (2007) All beta cells contribute equally to islet growth and maintenance. PLoS Biol 5(7):e163
Nir T, Melton DA, Dor Y (2007) Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 117(9):2553–2561
Herrera PL (2000) Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127(11):2317–2322
Vincent M, Guz Y, Rozenberg M, Webb G, Furuta M, Steiner D, Teitelman G (2003) Abrogation of protein convertase 2 activity results in delayed islet cell differentiation and maturation, increased alpha-cell proliferation, and islet neogenesis. Endocrinology 144(9):4061–4069
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Prasadan, K., Shiota, C., Xiangwei, X. et al. A synopsis of factors regulating beta cell development and beta cell mass. Cell. Mol. Life Sci. 73, 3623–3637 (2016). https://doi.org/10.1007/s00018-016-2231-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2231-0